Quantitative Proteomics Characterization Deciphers New Rules of Human Th17 cells Differentiation Regulation
A combination of regulated responses toward pathogens and minimized autoimmune reactions is needed for the balanced function of the immune system. Amongst the immunologically important CD4+ lymphocytes, T helper 17 (Th17) cells help maintain homeostasis and provide protection against pathogens of fungal or bacterial origins [1,2]. Th17 cells are characterized by their ability to secrete interleukin (IL)-17, a pro-in lammatory cytokine, which has been associated with the pathogenesis of several in lammatory diseases. The latter include psoriasis, rheumatoid arthritis, in lammatory bowel disease and multiple sclerosis. Accordingly, the targeting of Th17 cells for the treatment of the human autoimmune diseases has received much interest [3-9]. Therefore, an understanding of the molecular mechanism of Th17 cell differentiation is important when developing new therapeutics against human in lammatory diseases.
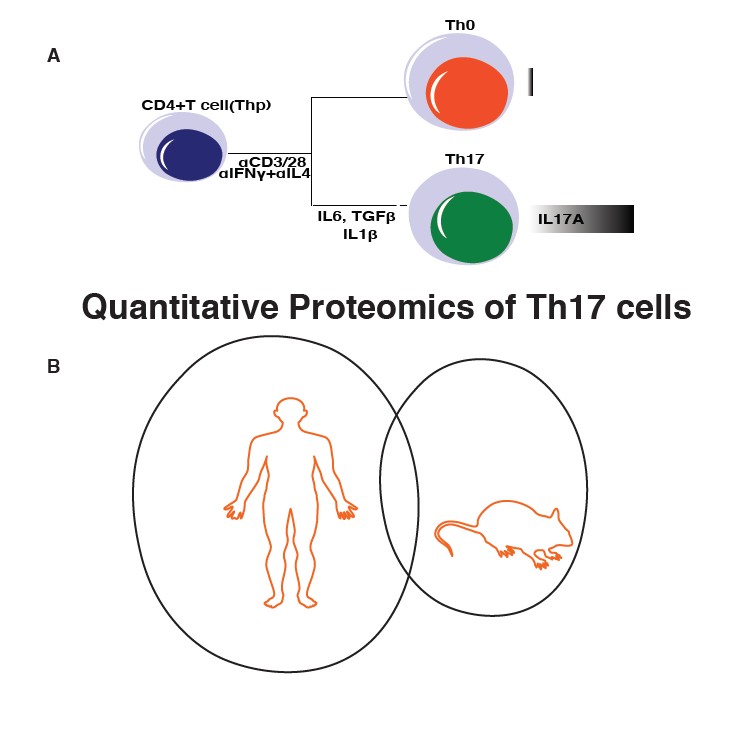
To date, the characteristics of Th17 cells and changes related to Th17 differentiation have been described in transcriptome data sets from both human and mouse [10-16]. These studies revealed important candidates associated with the pathologies caused by Th17 cells, some of which were further studied [3,6,15,17-20]. Notably, our previous comparative careful analysis of RNA-sequencing data of 10 longitudinal samples taken during early Th17 differentiation in human and mouse showed very little overlap between the species [14]. To complement our previous study, we exploited quantitative proteomics to study dynamics of protein expression during early stages of human Th17 cell differentiation. Further, we looked into concurrence of Th17 proteomics with Th17 transcriptome as well as compared Th17 proteomics results between human and mouse.
In the January 2019 issue of iScience, we presented results from a combined analysis of the Th17 cell proteome and transcriptome in human [21]. Importantly, this novel proteomic study of human Th17 cells revealed notable differences and correlations previously not reported. In keeping with an earlier report, there were more striking differences in Th17 regulation between the mouse and human proteins [21]. For example, Tripathi et al. [21] demonstrated that the role of SATB1 in human Th17 cell differentiation was opposite to that observed in the mouse [22-24]. In human Th17 cells, SATB1 was found to negatively regulate differentiation, further emphasising the importance of human studies for translational research. Also, regarding the changes in the human Th17 proteome and transcriptome, comparison of the cells after 72 hours of polarization revealed a high level of overlap; for 93% of the proteins differentially abundant in Th17 cells relative to Th0 cells the detected mRNA transcripts were changed in a similar fashion.
