Abstract
Epithelial-mesenchymal transition (EMT), a lineage transition from epithelium to mesenchyme, is induced by a convergence of signaling pathways and often driven by TGFß/SMAD signaling. Except for its critical roles on cell shape and motility during cancer invasion and metastasis, TGFß/SMAD activation-induced EMT has also been reported to be involved in cancer stem cell (CSC) generation. We studied the role of PRMT1 in TGFß/SMAD signaling and found that PRMT1-catalyzed arginine methylation of SMAD7 is required for TGFß/SMAD signaling, and thus critical for TGFß-induced EMT and maintenance of epithelial stemness. Together with the emerging associations between SMAD7, EMT and the sustenance of CSC, PRMT1-catalyzed SMAD7 arginine methylation is not only required for TGFß/SMAD signaling, more importantly, it also bridges EMT and stemness. Here, we offer perspectives on PRMT1 and SMAD7 methylation as potential therapeutic targets for tumor dissemination and relapse.
Keywords
Arginine methylation, PRMT1, SMAD7, TGFβ, EMT, Stemness
Commentary
Epithelial-mesenchymal transition (EMT) is a process that cells transition from epithelial to mesenchymal status, which allow the cells to progressively adopt the elongated appearance, migratory behavior, and invasive capacity. Cells acquire plasticity during EMT, which is crucial for embryonic development, wound healing in the skin, organ fibrosis, and cancer metastasis [1]. EMT is orchestrated by a set of core EMT-inducing transcriptions factors (EMTTFs), including the zinc-finger E-box-binding homeobox factors ZEB1 and ZEB2, SNAIL (also known as SNAI1), SLUG (also known as SNAI2) and the basic helix-loophelix factors TWIST1 and TWIST2 [2]. EMT can be induced by pleiotropic signaling factors through inducing the expression of the EMT-TFs. Among all the EMT activators, the transforming growth factor-β (TGFβ) plays a central role in inducing EMT during almost all the above mentioned physiological and pathological processes [3].
In the canonical TGFβ/Smad signaling pathway, once TGFβ interacts with the TGFβ receptor hetero-tetrameric complex, the type I TGFβ receptors (TbRIs) will be phosphorylated in the cytoplasmic domain and activated by type II TGFβ receptors (TbRIIs). The phosphorylated TbRIs thus interact with and phosphorylate receptorregulated Smads (R-Smads), which then form heteromeric complexes with the common Smad4 (co-Smad) and translocate into the nucleus where they regulate the expression of TGFβ/Smad target genes [4,5]. Among the TGFβ target genes, SMAD7 encodes the inhibitory Smad (I-Smad), Smad7, which establishes a negative feedback loop to inhibit TGFβ signaling. Multiple strategies are employed by I-Smad to repress TGFβ/SMAD signaling, including interference with activation of R-Smad by competitive binding to type I receptors [6,7], facilitating TbRI ubiquitylation and degradation through recruiting of E3 ubiquitin ligases [8] and reversing the activation of TbRI by recruiting phosphatase [9].
PRMT1 Mediated Arginine Methylation of SMAD7 is Required for TGFβ/Smad Signaling
Post-translational modifications (PTMs) of the Smads allow for orchestrated regulation and fine-tuning of the TGFβ signaling system. The phosphorylation and dephosphorylation of R-Smads have been extensively studied and documented as a symbolic feature of TGFβ signaling [4]. The best known PTMs of Smad7 include ubiquitylation by SMURF2, as well as deubiquitylation, acetylation, deacetylation and phosphorylation [10], governing the stability and activity of SMAD7, which further control TGFβ/SMAD signaling [4,11-13] (Figure 1).
