Abstract
Proto-oncogenes like C-MYC, EGFR and others have physiological function in regeneration, wound and any stressfully injury to maintain tissue echotexture and healing. Notably, these growth factors work together and had life span to retain to basal level after tissue remodeling and retain its function like what happen in partial hepatectomy. While in cancer, as we work out in 2 transgenic model of liver cancer, we notice that oncogenes do not like each other and just one of them was highly expressive, it keeps other ones at basal level or degraded. Moreover, if one oncogene was inhibited or silenced, other oncogenes become active or expressed and result in anticancer drug resistance. So bispecific antibody could successfully reduce anti-cancer drug resistance.
Introduction
Cross talk of tyrosine growth factors and c-Myc in liver regeneration
The liver regeneration in adult mice requires c-Myc [1], postnatal hepatocyte proliferation does not. Our findings demonstrate unexpected physiological roles for c-Myc in hepatocyte growth and proliferation throughout liver development. Here, we demonstrate that decreased hepatocyte size, cell ploidy, and disordered organ architecture are all results of perinatal c-Myc inactivation in the liver [2,3].
Under physiological circumstances, the loss of c-met did not seem to have a negative impact on hepatocyte function [4]. Yet, there was a significant impact on the liver's adaptive reactions to injury [5-7]. Moreover, the contribution of c-Met in the control of HSC response and support a distinct function for HGF/c-Met as a growth-factor-signaling pathway necessary for the regeneration of damaged liver [7-9].
The neuronal and epithelial organs of mice lacking the epidermal growth factor receptor (EGFR) exhibit a variety of abnormalities between mid-gestation and postnatal day 20 [10]. The transmembrane receptor tyrosine kinase known as the EGFR is activated by a number of ligands and results in the activation of several signaling pathways that primarily regulate proliferation, differentiation, and survival. It has been demonstrated that the EGFR signaling axis is crucial for liver regeneration after acute and long-term liver damage, as well as in cirrhosis [11]. While combined disturbance of EGFR and c-Met caused liver failure in normal mice [12].
Notably, High-level data evidence the share of growth factor like c-Met, c-Myc, EGFR in liver regeneration following resection and cytokine serial assessments in blood samples used to forecast liver [13].
Cross talk of tyrosine growth factors and c-Myc in liver cancer
Loss of c-met enhanced liver cancer progression in c-Met conditional knockout mice as it extended phosphorylation and EGFR activity were linked to increased hepatocarcinogenesis, and EGF-induced liver cancer over-expression in a transgenic mouse model [14,15].
In liver cancer, there is a synergy between c-Met and c-Myc, and c-Met transgenic animals have a defective Fas-mediated apoptotic pathway [16]. Although removal or reduction of HGF/c-Met signaling enhanced the tumor progression in both c-Myc transgenic mice and/or c-Myc/TGF transgenic mice, which is a typical function of liver cancers [17].
In our study in both EGF and c-Myc transgenic mouse model (submit article), we notice that in EGF transgenic model, the prolonged activation of EGFR enhanced loss of c-Met and keep the Myc protein at basal level similar to control non transgenic mice. While in ATT-myc model, we notice that both c-Met and EGFR were reduced and degraded with advanced stage of liver cancer [18].
