Abstract
Background: Cutaneous T cell lymphoma (CTCL) is an incurable cancer characterized by elevated p38β and p38γ and downregulated tumor-suppressive p38α.
Objectives: We aimed to identify selective p38β inhibitors and investigate their mechanisms and therapeutic implications in hematologic malignancies.
Methods: A high-throughput screen of Food and Drug Administration (FDA)-approved compounds was conducted to identify p38β inhibitors. In vitro kinase assays, Western blots, scRNA-seq, synergy tests, and mass spectrometry were used. Clinical trial and public datasets were analyzed.
Results: Nilotinib was identified as a selective p38β inhibitor (~25-fold over p38α). Its inhibition of p38β led to compensatory activation of p38γ, driving Epithelial-to-Mesenchymal Transition (EMT) and potential toxicities. Combining Nilotinib with p38γ inhibitors (e.g., CSH71 or Paclitaxel) produced synergistic anti-tumor effects in CTCL and leukemia cells. Clinical and FDA Adverse Event Reporting System (FAERS) data revealed higher toxicity rates for Nilotinib in Ph+ CML and off-target effects in non-CML cancers.
Conclusion: Nilotinib selectively inhibits p38β but activates p38γ, potentially reducing therapeutic selectivity. Combining it with p38γ inhibitors may enhance efficacy and reduce toxicity across hematologic malignancies.
Keywords
Chronic myeloid leukemia, Clinical trials, Cutaneous T cell lymphoma, Nilotinib, Paclitaxel
Abbreviation
CTCL: Cutaneous T Cell Lymphoma; CML: Chronic Myeloid Leukemia; Ph+ / Ph-: Philadelphia chromosome positive/negative; FDA: Food and Drug Administration; MAPK: Mitogen-Activated Protein Kinase; IC50: Half-Maximal Inhibitory Concentration; TKI: Tyrosine Kinase Inhibitor; PBMC: Peripheral Blood Mononuclear Cell; EMT: Epithelial-to-Mesenchymal Transition; LBS: Lipid Binding Site; AAD: All-Around Docking; MMR: Major Molecular Response; MR4.0/MR4.5: Deep/Very Deep Molecular Response - (4-log)/(4.5-log) reduction; SAE: Serious Adverse Event; AE: Adverse Event; ROR: Reporting Odds Ratio; NIH: National Institutes of Health; GIST: Gastrointestinal Stromal Tumor; CSC: Cancer Stem Cell; TGF-β: Transforming Growth Factor Beta; SMAD: SMAD is a gene family and protein domain name, derived from C. elegans Sma and Drosophila Mad; ENESTnd: Evaluating Nilotinib Efficacy and Safety in Clinical Trials—Newly Diagnosed; ECOG-ACRIN: Eastern Cooperative Oncology Group - American College of Radiology Imaging Network; NCI: National Cancer Institute; NCT: National Clinical Trial (identifier from ClinicalTrials.gov)
Introduction
The p38 kinase family comprises four isoforms—p38α(MAPK14), p38β(MAPK11), p38γ(MAPK12), and p38δ(MAPK13)—each playing a unique role in cellular processes including inflammatory responses and cell differentiation. These isoforms have garnered attention in clinical research, particularly in the context of cancer, due to their involvement in tumor progression and response to stress. Among them, the role of p38β role in Cutaneous T cell lymphoma (CTCL), an incurable cancer, is of particular interest. Our previous studies have highlighted a significant increase in p38β gene expression in CTCL patient samples as compared to normal CD4+ T cells [1], suggesting it may be a promising therapeutic target.
A challenge in targeting the p38 pathway for cancer treatment lies in the structural similarity between p38α and p38β. The isoforms share approximately 80% of their amino acids and are about 75% structurally similar [2,3]. Their functionalities, however, are likely to be quite different. p38α is recognized as a tumor suppressor [2–5] and is essential for the survival of many cell types. In contrast, p38β levels increase in T malignancy CTCL. The precise role of p38β in carcinogenesis and progression of cancer, however, is still unclear.
Clinical trials involving non-selective p38 inhibitors that target both p38α and p38β have been hampered by adverse effects and drug resistance. Non-selective p38 inhibitors target both p38α and p38β, leading to adverse effects such as rash, fatigue, nausea, constipation, pruritus, and vomiting, as seen in the first Phase I clinical trial with the oral p38 MAPK inhibitor Ralimetinib (LY2228820 Dimesylate) [6], and in this trial, none of the 89 patients with advanced cancer achieved a complete or partial response. It is likely due to Ralimetinib's targeting of p38α, which is recognized as a tumor suppressor and is essential for the survival of many cell types [2–5].
This underscores the necessity of developing p38β-specific inhibitors for effective CTCL treatments. In our quest for effective treatments for CTCL, we screened a library of Food and Drug Administration (FDA)-approved drugs to identify p38β-specific inhibitors to investigate the impacts of selected drugs in the clinical trials and CTCL cells. In addition, future p38β-targeted therapies should be rigorously evaluated and optimized to minimize pathway-related toxicities and overcome the limitations observed with non-selective p38 inhibition.
Results
Identification of Nilotinib as a potent p38β inhibitor
Given that p38β expression is elevated in CTCL cells, it is a promising therapeutic target. We conducted high-throughput screening of 1,443 FDA-approved compounds to identify specific inhibitors of p38β (Figure 1A). Using an in vitro p38β kinase assay, we identified several inhibitors, focusing on Linifanib, Ponatinib, and Nilotinib for further analysis (Figure 1B). To assess the specificity of these compounds against p38β kinase, we also determined the half-maximal inhibitory concentration (IC50) for p38α, p38γ, and p38δ, respectively (Table 1).
|
|
Flavopiridol |
Linifanib |
Nilotinib |
Ponatinib |
|
P38α |
1.34 |
2.18 |
0.35 |
0.003 |
|
P38β |
1.82 |
0.12 |
0.014 |
0.015 |
|
P38γ |
0.65 |
2.54 |
>10 |
0.23 |
|
P38δ |
0.45 |
1.21 |
4.45 |
0.18 |
We found that Nilotinib is the most potent p38β inhibitor among four p38 isoforms, with an IC50 value of 0.014 µM, showing it is 25-fold more potent against p38β than p38α. For comparison, we included the FDA orphan drug Flavopiridol, previously identified as a pan p38 isoform inhibitor [7]. Nilotinib inhibited p38γ least effectively (IC50>10 µM) and had an IC50 over 4.45 µM for p38δ (Figure 1C).
Nilotinib, an FDA-approved drug for treating imatinib-resistant Chronic Myelogenous Leukemia (CML) [8–10], has been thoroughly studied and been found to bind p38β which was validated by a curated X-ray crystallography study [PDB: 3GP0, DOI: 10.2210/pdb3GP0/pdb]. Based on this structural data, we applied our All-Around Docking (AAD) method [11] to predict the optimal binding site of Nilotinib on p38β. The docking pose revealed three key hydrogen bonds formed with residues E71, M109, and D168 (Figure 1D, left). The analysis confirmed that Nilotinib, along with five other tyrosine kinase inhibitors (TKIs), binds within the ATP-binding pocket of p38β (Figure 1D, right).
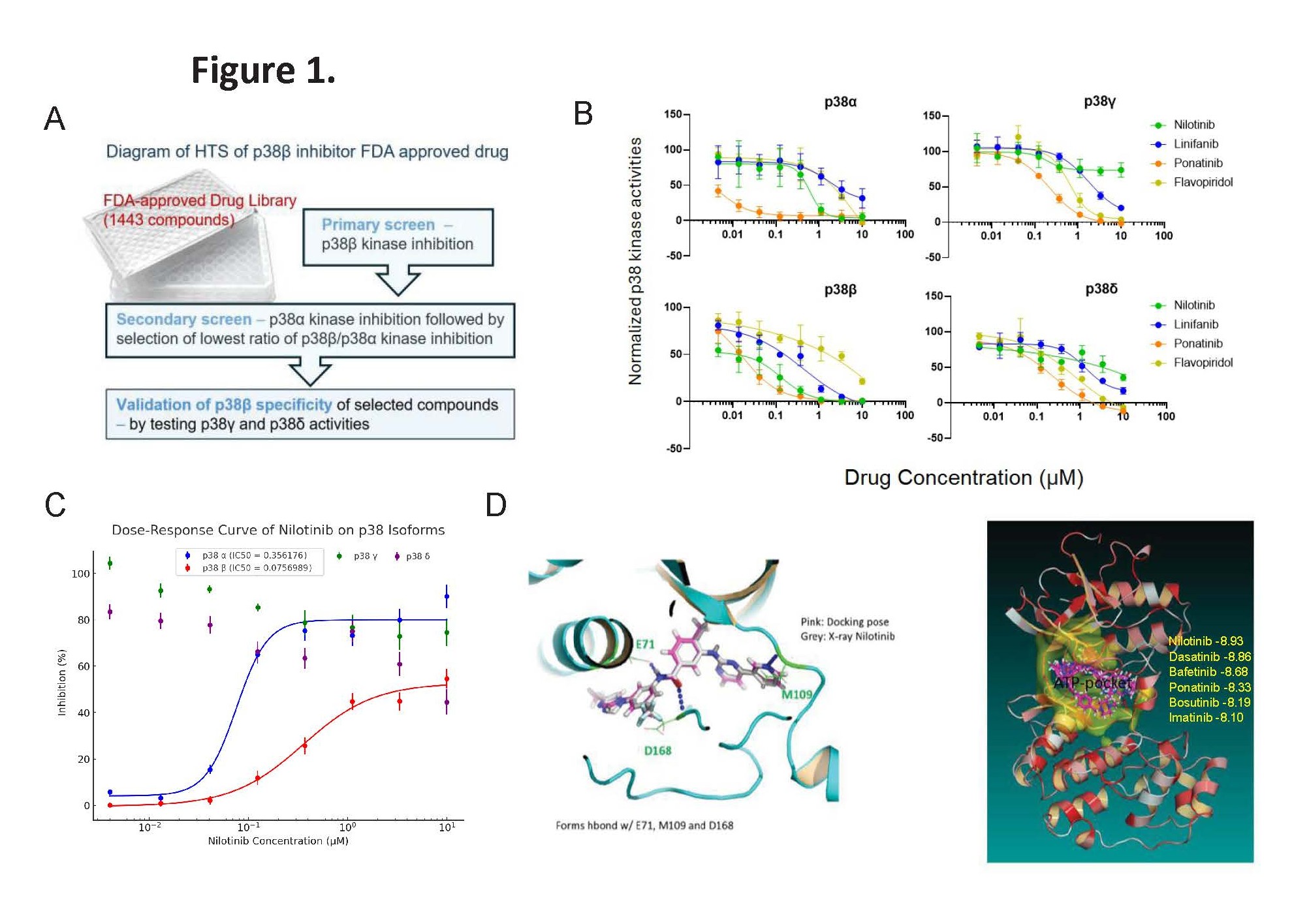
Figure 1. Nilotinib is selected as p38β inhibitor. (A)Diagram of High throughput screening (HTS) of p38β inhibitor from an FDA approved drug library (Selleck). (B) Effects of 4 FDA approved drugs (Flavopiridol, Linifanib, Ponatinib and Nilotinib) on kinase activities of 4 p38 isoforms. Nilotinib, a Bcr-Abl inhibitor, stood out as a potent p38β specific inhibitor, with IC50=14nM (red color) compared to other p38 isoforms. Inhibitory IC50 (µM) of Nilotinib for other 3 isoforms of p38 kinase activity listed in the Table 1. Nilotinib, a Bcr-Abl inhibitor, a potent p38β specific inhibitor in comparison of 3 other selected compounds. (C) Kinase activity assays of p38 MAPK isoforms. Nilotinib selectively inhibits p38β kinase activity (IC50=0.014–0.075 µM) with approximately 25-fold greater potency than p38α (IC50=0.356 µM), while showing minimal inhibition of p38γ and p38δ. These results demonstrate that Nilotinib is a more specific inhibitor of p38β compared to p38α. (D) Docking pose on p38β of Tyrosine Kinase inhibitors (TKI). Left: Docking pose of Nilotinib with p38β superimposed on X-ray crystallography data. Right: Nilotinib -8.93 showed the best docking score on p38β (-8.93 kcal/mol) in comparison of the other 5 TKIs: Dasatinib, Bafetinib, Ponatinib, Bosutinib and Imatinib, all of which binds p38β in the ATP-binding pocket. Imatinib is -8.10kcal/mol.