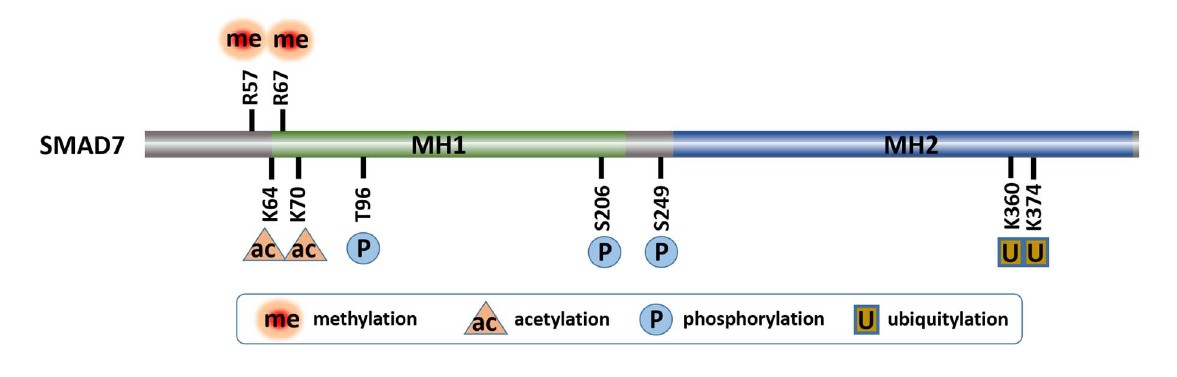
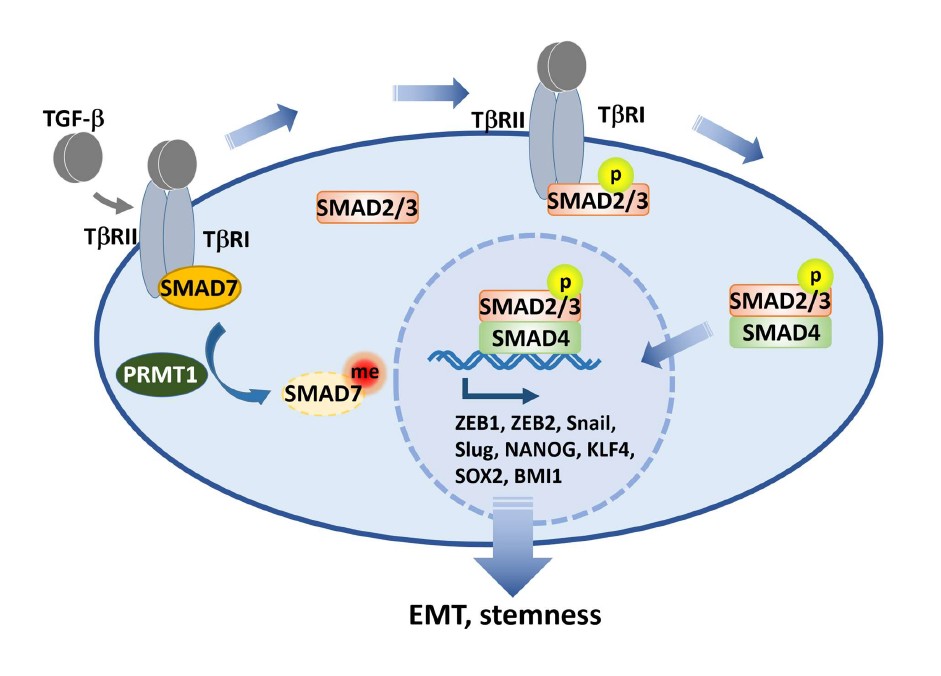
We reported a new PTM of SMAD7, methylation by protein arginine methyltransferase 1 (PRMT1) at arginine (R) 57 and 67 [5]. PRMT1 is the major arginine methyltransferase in the PRMT family, which is responsible for 75% of all arginine methylation in mammalian cells [14]. We found that TGFβ-induced PRMT1-catalyzed arginine methylation of SMAD7 decreased its binding efficiency to TbRI and promoted its dissociation from the activated TbRI. In accordance with the previously mentioned repressing function of I-SMAD, our findings suggested that SMAD7 methylation has the capacity to enhance TbRI availability for R-SMAD phosphorylation and activation [15]. Accordingly, through methylating SMAD7, PRMT1 promoted TGFβ/SMAD signaling activation and subsequent transcription activation of EMT transcription factors. In addition, the methylation of SMAD7 dramatically extended its half-life from half an hour to 4 hours, indicating PRMT1-mediated methylation decreased stability and increased degradation of SMAD7. When PRMT1 expression was suppressed, SMAD7 methylation was suppressed, TGFβ/SMAD signaling was impaired and its target genes could not be induced. These observations indicated that PRMT1 is required for TGFβ/ SMAD signaling [5] (Figure 2).
PRMT1-Smad7 Signaling Bridges EMT and Stemness
Consequential to the modulation of TGFβ/SMAD signaling, PRMT1 is critical for TGFβ-induced expression of core EMT-TF, including ZEB1 and ZEB2, Snail and Slug [5]. The roles of EMT on cell motility and cell shape have been extensively studied, especially in cancer metastasis and invasion. More recently, the relationship and underlying mechanism between EMT and cancer stem cells (CSCs) are emerging. The CSC has been defined as a population of cells with stem cell-like properties that has the ability to initiate tumor growth and give rise to a tumor mass [16]. It has been reported that EMT, or a partial EMT, promote the acquisition of stem-like properties in many types of cancers, including breast, lung, pancreatic and colon carcinomas [3]. EMT-TF ZEB1 was first found to be required for stemness in pancreatic cancer by repressing expression of stemness-inhibiting miRNAs [17]. In mammary gland, together with ZEB1, EMT-TFs Slug and Snail have been shown to promote the stemness of mammary epithelial cells and mammary tumor initiating cells [18-21].