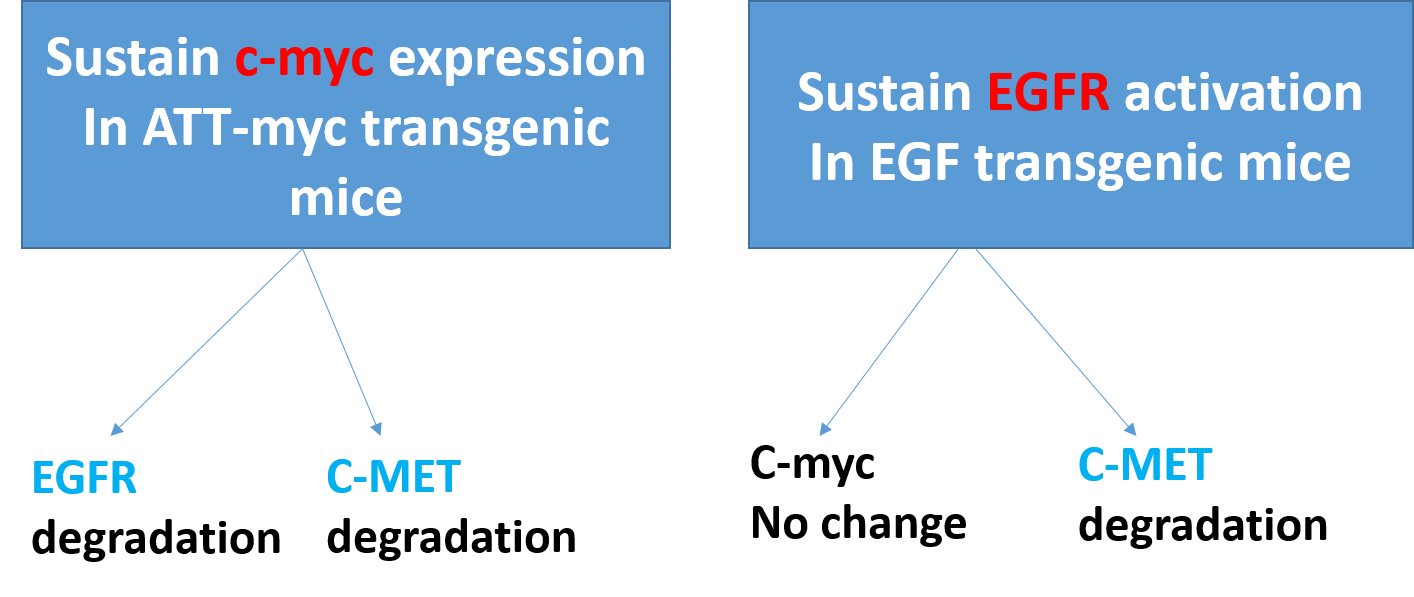
Figure 1.
Anti-cancer drugs resistance
Importantly, integrin 1 activation induced by ligands could result in c-Met activation, which would result in EGFR-TKI resistance [19]. In contrast, prolonged contact with the c-Met inhibitor (PF-2341066) caused the cells' EGFR to become activated [20]. Moreover, cells that are resistant to EGFR activation become vulnerable to PF-2341066 and an EGFR inhibitor when used together [21]. This implies that in resistant cells, the c-Met signaling may be balanced out by the EGFR pathway. Significantly, a recent study found that in NSCLC models, the selective c-Met inhibitor tepotinib can overcome resistance to epidermal growth factor receptor inhibitors caused by aberrant c-Met activation [22]. Also, EGFR tyrosine kinase inhibitor (TKI) resistance on-small cell lung cancer (NSCLC) result from aberrant c-Met activity [23,24] which mean that inhibition of EGFR leads to upregulation of c-Met or inhibition of c-Met could be compensated by activation of EGFR, and thus, this explains the resistance of TKI of both anti-EGFR or anti-c-Met.
Anti-EGFR resistance in metastatic colorectal cancer may be caused by changes to the transcriptional factor c-MYC (mCRC). Also, we discovered a strong relationship between the expression of the c-MYC associated miRNAs indicated above, the level of c-MYC, and anti-EGFR resistance. Moreover, c-critical MYC's function in CRC-related cell-cycle, apoptosis, signal transduction, and cell-growth pathways was highlighted by expression gene profiling [25,26].