Inhibition of p38β induces classical dual phosphorylation of p38γ
To understand the functional consequences of p38β inhibition by Nilotinib in CTCL cells, we performed Western blot analysis and found that Nilotinib treatment induces dual phosphorylation of p38γ, a pro-oncogenic isoform in its active state. In Hut78 cells, Nilotinib treatment led to the appearance of an additional higher molecular weight band (>40 kDa), consistent with the dually phosphorylated form of p38γ (Figure 2A, shCtr Hut78 cell panel).
To confirm the identity of the top band, we employed a lentiviral shRNA system to knock down p38γ. The band disappeared in p38γ-silenced cells (shp38γ Hut78 cell panel), validating it as phosphorylated p38γ (blue arrow, Figure 2A). Notably, due to its slightly larger size (367 amino acids), p38γ migrates higher than other p38 isoforms (~360 amino acids, orange arrow in Figure 2A), and its phosphorylated form is clearly resolved from non-phosphorylated species on SDS-PAGE gels which was then detected using a phospho-p38 MAPK (Thr180/Tyr182) antibody (CST #4511). In addition, similar higher-molecular-weight phospho-p38γ bands have been observed in other p38β inhibitors SB203580 in myoblast, a progenitor cell for muscle cells [12], although the higher-molecular-weight bands were not further characterized in those studies.
To further confirm that this p38γ phosphorylation event is specifically due to p38β inhibition rather than off-target effects on other kinases, we treated Hut78 cells with Ralimetinib (LY2228820), a known ATP- competitive inhibitor of p38β (IC50=3.2 nM) [6]. Similar to Nilotinib, Ralimetinib (LY2228820) induced the appearance of the dual-phosphorylated p38γ band, though to a lesser extent (Figure 2B). Likewise, we validated this weaker band as p-p38γ using p38γ knock down approach. Ralimetinib also inhibits p38α (IC50=5.8 nM), though Nilotinib shows approximately 25-fold selectivity for p38β over p38α, as demonstrated by our in vitro kinase assay (Figure 1C).
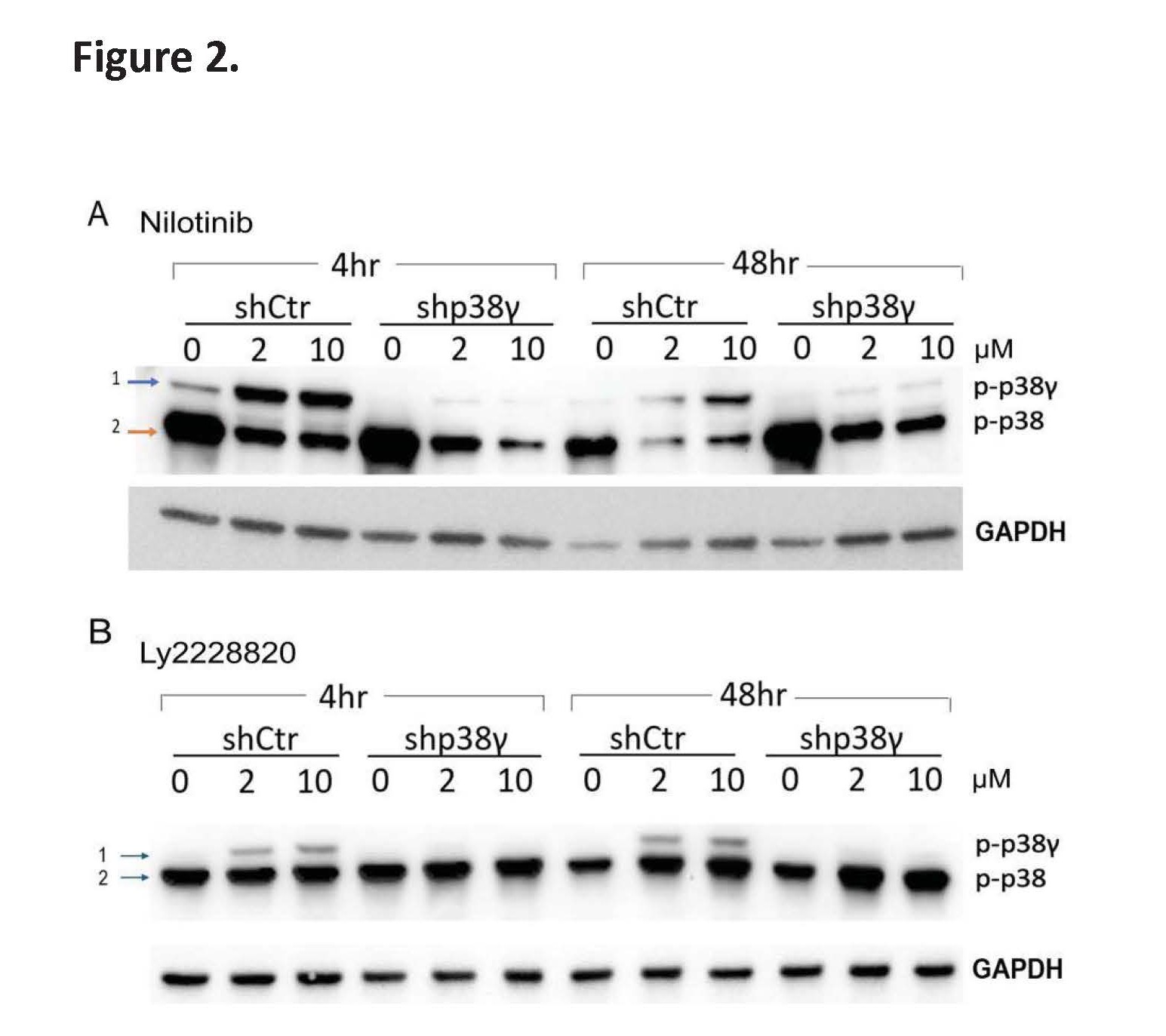
Figure 2. Nilotinib induces dual phosphorylation of p38γ in CTCL cells. (A) Scramble control (shCtrl) and p38γ knockdown Hut78 cells were treated with Nilotinib for 4 hours and 48 hours. A higher molecular weight band (Band 1), corresponding to the dually phosphorylated form of p38γ (p-p38γ), was detected in shCtrl cells but disappeared in p38γ knockdown cells, confirming its identity. (B) A similar experiment was conducted using the p38β/α inhibitor LY2228820. Treatment induced a weaker but distinct Band 1 in shCtrl cells, which was also abolished in p38γ knockdown cells. Note: p38γ has a higher molecular weight (367 amino acids) compared to other p38 isoforms (~360 amino acids), allowing clear resolution of p-p38γ (Band 1) from other isoforms (Band 2).
Potential EMT signal signature of Nilotinib in healthy PBMCs by single-cell RNA analysis
Given that compensatory p38γ activation can drive epithelial–mesenchymal transition (EMT) [13,14], we next examined whether Nilotinib triggers EMT-related pathways in healthy peripheral blood mononuclear cells (PBMCs). Single-cell RNA-sequencing of PBMCs treated with Nilotinib revealed simultaneous activation of KRAS signaling, EMT, and xenobiotic metabolism pathways (Figure 3A). We observed that EMT induction was confined to Treg and γδ T cell subsets.
To determine whether this p38γ-mediated EMT response extends beyond our experimental model, we next analyzed public datasets to assess the association between p38γ expression and EMT in clinical CTCL samples with large cell transformation. Public dataset [GSE113113] [15] analyses also support a role for p38γ—but not other p38 isoforms—in promoting EMT in CTCL with large-cell transformation (Figure 3B and Supplementary Figure S1). Our results implicate Nilotinib-induced EMT as a potential mechanism driving its immunotoxic or off-target effects.