Tripathi et al. [21] used a label-free liquid chromatography-tandem mass spectrometry (LCMS/ MS) approach to profile Thp, Th0, and Th17 cell subsets. Statistical analysis was made to identify proteins differentially expressed (DE) amongst these subsets. Consistent with our previous transcriptomics results [13,14] activation induced the greatest effect on abundance change, with the detection of 2050 and 3470 differential abundant proteins at 24h and 72h, respectively. The comparison between Th0 and Th17 cells identified 148 and 175 differentially regulated proteins at 24h and 72h, respectively. Several proteins previously reported to be differentially regulated during Th17 differentiation were confirmed. Importantly, changes were detected in a number of proteins with no previously known functions in Th17 cells. The latter included the upregulation of UHRF1, GK, ICAM1, KDSR, ATF3, IL4IT, APOD, VIM, PALLD, PTGFRN and KDM6A, and downregulation of ELF1, IL16, BTLA, LAIR1, UHRF1, ANXA1, NDFIP1 and NDFIP2. Future studies could aim at addressing the role of this unique set of proteins in Th17 cell differentiation and function.
Validation measurements, using the combined versatility of immunoblotting, flow cytometry and selected reaction monitoring mass spectrometry (SRM-MS), were made in additional samples to confirm changes in abundance. For example, ACSL4, FHOD1, radixin (RDX), smoothelin (SMTN) and PALLD expression were validated by SRMMS, and OASL and ATF3 by flow cytometry. Western blot analysis confirmed upregulation of BASP1, LMNA, ATP1B1 JUNB, SIRT1, CTNNA1, FAS, VIM, and IRF7, and downregulation of SATB1, SMAD3, STAT4, and ETS1 in Th17 cells. Amongst these, of particular interest for future studies on Th17 cell regulation and Th17 mediated immunopathology is for example ACSL4, a lipid metabolic enzyme that catalyzes addition of a coenzyme-A group to free long-chain fatty acids (Figure 1). Such are also the cytoskeleton proteins SMTN, RDX, and FHOD1, which are already known to play a key role in T cell activation and homeostasis by regulating cell adhesion, migration and T cell interaction with antigen presenting cells [25,26]
The proteomic analysis was made using whole cell lysates, and accordingly analysis of the Gene Ontology (GO) of the DE proteins indicated enrichment of a range of ontologies. The most enriched biological processes reflected cell stimulation and differentiation, i.e. response to external stimulus and cell surface receptor signaling pathway, as well as regulation of the immune system process and immune system development. Lipid metabolic processes were also highly enriched and represented with the previously reported differential regulation of VDR, MSMO1 and CYP51A1 [27,28], as well as other proteins previously not reported in relation to Th17 cells.
Detailed comparison was made between the proteomics of human and mice Th17 cells further emphasizing the dissimilarity of the two. Amongst the top 25 proteins DE in human and mouse only IL2RG was detected in both species. The protein IL2RG is a common receptor gamma chain, and a component of several interleukin receptors, including those of interleukin -2, -4, -7 and -21. Moreover, of altogether 758 differentially regulated proteins in human and 397 in mouse, only 33 were differentially regulated in the same direction, whereas 18 were regulated in an opposite fashion between the two species.
Overall, the study highlights the importance of comprehensive analysis of human cells to provide basis for translational research. Identification of targets that are regulated in a similar fashion in human and mouse points out the candidate human targets worth studying by using in vivo mouse models.
Acknowledgements
We would like to thank Academy of Finland (grants 292335, 294337, 319280, 292482 and 31444) and the Sigrid Jusélius Foundation for their support.
References
2. Hernández-Santos N, Huppler AR, Peterson AC, Khader SA, McKenna KC, Gaffen SL. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal immunology. 2013 Sep;6(5):900.
3. Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, Sloan-Lancaster J. LY2439821, a humanized anti–interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis & Rheumatism. 2010 Apr;62(4):929-39.
4. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, Wehkamp J, Feagan BG, Yao MD, Karczewski M, Karczewski J. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012 Dec 1;61(12):1693-700.
5. Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, Chow J, Manel N, Ciofani M, Kim SV, Cuesta A. Digoxin and its derivatives suppress T H 17 cell differentiation by antagonizing RORγt activity. Nature. 2011 Apr;472(7344):486.
6. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic T H 17 cells. Nature. 2013 Apr;496(7446):518.
7. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA. Induction and molecular signature of pathogenic T H 17 cells. Nature immunology. 2012 Oct;13(10):991.
8. Miossec P, Kolls JK. Targeting IL-17 and T H 17 cells in chronic inflammation. Nature reviews Drug discovery. 2012 Oct;11(10):763.
9. Xiao S, Yosef N, Yang J, Wang Y, Zhou L, Zhu C, Wu C, Baloglu E, Schmidt D, Ramesh R, Lobera M.Small-molecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity. 2014 Apr 17;40(4):477-89.
10. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, Newberry KM. A validated regulatory network for Th17 cell specification. Cell. 2012 Oct 12;151(2):289-303.
11. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, Kuchroo VK. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015 Dec 3;163(6):1400-12.
12. Hu D, Notarbartolo S, Croonenborghs T, Patel B, Cialic R, Yang TH, Aschenbrenner D, Andersson KM, Gattorno M, Pham M, Kivisakk P. Transcriptional signature of human pro-inflammatory TH 17 cells identifies reduced IL10 gene expression in multiple sclerosis. Nature communications. 2017 Nov 17;8(1):1600.
13. Tuomela S, Salo V, Tripathi SK, Chen Z, Laurila K, Gupta B, Äijö T, Oikari L, Stockinger B, Lähdesmäki H, Lahesmaa R. Identification of early gene expression changes during human Th17 cell differentiation. Blood. 2012 Jun 7;119(23):e151-60.
14. Tuomela S, Rautio S, Ahlfors H, Öling V, Salo V, Ullah U, Chen Z, Hämälistö S, Tripathi SK, Äijö T, Rasool O. Comparative analysis of human and mouse transcriptomes of Th17 cell priming. Oncotarget. 2016 Mar 22;7(12):13416.
15. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Zu Horste GM, Pawlak M, Kishi Y. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell. 2015 Dec 3;163(6):1413-27.
16. Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D. Dynamic regulatory network controlling T H 17 cell differentiation. Nature. 2013 Apr;496(7446):461.
17. Huang W, Thomas B, Flynn RA, Gavzy SJ, Wu L, Kim SV, Hall JA, Miraldi ER, Ng CP, Rigo F, Meadows S.DDX5 and its associated lncRNA Rmrp modulate T H 17 cell effector functions. Nature. 2015 Dec;528(7583):517.
18. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, Blumenschein WM, Sukumar S, Haines CJ, Sadekova S, McClanahan TK. Interleukin-23-induced transcription factor Blimp-1 promotes pathogenicity of T helper 17 cells. Immunity. 2016 Jan 19;44(1):131-42.
19. Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH. T-bet represses T H 17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nature immunology. 2011 Jan;12(1):96.
20. Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, Lee YK, Weaver CT, Yagi R, Lazarevic V. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity. 2014 Mar 20;40(3):355-66.
21. Tripathi SK, Välikangas T, Shetty A, Khan MM, Moulder R, Bhosale SD, et al. Quantitative Proteomics Reveals the Dynamic Protein Landscape during Initiation of Human Th17 Cell Polarization. iScience. 2019 Jan 26; 11:334–55.
22. Akiba Y, Kuwabara T, Mukozu T, Mikami T, Kondo M. Special AT- rich sequence binding protein 1 is required for maintenance of T cell receptor responsiveness and development of experimental autoimmune encephalomyelitis. Microbiology and immunology. 2018 Apr;62(4):255-68.
23. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, Newberry KM. A validated regulatory network for Th17 cell specification. Cell. 2012 Oct 12;151(2):289-303.
24. Mohammad I, Nousiainen K, Bhosale SD, Starskaia I, Moulder R, Rokka A, Cheng F, Mohanasundaram P, Eriksson JE, Goodlett DR, Lähdesmäki H. Quantitative proteomic characterization and comparison of T helper 17 and induced regulatory T cells. PLoS biology. 2018 May 31;16(5):e2004194.
25. Faure S, Salazar-Fontana LI, Semichon M, Tybulewicz VL, Bismuth G, Trautmann A, Germain RN, Delon J. ERM proteins regulate cytoskeleton relaxation promoting T cell–APC conjugation. Nature immunology. 2004 Mar;5(3):272.
26. Müller N, Fischer HJ, Tischner D, van den Brandt J, Reichardt HM. Glucocorticoids induce effector T cell depolarization via ERM proteins, thereby impeding migration and APC conjugation. The Journal of Immunology. 2013 Apr 15;190(8):4360-70.
27. Chang SH, Chung Y, Dong C. Vitamin D suppresses Th17 cytokine production by inducing C/EBP homologous protein (CHOP) expression. Journal of Biological Chemistry. 2010 Dec 10;285(50):38751-5
28. Santori FR, Huang P, van de Pavert SA, Douglass Jr EF, Leaver DJ, Haubrich BA, Lorbek G, Konijn T, Rosales BN, Rozman D, Horvat S. Identification of natural RORγ ligands that regulate the development of lymphoid cells. Cell metabolism. 2015 Feb 3;21(2):286-98.