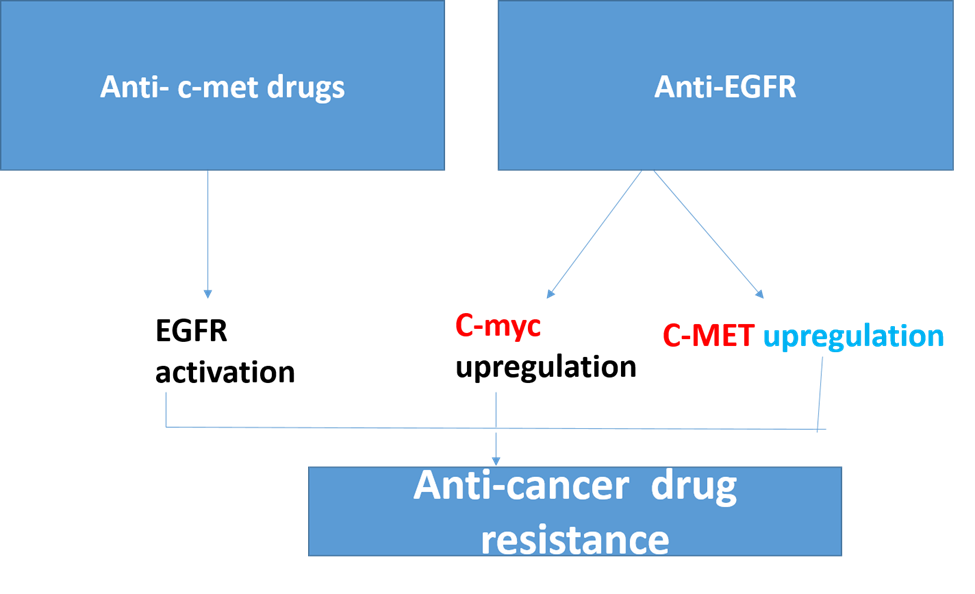
Figure 2.
Conclusions
The oncogene interaction and feedback expression play important role in tumor progression, and resistance against anti-cancer therapy.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through large group Research Project under grant number RGP2/291/44.
References
2. Baena E, Gandarillas A, Vallespinós M, Zanet J, Bachs O, Redondo C, et al. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proceedings of the National Academy of Sciences. 2005 May 17;102(20):7286-91.
3. Zheng K, Cubero FJ, Nevzorova YA. c-MYC—Making liver sick: Role of c-MYC in hepatic cell function, homeostasis and disease. Genes. 2017 Apr 19;8(4):123.
4. Nakamura T, Mizuno S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proceedings of the Japan Academy, Series B. 2010 Jun 11;86(6):588-610.
5. Factor VM, Seo D, Ishikawa T, Kaposi-Novak P, Marquardt JU, Andersen JB, et al. Loss of c-Met disrupts gene expression program required for G2/M progression during liver regeneration in mice. PloS one. 2010 Sep 16;5(9):e12739.
6. Huh CG, Factor VM, Sánchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proceedings of the National Academy of Sciences. 2004 Mar 30;101(13):4477-82.
7. Ishikawa T, Factor VM, Marquardt JU, Raggi C, Seo D, Kitade M, et al. Hepatocyte growth factor/c?met signaling is required for stem?cell–mediated liver regeneration in mice. Hepatology. 2012 Apr;55(4):1215-26.
8. Zhang XJ, Olsavszky V, Yin Y, Wang B, Engleitner T, Öllinger R, et al. Angiocrine hepatocyte growth factor signaling controls physiological organ and body size and dynamic hepatocyte proliferation to prevent liver damage during regeneration. The American Journal of Pathology. 2020 Feb 1;190(2):358-71.
9. Zhao Y, Ye W, Wang YD, Chen WD. HGF/c-Met: a key promoter in liver regeneration. Frontiers in Pharmacology. 2022;13.
10. Natarajan A, Wagner B, Sibilia M. The EGF receptor is required for efficient liver regeneration. Proceedings of the National Academy of Sciences. 2007 Oct 23;104(43):17081-6.