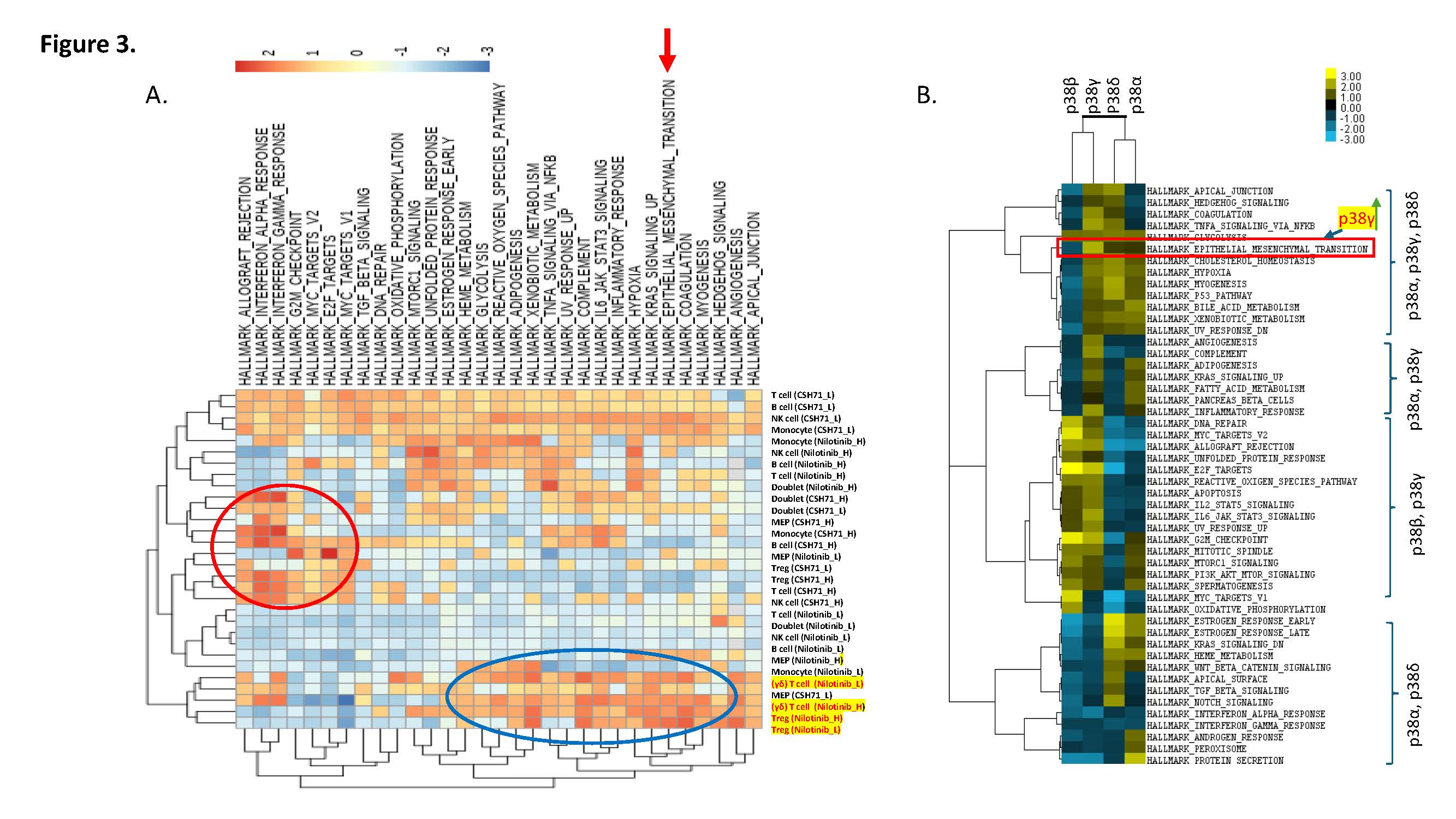
Figure 3. Single-cell RNA sequencing reveals distinct immune cell responses to Nilotinib and CSH71 treatments. (A) Single-cell transcriptomic analysis of PBMCs treated with Nilotinib or CSH71 at low (L) and high (H) concentrations identifies major immune cell populations, including T cells, B cells, NK cells, monocytes, Tregs, γδ T cells, and megakaryocyte-erythroid progenitors (MEPs). Treatment-specific clustering highlights transcriptional shifts, including enrichment of EMT-like signatures in Tregs and γδ T cells upon Nilotinib treatment and interferon response pathways with CSH71. Doublet populations and cell-type-specific responses are annotated by treatment condition. (B) A Public dataset analyses [GSE113113, https://www.ncbi.nlm.nih.gov/geo/] indicated that it is p38γ—but not other p38 isoforms—promoting EMT in CTCL with large cell transformation (LCT).
To mitigate this potential toxicity, we sought a synergistic partner for Nilotinib. We first determined the cytotoxic IC50 of Nilotinib in Hut78 cells (Supplementary Figure S2A). Next, we evaluated its combination treatments with CSH71 [16], a selective p38γ inhibitor that targets the lipid-binding site (LBS).
Combination index (CI) analysis revealed strong synergy in CTCL cells (CI=0.69–0.74, Supplementary Figure S2B), but antagonism in healthy PBMCs (Supplementary Figure S2C), suggesting that dual inhibition of p38β and p38γ preferentially targets malignant cells. In contrast, combining Nilotinib with the ATP-site p38γ inhibitor F7/PIK75 [11] yielded only additive effects, but showed synergy when F7 in a higher dosage (Supplementary Figure S2).
Clinical trials for Nilotinib in CML patients
To contextualize our preclinical findings, we reviewed the clinical trial landscape of Nilotinib in chronic myeloid leukemia (CML). A comprehensive overview of Phase I–IV studies (Table 2) demonstrates that Nilotinib has been extensively evaluated for efficacy and safety in both adult and pediatric CML populations [17–26]. Across multiple Phase II and III trials, Nilotinib consistently achieved higher rates of major molecular response (MMR) compared to Imatinib, particularly in newly diagnosed chronic-phase CML patients. In treatment-resistant or -intolerant cohorts, Nilotinib also produced meaningful cytogenetic and molecular responses. Pediatric trials revealed age-dependent pharmacokinetics and efficacy, while long-term follow-up studies confirmed durable responses with manageable toxicity [20–27]. Phase IV studies further confirmed long-term benefits, including sustained deep molecular responses (MR4.0 and MR4.5) and treatment-free remission in selected patients. Together, these data support Nilotinib as a potent second-generation tyrosine kinase inhibitor with an overall favorable benefit-risk profile in CML. Nevertheless, SAE rates ranged widely—from 3% to 60%—with higher toxicity observed in studies involving advanced disease. Notably, SAE rates are even higher in non-CML cancers such as gastrointestinal stromal tumors (GIST, Table 3), where Nilotinib acts as a multi-kinase inhibitor in the absence of BCR-ABL, increasing the risk of off-target toxicities driven by unintended pathway activation.
|
Clinical Trial |
Phase |
Aim |
Result |
Cancer Type |
|
NCT00109707 |
I/II |
Evaluate efficacy, safety, and PK of Nilotinib in CML/ALL patients across six groups |
Major cytogenetic response after 7.5 years: 59.5% (CML-CP, prior Imatinib), 32.1% (CML-AP, prior Imatinib) |
AML, CML |
|
NCT01077544 |
I |
Assess PK, safety, and activity of Nilotinib in pediatric Ph+ CML or Ph+ ALL patients |
MMR achieved in 1/5 patients <10 years, and 2/6 patients aged 10–18 |
AML, CML |
|
NCT01698905 |
II |
Evaluate if Nilotinib can be safely discontinued in CML patients |
57.9% achieved treatment-free remission within 48 weeks |
CML |
|
NCT02353728 |
II |
Assess nilotinib's effect on leukemic stem cells in newly diagnosed CML |
— |
CML |
|
NCT00129740 |
II |
Determine if Nilotinib controls chronic phase CML |
131/148 achieved complete cytogenetic response; 23/148 achieved complete molecular response |
CML |
|
NCT01784068 |
II |
Evaluate safety of Nilotinib cessation in CML |
51.6% achieved MMR at 48 weeks (n=190) |
CML |
|
NCT01744665 |
II |
Assess relapse-free survival post- Nilotinib in MR4.5 patients |
Median relapse-free survival: 21.4 weeks |
CML |
|
NCT01844765 |
II |
Evaluate long-term safety and PK of Nilotinib in pediatric Ph+ CML |
MMR at 6 cycles in resistant pediatric CML: 39.4%; at 12 cycles in newly diagnosed: 64% |
CML |
|
NCT01274351 |
II |
Study Nilotinib as first-line therapy in adult Ph+ CML-CP |
66.1% achieved MMR at 12 months |
CML |
|
NCT03578367 |
II |
Compare Nilotinib vs Asciminib ± Imatinib in pretreated CML-CP |
MR4.5 at 48 weeks: Asciminib60+Imatinib400=19%, Asciminib40+Imatinib400=28.6%, Imatinib alone=0%, Nilotinib300=14.3% |
CML |
|
NCT01254188 |
III |
Evaluate efficacy and safety of Nilotinib in newly diagnosed CML |
70.8% achieved MMR at 12 months |
CML |
|
NCT01743989 |
III |
Assess optimal Nilotinib treatment duration in Ph+ CML |
Sustained remission: 31.9% (24 months), 37.5% (36 months) |
CML |
|
NCT00802841 |
III |
Compare efficacy of Nilotinib vs Imatinib in CML |
Complete cytogenetic response: 50% (Nilotinib) vs. 42.1% (Imatinib) |
CML |
|
NCT01275196 |
III |
Compare Nilotinib vs Imatinib in Chinese CML-CP patients |
MMR: 52.2% (Nilotinib) vs. 27.8% (Imatinib) |
CML |
|
NCT00471497 |
III |
Compare two Nilotinib doses vs Imatinib in newly diagnosed Ph+ CML-CP |
MMR at 12 months: 44.3% (Nilotinib 300 mg), 42.7% (400 mg), 22.3% (Imatinib) |
CML |
|
NCT02272777 |
III |
Extended comparison of Nilotinib vs Imatinib in Chinese CML-CP patients |
AEs: 84/113 (Nilotinib) vs. 82/112 (Imatinib); SAEs: 3/113 vs. 1/112 |
CML |
|
NCT00760877 |
III |
Compare cumulative CMR between Nilotinib and Imatinib |
CMR at 12 months: 13/104 (Nilotinib) vs. 6/103 (Imatinib) |
CML |
|
NCT01061177 |
IV |
Evaluate Nilotinib efficacy in newly diagnosed Ph+ CML |
MR4.0 at 18 months: 38.3%; MMR: 56.2% (12 months), 61.1% (24 months) |
CML |
|
NCT00980018 |
IV |
Assess low-grade AEs upon switching from Imatinib to Nilotinib |
AE reduction after switching from Imatinib to Nilotinib: 71.2% (cycle 1), 82.7% (cycle 2), 84.6% (cycle 3) |
CML |
|
NCT01227577 |
IV |
Evaluate deep molecular responses with Nilotinib over 4 years |
Complete molecular response at 4 years: 34/128 |
CML |
|
NCT01043874 |
IV |
Assess MMR in Ph+ CML-CP patients with suboptimal response to Imatinib |
MMR at 12 months: 51.1% |
CML |
|
NCT02546674 |
IV |
Evaluate MR4.5 after 24 months Nilotinib in newly diagnosed CML |
MR4.5 at 24 months: 35.3% (n=156) |
CML |
|
NCT01735955 |
IV |
Long-term follow-up of Nilotinib-treated patients in extension study |
Long-term clinical benefit maintained in 26/33 patients at week 192; 4 patients remained on treatment at week 528 |
CML, ALL, GIST, KIT melanoma |
|
Clinical Trial |
Phase |
Aim |
Result |
Cancer Type |
SAE |
|
NCT01863745 |
II |
Evaluate long-term safety of nilotinib |
Safety monitoring: assessed SAEs and AEs in GIST patients |
GIST |
0.6 |
|
NCT01289028 |
III |
Evaluate nilotinib efficacy in pretreated metastatic GIST |
Stable disease for ≥4 months in 48.8% of GIST patients |
GIST |
0.48 |
|
NCT00471328 |
III |
Compare nilotinib vs current treatments in GIST post- 1st/2nd line failure |
Median progression-free survival (PFS) in GIST: 109 days |
GIST |
0.491 |
Given that p38β is upregulated in CML, its inhibition by Nilotinib may confer additional therapeutic benefit. However, clinical data in Table 4 (NCT02272777) [28,29] indicate a higher incidence of serious adverse events (SAEs) in Nilotinib-treated patients compared to those receiving Imatinib. This elevated toxicity likely reflects the broader impact of Nilotinib on dysregulated signaling networks involving both p38β and p38γ. Our findings show that Nilotinib-induced activation of p38γ occurs through inhibition of p38β (Figure 2A and 2B), offering a mechanistic explanation for adverse signaling and reduced therapeutic selectivity.