As a pluripotent cytokine, TGFβ is involved in cancer stem cells generation and thus tumor initiation in breast cancer and skin squamous cell carcinomas [18,19,22]. We observed that PRMT1-mediated SMAD7 methylation promoted EMT-TF expression, and was needed in stem cell generation induced by TGFβ. The suppression of PRMT1 significantly repressed the rise of CD44highCD24low cell population, the signature for epithelial stem cells, in human mammary epithelial cells [5]. The expression of the protein markers of mammary epithelial stem cells were examined, including CD44, KLF4, BMI1, POU5F1, and NANOG. The result showed that PRMT1 silencing dramatically inhibited the induction of the expression of those stemness genes and mammosphere formation by TGFβ [5]. Our results demonstrated that SMAD7 methylation by PRMT1 enabled TGFβ/SMAD signaling, and thus promote EMT-TF expression, EMT progression, and cancer stem-cell formation. These observations also provide additional evidence bridging EMT and stemness.
As a key intrinsic negative regulator of TGFβ/ SMAD signaling pathway, reduction of SMAD7 expression leads to enhancement in TGFβ-induced EMT and tumor metastasis. The reduction of SMAD7 expression has been correlated with shorter recurrence-free survival and poor overall survival in hepatocellular carcinoma (HCC), breast cancer, melanoma, glioblastoma, and renal cancer [23-28]. However, there are also studies that reported the opposite observations whereby increased expression of SMAD7 indicated poor prognosis in cancer types, such as melanoma and acute myeloid leukemia (AML) [29,30]. In colorectal cancer (CRC), SMAD7 was observed to play a dual role that it could be associated with either poor prognosis or better outcome [31]. The association between SMAD7 expression and cancer prognosis may ascribe to TGFβ’s bidirectional regulation on cancer [32].
Despite the debated role of SMAD7 in cancer prognosis, it is noteworthy that the role of SMAD7 in the maintenance of CSCs stemness is emerging. It has been reported that suppressing SMAD7 expression enhances CSCs stemness in HCC, breast cancer, nasopharyngeal carcinoma (NPC), and glioma [33-36]. More importantly, SMAD7 ubiquitination, one of the best known PTMs of SMAD7, was found to promote CSCs stemness through promoting its degradation [34,36]. We demonstrated that PRMT1- catalyzed methylation decreased SMAD7 stability and regulated epithelial stemness. Taken together, these findings indicate a role of SMAD7 in maintaining CSCs stemness.
Perspective
In multiple cancer types, PRMT1 has been found to be aberrantly expressed, such as in breast, prostate, colon cancer, and leukemia [37]. It has documented its roles in DNA repair, telomere maintenance, apoptosis and oxidative stress response in cancer formation [38-41]. PRMT1 expression is mostly reported to be increased and associated with poor prognosis [42-46]. In accordance with our observations, PRMT1 is promotive for EMT during tumor progression in many cancer types [47-50]. Recently, we found that by interfering with p53 stability, PRMT1 is also involved in heart development through regulating epicardial differentiation and EMT, further attesting critical roles of PRMT1 in EMT and stemness [51]. EMT has the potential to promote CSC stemness, which is crucial for tumorigenesis, therapy resistance and thus tumor relapse post-treatment [52]. We have demonstrated that PRMT1 induced SMAD7 methylation is required for TGFβ/SMAD signaling and TGFβ induced EMT, which offer a perspective on the roles of PRMT1 and the corresponding SMAD7 methylation in CSCs-related tumorigenesis, therapy resistance and thus tumor relapse post-treatment. Based on our observations, PRMT1 and SMAD7 methylation are potential therapeutic targets for tumor dissemination and relapse.
References
2. Stemmler MP, Eccles RL, Brabletz S, Brabletz T. Nonredundant functions of EMT transcription factors. Nat Cell Biol. 2019 Jan;21(1):102-12.
3. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019 Feb;20(2):69-84.