11. Komposch K, Sibilia M. EGFR signaling in liver diseases. International Journal of Molecular Sciences. 2015 Dec 29;17(1):30.
12. Tsagianni A, Mars WM, Bhushan B, Bowen WC, Orr A, Stoops J, et al. Combined systemic disruption of MET and epidermal growth factor receptor signaling causes liver failure in normal mice. The American Journal of Pathology. 2018 Oct 1;188(10):2223-35.
13. Hoffmann K, Nagel AJ, Tanabe K, Fuchs J, Dehlke K, Ghamarnejad O, et al. Markers of liver regeneration—the role of growth factors and cytokines: a systematic review. BMC Surgery. 2020 Dec;20:1-15.
14. Borlak J, Meier T, Halter R, Spanel R, Spanel-Borowski K. Epidermal growth factor-induced hepatocellular carcinoma: gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene. 2005 Mar;24(11):1809-19.
15. Takami T, Kaposi-Novak P, Uchida K, Gomez-Quiroz LE, Conner EA, Factor VM, et al. Loss of hepatocyte growth factor/c-Met signaling pathway accelerates early stages of N-nitrosodiethylamine–induced hepatocarcinogenesis. Cancer Research. 2007 Oct 15;67(20):9844-51.
16. Amicone L, Terradillos O, Calvo L, Costabile B, Cicchini C, Rocca CD, et al. Synergy between truncated c-Met (cyto-Met) and c-Myc in liver oncogenesis: importance of TGF-? signalling in the control of liver homeostasis and transformation. Oncogene. 2002 Feb;21(9):1335-45.
17. Thorgeirsson SS, Santoni-Rugiu E. Interaction of c-myc with transforming growth factor ? and hepatocyte growth factor in hepatocarcinogenesis. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 1997 May 12;376(1-2):221-34.
18. Elalfy MM. Studies on hepatocell carcinoma due to misuse of some preservatives (Doctoral dissertation, Hannover Medical school-Hanover-Germany).
19. Ju L, Zhou C. Association of integrin beta1 and c-MET in mediating EGFR TKI gefitinib resistance in non-small cell lung cancer. Cancer cell international. 2013 Dec;13:1-8.
20. McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance to MET kinase inhibition in MET-dependent non-small cell lung cancer cells mediated by a switch to EGFR dependency. Cancer Research. 2010 Feb 2;70(4):1625.
21. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010 Jan 19;17(1):77-88.
22. Friese-Hamim M, Bladt F, Locatelli G, Stammberger U, Blaukat A. The selective c-Met inhibitor tepotinib can overcome epidermal growth factor receptor inhibitor resistance mediated by aberrant c-Met activation in NSCLC models. American Journal of Cancer Research. 2017;7(4):962.
23. Hochart A, Leblond P, Le Bourhis X, Meignan S, Tulasne D. MET receptor inhibition: Hope against resistance to targeted therapies?. Bulletin du Cancer. 2016 Nov 15;104(2):157-66.
24. Wu YL, Yang JC, Kim DW, Su WC, Ahn MJ, Lee DH, et al. Safety and efficacy of INC280 in combination with gefitinib (gef) in patients with EGFR-mutated (mut), MET-positive NSCLC: A single-arm phase lb/ll study. American Society of Clinical Oncology Annual Meeting I. American Society of Clinical Oncology Annual Meeting I. Chicago, USA. 2014.
25. Strippoli A, Cocomazzi A, Basso M, Cenci T, Ricci R, Pierconti F, et al. c-MYC expression is a possible keystone in the colorectal cancer resistance to EGFR inhibitors. Cancers. 2020 Mar 10;12(3):638.
26. Yu Y, Guo M, Wei Y, Yu S, Li H, Wang Y, et al. FoxO3a confers cetuximab resistance in RAS wild-type metastatic colorectal cancer through c-Myc. Oncotarget. 2016 Dec 12;7(49):80888.