|
Adverse Events |
Imatinib (n/N, %) |
Nilotinib (n/N, %) |
|
Blood bilirubin increased |
2/112 (1.79%) |
24/113 (21.24%) |
|
Blood triglycerides increased |
12/112 (10.71%) |
17/113 (15.04%) |
|
Blood cholesterol increased |
3/112 (2.68%) |
11/113 (9.73%) |
|
Alanine aminotransferase increased |
4/112 (3.57%) |
9/113 (7.96%) |
|
Blood glucose increased |
9/112 (8.04%) |
8/113 (7.08%) |
|
Low density lipoprotein increased |
1/112 (0.89%) |
7/113 (6.19%) |
|
Lipase increased |
6/112 (5.36%) |
7/113 (6.19%) |
ROR assessment and SAEs by Nilotinib in long-term clinical trials
To further assess safety, we analyzed 2024 (Q1–Q4) FDA Adverse Event Reporting System (FAERS) data [30], for three BCR-ABL inhibitors—Nilotinib, Imatinib, and Dasatinib. We used the Reporting Odds Ratio (ROR), a pharmacovigilance method recommended by the NIH [31–33], to detect adverse events (AEs) that occur more frequently with specific drugs. Our integrated analysis of clinical trial data highlights a consistent safety concern with Nilotinib use across cancer types. In Ph+ CML patients, the rate of SAEs exceeded 25% in multiple clinical trials, underscoring the need to weigh therapeutic efficacy against toxicity (Table 2). Notably, in Nilotinib-repurposed GIST trials, the incidence of SAEs was even higher than in CML trials (Table 3, Figure 4A) [clinicaltrials.gov].
Nilotinib showed greater toxicity in Ph+ CML patients compared to the other two TKIs (Figure 4B). Venn diagram analysis indicated that Imatinib was associated with the broadest range of adverse events (314 total signals), while Nilotinib had fewer (39 unique) but more severe signals. Dasatinib exhibited the fewest (17 unique), with nine AEs shared across all three drugs, suggesting potential class-related toxicities (Figure 4C). Applying a threshold of ROR>2 with at least three reports, we found that Nilotinib consistently showed higher ROR values than Imatinib and Dasatinib—especially for gastrointestinal, neurological, and cardiac events (Figure 4D). While Imatinib is linked to a broader range of AEs, Nilotinib appears to carry a greater risk of severe toxicities within the BCR-ABL inhibitor class.
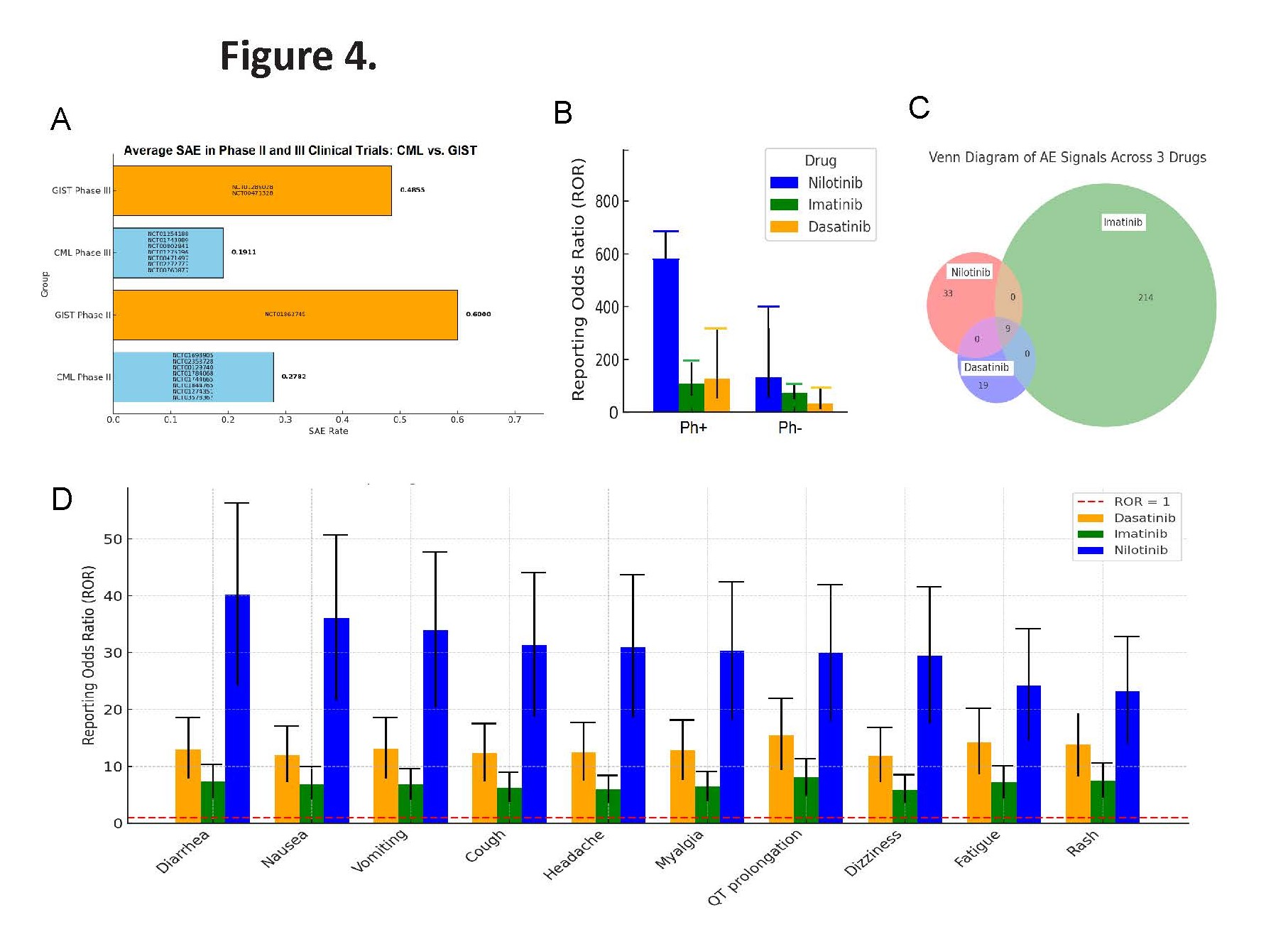
Figure 4. Comparative safety analysis of BCR-ABL inhibitors using clinical trial data and FAERS pharmacovigilance signals. (A) Incidence of serious adverse events (SAEs) across clinical trials for Nilotinib in CML and non-CML malignancies, showing a higher SAE rate in non-CML (e.g., GIST) settings. For example, a CML trial with 200 patients and 38 SAEs yields an SAE rate of 0.19, whereas a GIST trial with 150 patients and 62 SAEs yields a rate of 0.413. (B) Total number of adverse event (AE) signals associated with Nilotinib, Imatinib, and Dasatinib, based on 2024 Q1–Q4 FAERS data. The analysis shows that Imatinib is linked to the broadest range of AEs, while Nilotinib is associated with fewer but more severe events. (C) Venn diagram illustrating the overlap and uniqueness of reported AEs for Nilotinib, Imatinib, and Dasatinib. Nilotinib and Dasatinib share some common AEs with Imatinib but also exhibit distinct AE profiles, underscoring potential differences in off-target effects. (D) Reporting Odds Ratio (ROR) analysis comparing Nilotinib to Imatinib and Dasatinib. Nilotinib shows significantly elevated ROR values, particularly for gastrointestinal, neurological, and cardiac toxicities, indicating a higher relative risk of severe adverse events. Note: Reporting Odds Ratio (ROR) is a pharmacovigilance metric used to detect whether a particular adverse event (AE) is reported more frequently for a specific drug compared to others in a large database (like FAERS). Data indicates 10 AEs are reported much more frequently for Nilotinib than for Imatinib and Dasatinib.
Relocation of p38β and p38γ genes following the t (9;22) (q34; Q11) chromosomal translocation
To better understand the regulation of p38β and p38γ in hematologic malignancies, we examined their genomic architecture and expression patterns. Notably, both MAPK11 (p38β) and MAPK12 (p38γ) are originally located on chromosome 22q11 [34]. In Philadelphia chromosome–positive (Ph+) CML, the t(9;22)(q34;q11) translocation relocates these genes to chromosome 9 [35,36], as illustrated in Figure 5A, placing them (p38s) near dysregulated epigenetic regulators such as EP300, but away from EHMT1. This genomic repositioning may contribute to aberrant signaling and altered chromatin regulation in leukemic cells.
To determine whether this structural rearrangement translates into functional changes, we analyzed public RNA-Seq dataset of leukemia samples [37]. The results revealed elevated expression of p38β and p38γ in leukemia samples, supporting their potential roles in leukemogenesis and in mediating the cellular response to Nilotinib relative to non-cancerous samples (Figure 5B). The translocation also shifts epigenetic regulators such as EHMT1, which is upregulated together with its isoform EHMT2 in Ph+ CML (Figure 5C). In addition, EP300 expression level has no significant changes (Supplementary Figure S4).