4. Xu P, Lin X, Feng XH. Posttranslational Regulation of Smads. Cold Spring Harb Perspect Biol. 2016 Dec 1;8(12).
5. Katsuno Y, Qin J, Oses-Prieto J, Wang H, Jackson- Weaver O, Zhang T, et al. Arginine methylation of SMAD7 by PRMT1 in TGF-beta-induced epithelial-mesenchymal transition and epithelial stem-cell generation. J Biol Chem. 2018 Aug 24;293(34):13059-72.
6. Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, et al. The MAD-related protein Smad7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997 Jun 27;89(7):1165-73.
7. Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, et al. Identification of Smad7, a TGFbetainducible antagonist of TGF-beta signalling. Nature. 1997 Oct 9;389(6651):631-5.
8. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000 Dec;6(6):1365-75.
9. Shi W, Sun C, He B, Xiong W, Shi X, Yao D, et al. GADD34- PP1c recruited by Smad7 dephosphorylates TGFbeta type I receptor. J Cell Biol. 2004 Jan 19;164(2):291-300.
10. Ogunjimi AA, Briant DJ, Pece-Barbara N, Le Roy C, Di Guglielmo GM, Kavsak P, et al. Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol Cell. 2005 Aug 5;19(3):297-308.
11. Zhao Y, Thornton AM, Kinney MC, Ma CA, Spinner JJ, Fuss IJ, et al. The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor beta (TGFbeta) signaling and the development of regulatory T cells. J Biol Chem. 2011 Nov 25;286(47):40520-30.
12. Gronroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002 Sep;10(3):483-93.
13. Seong HA, Jung H, Ha H. Murine protein serine/ threonine kinase 38 stimulates TGF-beta signaling in a kinase-dependent manner via direct phosphorylation of Smad proteins. J Biol Chem. 2010 Oct 1;285(40):30959- 70.
14. Blanc RS, Richard S. Arginine Methylation: The Coming of Age. Mol Cell. 2017 Jan 5;65(1):8-24.
15. Miyazawa K, Miyazono K. Regulation of TGF-beta Family Signaling by Inhibitory Smads. Cold Spring Harb Perspect Biol. 2017 Mar 1;9(3).
16. Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol. 2015 Apr;31:28-35.
17. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009 Dec;11(12):1487-95.
18. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013 Jul 3;154(1):61-74.
19. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008 May 16;133(4):704-15.
20. Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012 Mar 2;148(5):1015-28.
21. Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015 Sep 10;525(7568):256-60.
22. Oshimori N, Oristian D, Fuchs E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015 Feb 26;160(5):963-76.
23. Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013 Aug;58(2):629-41.
24. Tong L, Shen S, Huang Q, Fu J, Wang T, Pan L, et al. Proteasome-dependent degradation of Smad7 is critical for lung cancer metastasis. Cell death and differentiation. 2020 Jun;27(6):1795-806.
25. Tuncer E, Calcada RR, Zingg D, Varum S, Cheng P, Freiberger SN, et al. SMAD signaling promotes melanoma metastasis independently of phenotype switching. The Journal of clinical investigation. 2019 Apr 30;129(7):2702- 16.
26. Kit Leng Lui S, Iyengar PV, Jaynes P, Isa Z, Pang B, Tan TZ, et al. USP26 regulates TGF-beta signaling by deubiquitinating and stabilizing SMAD7. EMBO reports. 2017 May;18(5):797-808.
27. Zhai W, Li S, Zhang J, Chen Y, Ma J, Kong W, et al. Sunitinib-suppressed miR-452-5p facilitates renal cancer cell invasion and metastasis through modulating SMAD4/ SMAD7 signals. Molecular cancer. 2018 Nov 12;17(1):157.
28. Eckhardt BL, Cao Y, Redfern AD, Chi LH, Burrows AD, Roslan S, et al. Activation of Canonical BMP4-SMAD7 Signaling Suppresses Breast Cancer Metastasis. Cancer research. 2020 Mar 15;80(6):1304-15.
29. Kaczorowski M, Biecek P, Donizy P, Pieniazek M, Matkowski R, Halon A. SMAD7 is a novel independent predictor of survival in patients with cutaneous melanoma. Translational research : the journal of laboratory and clinical medicine. 2019 Feb;204:72-81.
30. Zhang J, Zhang L, Cui H, Zhang X, Zhang G, Yang X, et al. High expression levels of SMAD3 and SMAD7 at diagnosis predict poor prognosis in acute myeloid leukemia patients undergoing chemotherapy. Cancer gene therapy. 2019 May;26(5-6):119-27.