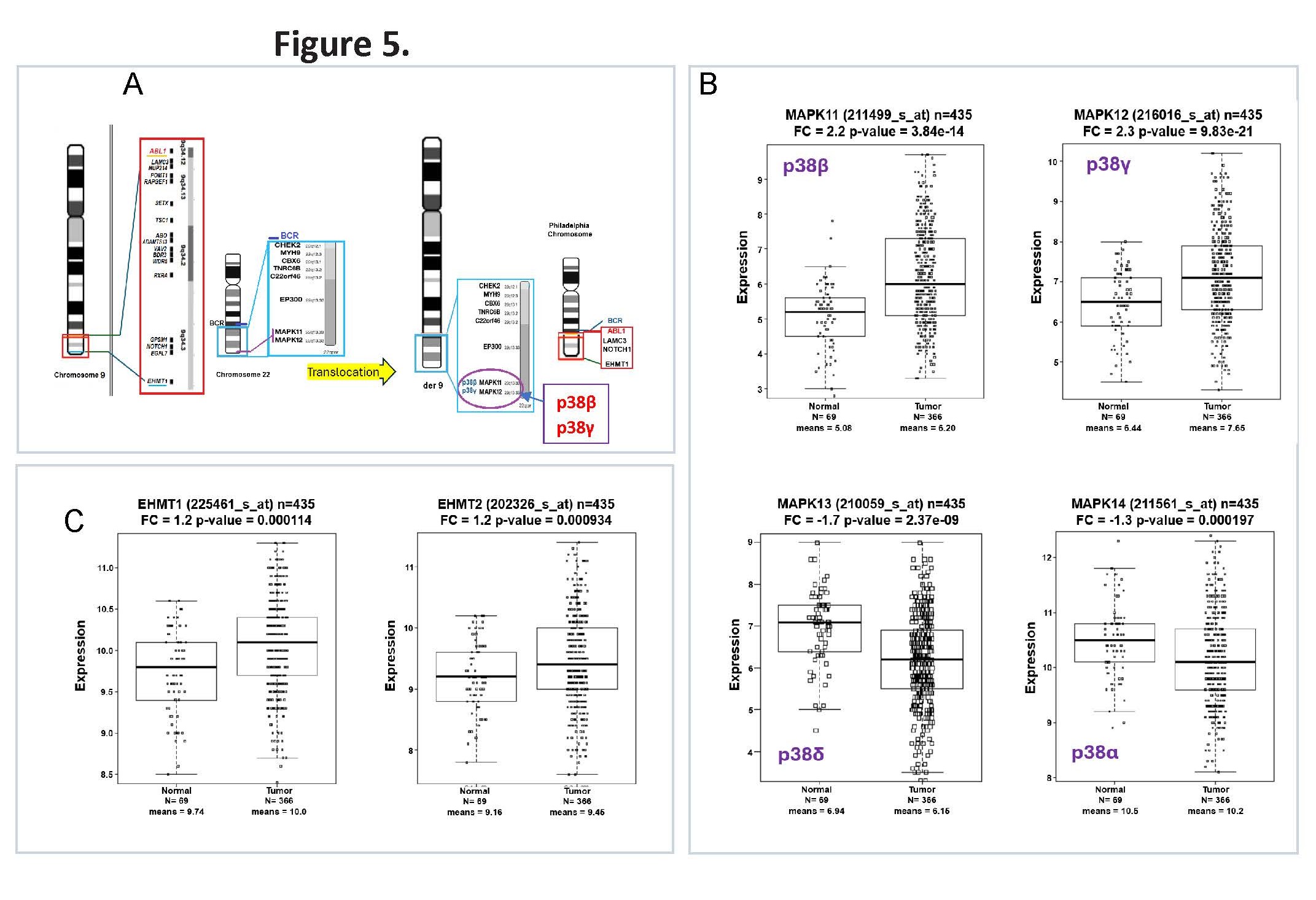
Figure 5. Chromosomal translocation t(9;22)(q34;q11) alters genomic positioning and expression of p38 isoforms and epigenetic regulators in Ph+ CML. (A) Schematic representation of the t(9;22)(q34;q11) translocation in chronic myeloid leukemia (CML), illustrating the repositioning of genes including MAPK11 (p38β), MAPK12 (p38γ), EP300, and EHMT1. The rearrangement relocates these genes from chromosome 22 to chromosome 9, placing them near additional oncogenic loci such as CHEK2, CBX6, LAMC3, and NOTCH1, potentially altering transcriptional regulation and chromatin architecture. (B) Public database analysis for RNA-Seq in leukemia samples (Mills et al. 2009 [37], GSE15061) showing significantly elevated expression of p38β (MAPK11) and p38γ (MAPK12), and reduced expression of p38α (MAPK14) and p38δ (MAPK13) in leukemia samples compared to non- cancerous samples. These findings suggest that the chromosomal translocation may contribute to dysregulated MAPK signaling in Ph+ CML. (C) Expression profiling of epigenetic regulators shows marked upregulation of EHMT1 and EHMT2 (G9a) compared to non-cancerous samples. The elevated EHMT1/2 expression supports a repressive chromatin environment conducive to leukemic stemness and therapy resistance.
Clinical trial of combination of Nilotinib and Paclitaxel
Paclitaxel selectively inhibits angiogenesis at ultra-low picomolar concentrations without disrupting microtubule assembly, supporting its clinical potential as a continuous low-dose anti-cancer strategy [38]. Table 6 indicated this promising combination (Nilotinib + Paclitaxel) in a published Phase I clinical trial (NCT02379416) [39], which demonstrated both safety and preliminary efficacy, including tumor regression in patients resistant to prior taxane (e.g., Paclitaxel) therapy. Notably, the trial also reported a reduced incidence of peripheral neuropathy—a common and debilitating side effect of Paclitaxel—suggesting a potential neuroprotective benefit of Nilotinib [40]. Upon these encouraging results, a Phase II trial (NCT05554341), known as ComboMATCH (EAY191-E4) [41] and sponsored by the National Cancer Institute (NCI) through the Eastern Cooperative Oncology Group - American College of Radiology Imaging Network (ECOG-ACRIN) Cancer Research Group, is currently underway to further assess the combination’s therapeutic potential in advanced solid tumors. However, clinical trials evaluating the Nilotinib and Paclitaxel combination in hematologic malignancies have not yet been initiated—a gap our research aims to address.
|
Total Dose (μM) |
Fa (CTCL Hut78) |
CI (CTCL Hut78) |
Fa (AML Molm13-luc) |
CI (AML Molm13- luc) |
|
1.25 |
0.9999 |
510.744 |
0.9999 |
510.744 |
|
2.50 |
0.8167 |
1.45788 |
0.8167 |
1.45788 |
|
5.00 |
0.3276 |
0.57662 |
0.3276 |
0.57662 |
|
10.00 |
0.1872 |
0.65581 |
0.1872 |
0.65581 |
|
20.00 |
0.1072 |
0.87133 |
0.1072 |
0.87133 |
|
40.00 |
0.0738 |
1.31850 |
0.0738 |
1.31850 |
|
Common Adverse Events (All Grades) |
Grade ≥3 Adverse Events / Dose- Limiting Toxicities |
|
Fatigue (52%) |
Neutropenia (23%) |
|
Peripheral neuropathy (45%) |
Fatigue (11%) |
|
Nausea (43%) |
Hypertension (9%) |
|
Diarrhea (34%) |
Peripheral neuropathy (7%) |
|
Alopecia (34%) |
Hypophosphatemia (5%) |
|
Vomiting (30%) |
Febrile neutropenia (5%) |
|
Anorexia (27%) |
|
|
Neutropenia (27%) |
Preclinical studies using human tumor cell lines have shown that combining Nilotinib with Paclitaxel, a taxane-based chemotherapeutic agent, produces synergistic cytotoxic effects, killing cancer cells more effectively than either drug alone [39,40,42–54]. We also demonstrated that the IC50 of Paclitaxel in CTCL Hut78 cells is 2nM (Supplementary Figure S5), however with a higher dosage of Paclitaxel, Hut78 cells showed increase of cell proliferation. Consistent with this, our cytotoxicity assays demonstrated synergistic effects of Nilotinib + Paclitaxel in acute myeloid leukemia (AML) Molm13 cells (Supplementary Figure S6).
Moreover, we found that Paclitaxel directly binds to p38γ, as validated by mass spectrometry (Figure 6), suggesting that it may act as a p38γ inhibitor. Depending on experimental conditions, one to three Paclitaxel molecules were detected bound to each p38γ protein. By functioning as a p38γ inhibitor, Paclitaxel may counteract the elevated p38γ activity induced by Nilotinib’s p38β inhibition.
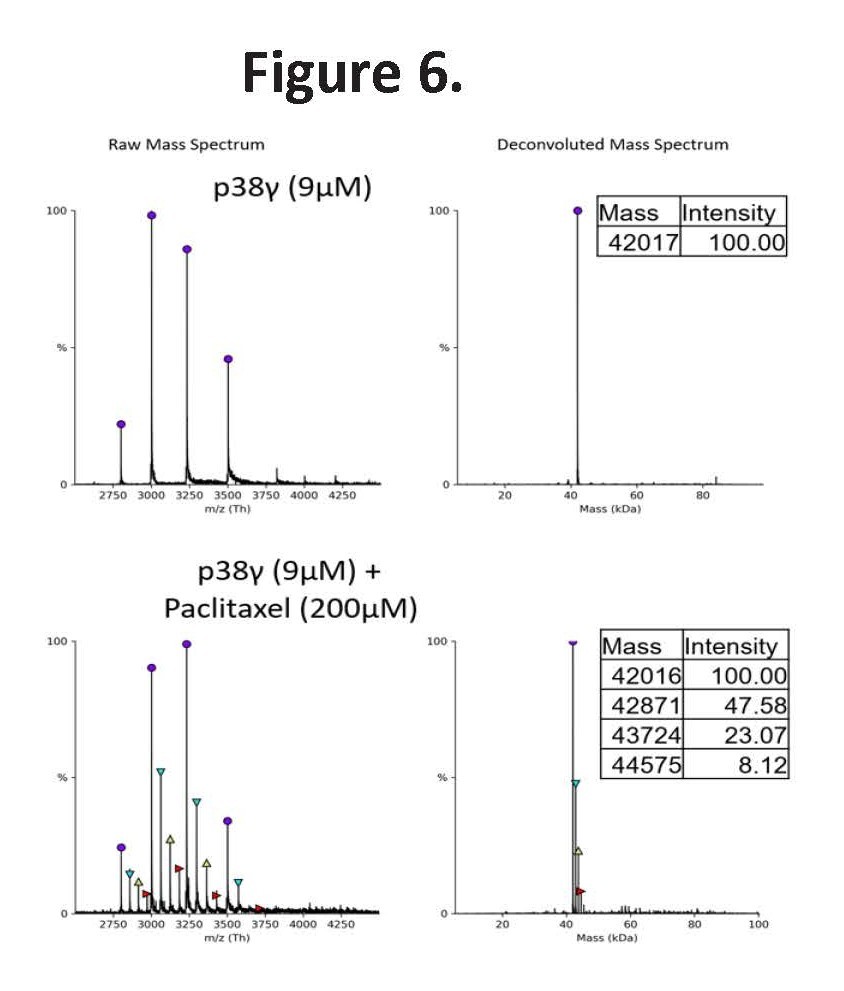
Figure 6. Native mass spectrometry confirms direct binding of Paclitaxel to p38γ. Left panels show raw mass spectra; right panels show corresponding deconvoluted spectra. Top panel: Deconvoluted native mass spectrum of recombinant p38γ (9 µM) alone displays the expected molecular mass of unbound protein. Bottom panel: Deconvoluted spectrum of p38γ incubated with Paclitaxel (200 µM) reveals a distinct mass shift consistent with the formation of a p38γ–Paclitaxel complex. Data validate Paclitaxel as a direct binder of p38γ and provide biophysical support for its potential role in counteracting Nilotinib-induced p38γ upregulation, as observed in synergy experiments.
Discussion
Nilotinib-induced activation of p38γ through p38β inhibition
The higher rate of SAEs observed with Nilotinib in non-CML malignancies compared to CML is rooted in differences in disease biology. In CML, the characteristic t(9;22)(q34;q11) translocation produces the BCR-ABL fusion kinase—a direct driver of leukemogenesis and an ideal drug target. The development of Imatinib (Gleevec), a selective BCR-ABL inhibitor, revolutionized CML treatment and became a landmark in pharmacology, establishing the paradigm of targeted cancer therapy. Nilotinib, a second-generation BCR-ABL inhibitor, builds upon this success. However, Nilotinib also inhibits other kinases, including p38β. In cancers lacking the BCR-ABL fusion, such as GIST, Nilotinib’s multi-kinase activity may engage non-malignant signaling proteins, reducing efficacy and increasing off-target toxicities. Importantly, parallel elevation of p38β and p38γ expression is observed in cutaneous T-cell lymphoma (CTCL) [11] as well as in leukemia (Figure 5B). Our findings show that Nilotinib modulates oncogenic pathways in CTCL by targeting p38β and indirectly promoting p38γ activation—independent of the t(9;22)(q34;q11) translocation—highlighting a novel mechanistic axis that contributes to Nilotinib-associated toxicities across diverse cell types through a general p38 isoform compensation mechanism.
This mechanism appears broadly applicable, as supported by public literature. Similar higher molecular weight bands—indicative of p38γ activation—have been observed in Western blots of myoblasts (non- hematologic cells) treated with either Nilotinib or the p38β inhibitor SB203580 [12]. These studies also reported that Nilotinib promotes myoblast proliferation, thereby preventing the formation of mature myotubes. Notably, p38γ is highly expressed in skeletal muscle cells, of which myoblasts serve as their precursors. Although the functional significance of these bands was not explored [12], these findings support our conclusion that in cells with high p38γ expression, p38β inhibition triggers compensatory activation and phosphorylation of p38γ. This mechanism likely contributes to both cancer progression and Nilotinib-associated SAEs. Notably, two long-term ENESTnd studies (Evaluating Nilotinib Efficacy and Safety in newly diagnosed Ph+ Chronic Myeloid Leukemia in chronic phase, >10 years follow-up) reported significantly higher rates of cardiovascular events (CVEs) and hepatotoxicity in Nilotinib-treated patients compared to those receiving Imatinib [55,56].
We further demonstrate that p38γ inhibitors synergize with Nilotinib to enhance anti-tumor activity across multiple cancer types [6], including AML (Supplementary Figure S6), suggesting a potential strategy to improve both the safety and efficacy of Nilotinib in CML and solid tumors. Clinical studies also support the promise of this approach. A Phase I trial (NCT02379416) reported favorable safety and preliminary efficacy of the Nilotinib + Paclitaxel combination, including reduced incidence of Paclitaxel-induced peripheral neuropathy. A follow-up Phase II study (NCT05554341, ComboMATCH) is currently evaluating this combination in advanced solid tumors. However, its clinical efficacy in hematologic malignancies remains unexplored, emphasizing the translational importance of our findings.
To assess whether Nilotinib is disproportionately associated with specific toxicities, we performed a pharmacovigilance analysis using the ROR, a standard metric that quantifies the likelihood of an adverse event being reported for a particular drug compared to others. A significantly elevated ROR flags a potential safety concern that warrants further investigation.
EMT
EMT is a transformative biological process where epithelial cells lose their apical-basal polarity and loosen cell–cell connections, adopting the shape and invasive traits of mesenchymal cells to migrate through the extracellular matrix [57]. KRAS activation can trigger EMT via pathways like MAPK/ERK and PI3K/AKT, influencing transcription factors such as SNAIL [13], SLUG, and TWIST to facilitate this transition. In our single-cell analysis (Figure 3A), we observed that only two T cell populations—Tregs and γδ T cells—are prone to exhibit EMT signatures among Nilotinib-treated PBMC population. While in general, Treg cells do not undergo full EMT, they can adopt EMT-like transcriptional programs to enable tissue infiltration and motility, largely via TGF-β/SMAD signaling. γδ T cells are particularly prone to EMT-like reprogramming, especially within tumor microenvironments rich in TGF-β, enabling tissue infiltration, matrix remodeling, and—depending on the context—either tumor progression or immune evasion [58–62]. In this context, Nilotinib-induced EMT in PBMCs is likely driven by its inhibition of p38β, which in turn leads to compensatory activation of p38γ. Notably, p38γ MAPK has been shown to promote EMT and the expansion of cancer stem cell (CSC) populations in breast cancer cells [14], supporting the relevance of this mechanism across cell types. EMT is associated with harmful outcomes in adult tissues, including its involvement in pathological processes such as fibrosis and cancer metastasis.
Chromosomal translocation and pathway dysregulation
Chromosomal rearrangements can profoundly alter gene proximity and regulatory landscapes, contributing to oncogenesis. In Ph+ CML, the characteristic BCR-ABL translocation (t(9;22)(q34;q11)) not only produces the BCR-ABL fusion kinase but also repositions the p38β and p38γ genes from chromosome 22 near genes on chromosome 9, including CHEK2, CBX6, and EP300. The BCR-ABL fusion locus is likewise positioned near additional leukemia-associated genes such as LAMC3, NOTCH1, and EHMT1 (Figure 5A). This complex rearrangement may foster an altered crosstalk between kinase signaling and epigenetic regulation. Although the precise mechanism by which p38β inhibition triggers compensatory p38γ phosphorylation remains under investigation, the chromosomal context suggests that these interactions are embedded within broader dysregulated networks. Our findings underscore the complexity of chromosomal rearrangements in cancer and highlight the importance of understanding their broader impact beyond fusion gene generation, particularly as it relates to therapeutic resistance and disease progression.
Among the affected epigenetic regulators, histone acetyltransferase EP300 [63] and euchromatic histone-lysine N-methyltransferase 1 (EHMT1) are also repositioned by the translocation (Figure 5A). Expression profiling reveals that while EP300—a well-established tumor suppressor [64]—is not significantly downregulated (Supplementary Figure S4), EHMT1, which promotes leukemic stemness [65,66], is markedly upregulated in Ph+ CML patients (Figure 5C). EHMT2 (G9a, located on chromosome 6), is similarly upregulated. EHMT1, functioning as a heterodimer with EHMT2, catalyzes H3K9 mono- and dimethylation, generating repressive chromatin marks that silence tumor suppressor genes and sustain leukemic stemness [67,68]. Our data confirmed elevated EHMT1 expression (p<0.001), further implicating epigenetic imbalance in CML pathogenesis. We hypothesize that the t(9;22)(q34;q11) translocation not only drives oncogenesis through BCR-ABL fusion kinase activity but also reshapes the genomic and epigenetic landscape of Ph+ leukemia. The repositioning and dysregulation of key signaling and epigenetic regulators, including p38γ/p38β, EHMT1, and others—create a permissive environment for aberrant pathway crosstalk and leukemic stemness.
Clinical implications
Our data further reveal that Paclitaxel directly binds to p38γ and may functionally act as a p38γ inhibitor, counteracting the Nilotinib-induced activation of p38γ and contributing to the observed synergy in CTCL and AML models. Importantly, our study uncovers a previously unrecognized mechanism: Nilotinib potently inhibits p38β, which in turn drives compensatory hyperactivation of p38γ. This unintended activation of p38γ may underlie many of the EMT- related phenotypes and serious adverse effects observed in Nilotinib-treated patients, especially in cancers lacking BCR-ABL. These findings offer a compelling mechanistic rationale for applying Nilotinib in combination with p38γ inhibitors to neutralize its p38γ-driven toxicities and enhance therapeutic efficacy across both hematologic malignancies and solid tumors. Although the Nilotinib + Paclitaxel combination exhibits strong synergistic anti-cancer activity, known risks of neurotoxicity and cardiotoxicity for both agents warrant caution. Our results suggest that combining Nilotinib with safer p38γ-selective inhibitors could offer a more favorable therapeutic window. In this regard, FDA-approved pirfenidone—a known p38γ inhibitor [69,70] —demonstrates synergy with Nilotinib in CTCL (Figure 7) and may help mitigate its adverse effects. Additionally, more selective alternatives such as the LBS- targeting p38γ inhibitor CSH71 [16], Pirfenidone [71], or the ATP-site inhibitor F7/PIK75 [11] represent promising strategies to optimize clinical outcomes while minimizing toxicity.
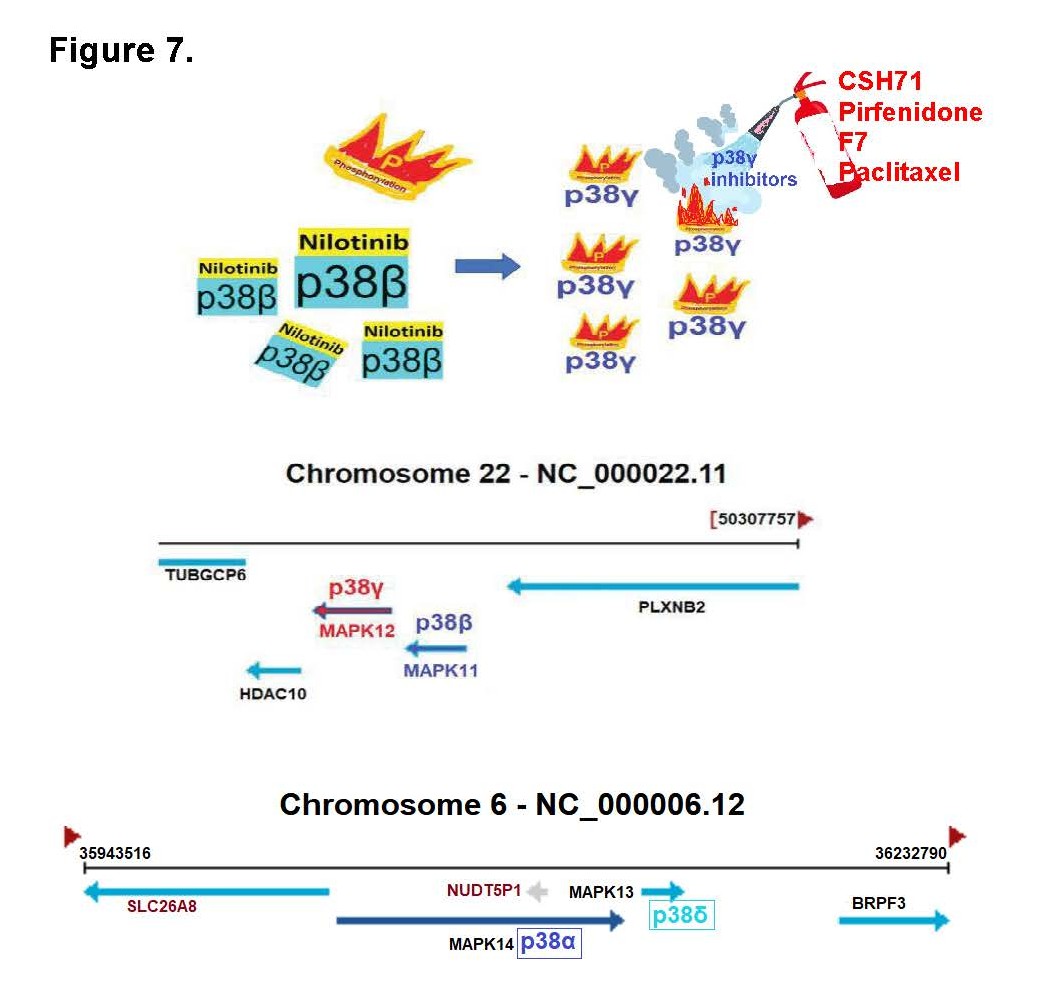
Figure 7. Illustration of compensatory activation of p38γ by Nilotinib and genomic localization of p38 MAPK isoforms. (A) Schematic depicting the compensatory activation of p38γ following p38β inhibition in cells expressed p38γ. Nilotinib binds to and inhibits p38β, leading to increased phosphorylation and activation of p38γ, a response that can be attenuated by p38γ-specific inhibitors to alleviate downstream stress signaling. (B) Genomic mapping shows that p38γ (MAPK12) and p38β (MAPK11) are located adjacent to each other on chromosome 22q13.33, suggesting potential transcriptional or regulatory linkage. (C) p38α (MAPK14) and p38δ (MAPK13) are clustered together on chromosome 6p21.31, indicating conserved genomic organization among p38 MAPK family members.