31. Troncone E, Monteleone G. Smad7 and Colorectal Carcinogenesis: A Double-Edged Sword. Cancers. 2019 May 1;11(5).
32. Akhurst RJ, Derynck R. TGF-beta signaling in cancer- -a double-edged sword. Trends in cell biology. 2001 Nov;11(11):S44-51.
33. Wang Z, Shen M, Lu P, Li X, Zhu S, Yue S. NEDD9 may regulate hepatocellular carcinoma cell metastasis by promoting epithelial-mesenchymal-transition and stemness via repressing Smad7. Oncotarget. 2017 Jan 3;8(1):1714-24.
34. Zhang Z, Fan Y, Xie F, Zhou H, Jin K, Shao L, et al. Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nature communications. 2017 Dec 13;8(1):2116.
35. Cai L, Long Y, Chong T, Cai W, Tsang CM, Zhou X, et al. EBV-miR-BART7-3p Imposes Stemness in Nasopharyngeal Carcinoma Cells by Suppressing SMAD7. Frontiers in genetics. 2019;10:939.
36. Liang H, Wang Q, Wang D, Zheng H, Kalvakolanu DV, Lu H, et al. RGFP966, a histone deacetylase 3 inhibitor, promotes glioma stem cell differentiation by blocking TGFbeta signaling via SMAD7. Biochemical pharmacology. 2020 Jun 23;180:114118.
37. Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013 Jan;13(1):37-50.
38. Boisvert FM, Rhie A, Richard S, Doherty AJ. The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle. 2005 Dec;4(12):1834-41.
39. Dejardin J, Kingston RE. Purification of proteins associated with specific genomic Loci. Cell. 2009 Jan 9;136(1):175-86.
40. Sakamaki J, Daitoku H, Ueno K, Hagiwara A, Yamagata K, Fukamizu A. Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc Natl Acad Sci U S A. 2011 Apr 12;108(15):6085-90.
41. Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. 2008 Oct 24;32(2):221-31.
42. Wang L, Jia Z, Xie D, Zhao T, Tan Z, Zhang S, et al. Methylation of HSP70 orchestrates its binding to and stabilization of BCL-2 mRNA and renders pancreatic cancer cells resistant to therapeutics. Cancer Res. 2020 Jul 22.
43. Song C, Chen T, He L, Ma N, Li JA, Rong YF, et al. PRMT1 promotes pancreatic cancer growth and predicts poor prognosis. Cell Oncol (Dordr). 2020 Feb;43(1):51-62.
44. Chen C, Zhou H, Zhang X, Liu Z, Ma X. PRMT1 potentiates chondrosarcoma development through activation of YAP activity. Mol Carcinog. 2019 Dec;58(12):2193-206.
45. Zhao Y, Lu Q, Li C, Wang X, Jiang L, Huang L, et al. PRMT1 regulates the tumour-initiating properties of esophageal squamous cell carcinoma through histone H4 arginine methylation coupled with transcriptional activation. Cell Death Dis. 2019 May 1;10(5):359.
46. Altan B, Yokobori T, Ide M, Mochiki E, Toyomasu Y, Kogure N, et al. Nuclear PRMT1 expression is associated with poor prognosis and chemosensitivity in gastric cancer patients. Gastric Cancer. 2016 Jul;19(3):789-97.
47. Zhang Y, Wang D, Zhang M, Wei H, Lu Y, Sun Y, et al. Protein arginine methyltransferase 1 coordinates the epithelial-mesenchymal transition/proliferation dichotomy in gastric cancer cells. Exp Cell Res. 2018 Jan 1;362(1):43-50.
48. Avasarala S, Van Scoyk M, Karuppusamy Rathinam MK, Zerayesus S, Zhao X, Zhang W, et al. PRMT1 Is a Novel Regulator of Epithelial-Mesenchymal-Transition in Non-small Cell Lung Cancer. J Biol Chem. 2015 May 22;290(21):13479-89.
49. Li B, Liu L, Li X, Wu L. miR-503 suppresses metastasis of hepatocellular carcinoma cell by targeting PRMT1. Biochem Biophys Res Commun. 2015 Sep 4;464(4):982-7.
50. Gao Y, Zhao Y, Zhang J, Lu Y, Liu X, Geng P, et al. The dual function of PRMT1 in modulating epithelialmesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB1. Sci Rep. 2016 Jan 27;6:19874.
51. Jackson-Weaver O, Ungvijanpunya N, Yuan Y, Qian J, Gou Y, Wu J, et al. PRMT1-p53 Pathway Controls Epicardial EMT and Invasion. Cell Rep. 2020 Jun 9;31(10):107739.
52. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell. 2019 Jan 3;24(1):25-40.