Materials and Methods
High-throughput screening for p38β inhibitor from FDA-approved drug library
We used an in vitro kinase assay to screen a library of 1,443 FDA-approved compounds (1,393 in DMSO, 50 in water) to identify p38β inhibitors. The library, in 96-well format, was purchased from Selleck Chemicals (Houston, TX, USA).
P38 kinase assays
In vitro kinase assays were performed using the ADP-Glo kit (Promega, WI) (Promega) as previously described [22]. Briefly, human recombinant p38 α, β or δ protein (active full-length) expressed by baculovirus in sf9 insect cells (Signal Chem, Vancouver, Canada) was homogenized in 96-well white opaque microplates with kinase buffer (40 mM Tris-HCl (pH 7.5), 20 mM MgCl2, 0.1 mg/mL BSA, and 50 µM DTT). p38 kinase was preincubated with compounds as indicated in a dose-dependent manner for 10 min. Next, synthetic peptide substrates (IPTTPITTTYFFFKKK) were added to the mixture at a final concentration of 0.2 µg/µL followed by ATP at various concentrations.
Viability assays using trypan blue exclusion and CellTiterGlo Cell Viability Assay
Cell viability was calculated by diluting cell suspensions 1:1 in 0.4% trypan blue solution (Sigma-Aldrich, St. Louis, MO) and counting the number of viable cells using a TC20 automated cell counter (Bio-Rad, Irvine, CA) that automatically excludes non-viable cells. CellTiterGlo Cell Viability Assay (Promega) was used as previously described [22]. All data points represent the average of three experiments. SS patients’ cells acquisition was approved by UMAS (IRB#204503, MTA#25468).
Single cell RNA sequencing analysis
Cell number and viability were measured using a TC20 Automated Cell Counter (Bio-Rad), and only samples with ≥80% viability were processed. Both CSH71- or Nilotinib-treated cells were loaded onto a Chromium Controller (10x Genomics), targeting 2,000–5,000 cells per lane. Single-cell 3′ RNA-seq libraries were prepared using the Chromium Single Cell 3′ v2 Reagent Kit (10x Genomics) following the manufacturer’s protocol and sequenced on a HiSeq 2500 system (Illumina) with a depth of 50,000–100,000 reads per cell. Raw sequencing data were processed using the Cell Ranger pipeline (v2.0, 10x Genomics) and aligned to the mm10 reference genome. Data analysis was performed with the Seurat package in R. Low-quality cells (greater than 10–20% mitochondrial reads and less than 200 detectable genes) were excluded. Data were normalized, scaled, and analyzed using principal component analysis of highly variable genes, followed by clustering and visualization with t-SNE or UMAP. Cluster-specific marker genes were identified to generate heatmaps and feature plots. Gene set enrichment analysis (GSEA) was performed using Hallmark gene sets from MSigDB.
Western blot analysis and evaluation of p38β and p38γ gene silencing by shRNA expression
Hut 78 cell culture and gene silencing by shRNA were performed as previously described [23]. MISSION® shRNA Lentiviral Transduction Particles (company validated) shRNAs in pLKO.1- puro shRNA vector that targets the human p38β (MAPK11), p38γ gene (MAPK12) and a scramble Transduction Particles (pLKO.1-puro shRNA vector only) were purchased from Sigma. All antibodies were procured from Cell Signaling, Danvers, MA, including p38α (Cat# 9218), p38β (2339), p38γ (2307), p38δ (2308), p-p38 T180/Y182 (4511), and GAPDH (2118).
Analysis of Paclitaxel binding to p38γ by native mass spectrometry
Recombinant p38γ (450 µM) was buffer-exchanged into 50 mM ammonium acetate (pH 7.5) using an Amicon Ultra 10 kDa cutoff filter and diluted to a final concentration of ~9 µM. Potential ligands were prepared at 10 mM in HPLC/MS-grade water (Vinorelbine ditartrate, Betaxolol HCl, DL-Carnitine HCl, Sildenafil citrate, myo-Inositol, Oclacitinib maleate) or ethanol (remaining ligands). Ligands (1 µL) were added to 50 µL aliquots of p38γ to achieve final concentrations of 9 µM p38γ and 200 µM ligand.
Samples were analyzed on an Agilent 6520 QTOF mass spectrometer operated in high mass mode, with flow injection at 100 µL/min using 50 mM ammonium acetate (pH 7.5) as carrier solvent. Instrument parameters were optimized for native mass spectrometry. Spectra were averaged, and background spectra (collected during protein-free elution) were subtracted. Data in the m/z 2500–4500 range were deconvoluted using UniDec.
Statistical analysis
All experimental data are presented as mean ± standard error of the mean (SEM), unless otherwise indicated. Statistical significance of differences, such as in cell viability assays and mRNA expression of target genes, was assessed using the Student’s t-test (SPSS; IBM, Armonk, NY) or one-way analysis of variance (ANOVA) (GraphPad PRISM, version 3.0, GraphPad). Differences were considered significant at P < 0.05.
Data Sharing Statement
For original results such as public dataset analysis, cell viability assays and other analysis, please contact Xu Hannah Zhang PhD xuzhang@coh.org. FDA drug library screening data, please contact Sangkil Nam (snam@coh.org). For single cell analysis, please contact Xiwei Wu (xwu@coh.org); For original Mass spectrometry data please contact Roger Moore (rgmoore@coh.org). Other raw data may be found in a data supplement available with the online version of this article.
Conflict of Interest
There are no conflicts to declare.
Author Contributions
XHZ contributed to conceptualization, design and investigation, writing—original draft, and writing—review, editing and corresponding, initializing several key assays and analysis such as Nilotinib toxicity assessments, etc.; HL contributed to conceptualization of docking/virtual screening; YCY contributed to public dataset mining; JH contributed to cell viability assays and synergistical assays of compounds pairs, summarized Table 2; XW contributed to microarray analysis; SH administrated lab protocols; RM contributed to experiments and data analysis; SN contributed to FDA library screening and kinase activity assays; STR contributed to overseeing the lab.
Acknowledgements
Research reported in this publication included work performed in City of Hope Cores (Integrative Genomics, Bioinformatics Core, Integrated Mass Spectrometry Core Facility) supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. Other support included 1R01CA233922-01 (ROSEN) and LLS Grant ID: 6576-19 (ROSEN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
2. Chen G, Hitomi M, Han J, Stacey DW. The p38 pathway provides negative feedback for Ras proliferative signaling. J Biol Chem. 2000 Dec 15;275(50):38973–80.
3. Matsuo Y, Amano S, Furuya M, Namiki K, Sakurai K, Nishiyama M, et al. Involvement of p38alpha mitogen-activated protein kinase in lung metastasis of tumor cells. J Biol Chem. 2006 Dec 1;281(48):36767–75.
4. Demidov ON, Kek C, Shreeram S, Timofeev O, Fornace AJ, Appella E, et al. The role of the MKK6/p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene. 2007 Apr 12;26(17):2502–6.
5. Loesch M, Chen G. The p38 MAPK stress pathway as a tumor suppressor or more? Front Biosci. 2008 May 1;13:3581–93.
6. Campbell RM, Anderson BD, Brooks NA, Brooks HB, Chan EM, De Dios A, et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol Cancer Ther. 2014 Feb;13(2):364–74.
7. Zhang XH, Hsiang J, Rosen ST. Flavopiridol (Alvocidib), a Cyclin-dependent Kinases (CDKs) Inhibitor, Found Synergy Effects with Niclosamide in Cutaneous T-cell Lymphoma. J Clin Haematol. 2021;2(2):48–61.
8. Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007 May;7(5):345–56.
9. Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010 Jun 17;362(24):2251–9.
10. Guilhot F, Hehlmann R. Long-term outcomes of tyrosine kinase inhibitors in chronic myeloid leukemia. Blood. 2025 Feb 27;145(9):910–20.
11. Zhang XH, Nam S, Wu J, Chen CH, Liu X, Li H, et al. Multi-Kinase Inhibitor with Anti-p38γ Activity in Cutaneous T-Cell Lymphoma. J Invest Dermatol. 2018 Nov;138(11):2377–87.
12. Contreras O, Villarreal M, Brandan E. Nilotinib impairs skeletal myogenesis by increasing myoblast proliferation. Skelet Muscle. 2018 Feb 20;8(1):5.
13. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014 Sep 23;7(344):re8.
14. Xu M, Wang S, Wang Y, Wu H, Frank JA, Zhang Z, et al. Role of p38γ MAPK in regulation of EMT and cancer stem cells. Biochim Biophys Acta Mol Basis Dis. 2018 Nov;1864(11):3605–17.
15. Di Raimondo C, Rubio-Gonzalez B, Palmer J, Weisenburger DD, Zain J, Wu X, et al. Expression of immune checkpoint molecules programmed death protein 1, programmed death-ligand 1 and inducible T-cell co-stimulator in mycosis fungoides and Sézary syndrome: association with disease stage and clinical outcome. Br J Dermatol. 2022 Aug;187(2):234–43.
16. Zhang XH, Chen CH, Li H, Hsiang J, Wu X, Hu W, et al. Targeting the non-ATP-binding pocket of the MAP kinase p38γ mediates a novel mechanism of cytotoxicity in cutaneous T-cell lymphoma (CTCL). FEBS Lett. 2021 Oct;595(20):2570–92.
17. Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W, Bochinski K, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006 Jun 15;354(24):2542–51.
18. Kantarjian HM, Giles FJ, Bhalla KN, Pinilla-Ibarz J, Larson RA, Gattermann N, et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase after imatinib resistance or intolerance: 24-month follow-up results. Blood. 2011 Jan 27;117(4):1141–5.
19. Mahon FX, Boquimpani C, Kim DW, Benyamini N, Clementino NCD, Shuvaev V, et al. Treatment-Free Remission After Second-Line Nilotinib Treatment in Patients With Chronic Myeloid Leukemia in Chronic Phase: Results From a Single-Group, Phase 2, Open-Label Study. Ann Intern Med. 2018 Apr 3;168(7):461–70.
20. Hijiya N, Maschan A, Rizzari C, Shimada H, Dufour C, Goto H, et al. Phase 2 study of nilotinib in pediatric patients with Philadelphia chromosome-positive chronic myeloid leukemia. Blood. 2019 Dec 5;134(23):2036–45.
21. Hijiya N, Maschan A, Rizzari C, Shimada H, Dufour C, Goto H, et al. A phase 2 study of nilotinib in pediatric patients with CML: long-term update on growth retardation and safety. Blood Adv. 2021 Jul 27;5(14):2925–34.
22. Ross DM, Masszi T, Gómez Casares MT, Hellmann A, Stentoft J, Conneally E, et al. Durable treatment-free remission in patients with chronic myeloid leukemia in chronic phase following frontline nilotinib: 96-week update of the ENESTfreedom study. J Cancer Res Clin Oncol. 2018 May;144(5):945–54.
23. Ross DM, Hughes TP. Treatment-free remission in patients with chronic myeloid leukaemia. Nat Rev Clin Oncol. 2020 Aug;17(8):493–503.
24. PDQ Cancer Information Summaries [Internet]. Bethesda (MD): National Cancer Institute (US); 2002-.
25. Saydam G, Haznedaroglu IC, Kaynar L, Yavuz AS, Ali R, Guvenc B, et al. Outcomes with frontline nilotinib treatment in Turkish patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase. Hematology. 2018 Feb 27:1–7.
26. Hughes TP, Saglio G, Geissler J, Kim DW, Lomaia E, Mayer J, et al. Asciminib add-on to imatinib demonstrates sustained high rates of ongoing therapy and deep molecular responses with prolonged follow-up in the ASC4MORE study. J Hematol Oncol. 2024 Dec 18;17(1):125.
27. Hijiya N, Zwaan CM, Rizzari C, Foà R, Abbink F, Lancaster D, et al. Pharmacokinetics of Nilotinib in Pediatric Patients with Philadelphia Chromosome-Positive Chronic Myeloid Leukemia or Acute Lymphoblastic Leukemia. Clin Cancer Res. 2020 Feb 15;26(4):812–20.
28. Mojtahedi H, Yazdanpanah N, Rezaei N. Chronic myeloid leukemia stem cells: targeting therapeutic implications. Stem Cell Res Ther. 2021 Dec 18;12(1):603.
29. Bylesjö EI, Forsgren L, Boman K. Epilepsy is difficult to treat during an attack of porphyria. Lakartidningen. 1996 Jun 26;93(26-27):2506–8.
30. Rothman KJ, Lanes S, Sacks ST. The reporting odds ratio and its advantages over the proportional reporting ratio. Pharmacoepidemiol Drug Saf. 2004 Aug;13(8):519–23.
31. Wysowski DK, Swartz L. Adverse drug event surveillance and drug withdrawals in the United States, 1969-2002: the importance of reporting suspected reactions. Arch Intern Med. 2005 Jun 27;165(12):1363–9.
32. Rodriguez EM, Staffa JA, Graham DJ. The role of databases in drug postmarketing surveillance. Pharmacoepidemiol Drug Saf. 2001 Aug-Sep;10(5):407–10.
33. Sakaeda T, Tamon A, Kadoyama K, Okuno Y. Data mining of the public version of the FDA Adverse Event Reporting System. Int J Med Sci. 2013 Apr 25;10(7):796–803.
34. Goedert M, Hasegawa J, Craxton M, Leversha MA, Clegg S. Assignment of the human stress-activated protein kinase-3 gene (SAPK3) to chromosome 22q13.3 by fluorescence in situ hybridization. Genomics. 1997 May 1;41(3):501–2.
35. Ortiz-Hidalgo C. History of Leukemia, Revisited. Curr Oncol Rep. 2025 Apr;27(4):472–82.
36. Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984 Jan;36(1):93–9.
37. Mills KI, Kohlmann A, Williams PM, Wieczorek L, Liu WM, Li R, et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood. 2009 Jul 30;114(5):1063–72.
38. Wang J, Lou P, Lesniewski R, Henkin J. Paclitaxel at ultra low concentrations inhibits angiogenesis without affecting cellular microtubule assembly. Anticancer Drugs. 2003 Jan;14(1):13–9.
39. Shin SJ, O'Sullivan Coyne G, Kummar S, Miller SB, Johnson BC, Anderson L, et al. A Phase I Study of Nilotinib in Combination with Paclitaxel in Patients with Advanced Solid Tumors. Clin Cancer Res. 2025 Jun 3;31(11):2124–33.
40. Hu Y, Girdenyté M, Roest L, Liukkonen I, Siskou M, Bällgren F, et al. Analysis of the contributing role of drug transport across biological barriers in the development and treatment of chemotherapy-induced peripheral neuropathy. Fluids Barriers CNS. 2024 Feb 8;21(1):13.
41. Targeted Therapy Directed by Genetic Testing in Treating Patients with Locally Advanced or Advanced Solid Tumors, The ComboMATCH Screening Trial [Internet]. ClinicalTrials.gov. Bethesda (MD): National Cancer Institute; 2025 [updated 2025 Sep 05]. Available from: https://www.clinicaltrials.gov/study/NCT05564377. Identifier: NCT05564377.
42. Weigel MT, Rath K, Alkatout I, Wenners AS, Schem C, Maass N, et al. Nilotinib in combination with carboplatin and paclitaxel is a candidate for ovarian cancer treatment. Oncology. 2014;87(4):232–45.
43. Leblanc AF, Sprowl JA, Alberti P, Chiorazzi A, Arnold WD, Gibson AA, et al. OATP1B2 deficiency protects against paclitaxel-induced neurotoxicity. J Clin Invest. 2018 Feb 1;128(2):816–25.
44. Boehmerle W, Hagenacker T, Leo M, Schmitt LI, Lehmann HC, Klein I, et al. Results of the preclinical multicenter randomized controlled paclitaxel-induced neuropathy prevention replication study (PINPRICS). BMC Res Notes. 2025 Apr 8;18(1):145.
45. Kim MJ, Lee JW, Oh KS, Choi CS, Kim KH, Han WS, et al. The tyrosine kinase inhibitor nilotinib selectively inhibits CYP2C8 activities in human liver microsomes. Drug Metab Pharmacokinet. 2013;28(6):462–7.
46. Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ, et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013 Jan 28;328(2):307–17.
47. Tiwari AK, Sodani K, Wang SR, Kuang YH, Ashby CR Jr, Chen X, et al. Nilotinib (AMN107, Tasigna) reverses multidrug resistance by inhibiting the activity of the ABCB1/Pgp and ABCG2/BCRP/MXR transporters. Biochem Pharmacol. 2009 Jul 15;78(2):153–61.
48. Shen T, Kuang YH, Ashby CR, Lei Y, Chen A, Zhou Y, et al. Imatinib and nilotinib reverse multidrug resistance in cancer cells by inhibiting the efflux activity of the MRP7 (ABCC10). PLoS One. 2009 Oct 20;4(10):e7520.
49. Navas T, Kinders RJ, Lawrence SM, Ferry-Galow KV, Borgel S, Hollingshead MG, et al. Clinical Evolution of Epithelial-Mesenchymal Transition in Human Carcinomas. Cancer Res. 2020 Jan 15;80(2):304–18.
50. Agarwal K, Katare DP, Jakhmola-Mani R. Foresee Novel Targets for Alzheimer's Disease by Investigating Repurposed Drugs. CNS Neurol Disord Drug Targets. 2023;22(8):1209–31.
51. Balachandran L, Haw TJ, Leong AJW, Croft AJ, Chen D, Kelly C, et al. Cancer Therapies and Cardiomyocyte Viability: Which Drugs are Directly Cardiotoxic? Heart Lung Circ. 2024 May;33(5):747–52.
52. Kurilov R, Haibe-Kains B, Brors B. Assessment of modelling strategies for drug response prediction in cell lines and xenografts. Sci Rep. 2020 Feb 18;10(1):2849.
53. Wang Y, Wang M, Qi H, Pan P, Hou T, Li J, et al. Pathway-dependent inhibition of paclitaxel hydroxylation by kinase inhibitors and assessment of drug-drug interaction potentials. Drug Metab Dispos. 2014 Apr;42(4):782–95.
54. Holbeck SL, Camalier R, Crowell JA, Govindharajulu JP, Hollingshead M, Anderson LW, et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017 Jul 1;77(13):3564–76.
55. Nakamae H, Yamamoto M, Sakaida E, Kanda Y, Ohmine K, Ono T, et al. Nilotinib vs. imatinib in Japanese patients with newly diagnosed chronic myeloid leukemia in chronic phase: 10-year follow up of the Japanese subgroup of the randomized ENESTnd trial. Int J Hematol. 2022 Jan;115(1):33–42.
56. Gugliotta G, Castagnetti F, Breccia M, Levato L, Intermesoli T, D'Adda M, et al. Treatment-free remission in chronic myeloid leukemia patients treated front-line with nilotinib: 10-year followup of the GIMEMA CML 0307 study. Haematologica. 2022 Oct 1;107(10):2356–64.
57. Amack JD. Cellular dynamics of EMT: lessons from live in vivo imaging of embryonic development. Cell Commun Signal. 2021 Jul 22;19(1):79.
58. Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994 Dec;127(6 Pt 2):2021–36.
59. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008 Dec 4;456(7222):593–8.
60. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016 Jun 30;166(1):21–45.
61. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019 Feb;20(2):69–84.
62. Klemm F, Maas RR, Bowman RL, Kornete M, Soukup K, Nassiri S, et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell. 2020 Jun 25;181(7):1643–1660.e17.
63. Eckner R, Ewen ME, Newsome D, Gerdes M, DeCaprio JA, Lawrence JB, et al. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994 Apr 15;8(8):869–84.
64. Zhang P, Zhang M. Epigenetic alterations and advancement of treatment in peripheral T-cell lymphoma. Clin Epigenetics. 2020 Nov 7;12(1):169.
65. Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006 Aug;79(2):370–7.
66. Xiong Y, Li F, Babault N, Dong A, Zeng H, Wu H, et al. Discovery of Potent and Selective Inhibitors for G9a-Like Protein (GLP) Lysine Methyltransferase. J Med Chem. 2017 Mar 9;60(5):1876–91.
67. Nachiyappan A, Gupta N, Taneja R. EHMT1/EHMT2 in EMT, cancer stemness and drug resistance: emerging evidence and mechanisms. FEBS J. 2022 Mar;289(5):1329–51.
68. Nachiyappan A, Soon JLJ, Lim HJ, Lee VK, Taneja R. EHMT1 promotes tumor progression and maintains stemness by regulating ALDH1A1 expression in alveolar rhabdomyosarcoma. J Pathol. 2022 Mar;256(3):349–62.
69. Qi X, Yin N, Ma S, Lepp A, Tang J, Jing W, et al. p38γ MAPK Is a Therapeutic Target for Triple-Negative Breast Cancer by Stimulation of Cancer Stem-Like Cell Expansion. Stem Cells. 2015 Sep;33(9):2738–47.
70. Tomás-Loba A, Manieri E, González-Terán B, Mora A, Leiva-Vega L, Santamans AM, et al. p38γ is essential for cell cycle progression and liver tumorigenesis. Nature. 2019 Apr;568(7753):557–60.
71. Moran N. p38 kinase inhibitor approved for idiopathic pulmonary fibrosis. Nat Biotechnol. 2011 Apr;29(4):301.