Abstract
HIV-1 has the capability to establish latency during early infection in CD4+ cells, posing a significant challenge to the efforts aimed at curing HIV-1/AIDS. One extensively explored strategy to address this viral latency is the "shock-and-kill" approach. This involves reactivating viral replication using latency reversal agents (LRAs) to induce the death of infected cells. Regrettably, no LRAs with proven effectiveness have been identified thus far. In this study, we investigated the impact of Azidothymidine (AZT) treatment interruption and the administration of Phorbol-12-Myristate-13-Acetate (PMA), a PKC activator, as shock-and-kill approaches in vitro. We employed the susceptible Jurkat cell line and utilized a sensitive real-time PCR assay along with Western blotting analysis. Our findings revealed that AZT inhibited HIV-1 replication, and its treatment interruption led to the reactivation of viral replication. This reactivation occurred through the recruitment of host transcription factors, including NFAT, NF-κBp65, Ap-1, and Sp-1. These factors facilitated HIV production via TCR-related pathways, activation of p-TEFb pathways for transcription elongation, and upregulation of Jak/Stat pathways for viral enhancement. Furthermore, we demonstrated that PMA treatment increased the levels of these transcription factors through the activation of TCR-related signaling pathways in HIV-1 infected Jurkat cells, irrespective of the AZT treatment status. PMA also induced cell death through both extrinsic and intrinsic apoptotic signaling pathways, as well as autophagy. These results suggest that PMA effectively employs the shock-and-kill approach in HIV-1 infected Jurkat cells and highlight the potential of PKC pathway activators as promising LRAs.
Keywords
HIV-1, Latency, AZT PMA, Apoptosis, Autophagy
Introduction
Human immunodeficiency virus type 1 (HIV-1) infects and destructs cells crucial for combating diseases, with over 40.1 million people having succumbed to HIV-1 infection thus far. While antiretroviral therapy (ART) can reduce HIV load to undetectable levels, it requires lifelong continuation, unable to achieve complete HIV eradication. During the early stage, HIV targets specific cell types like resting CD4+ T-cells, integrating into host chromosomal DNA to form latent reservoirs known as latent proviral reservoirs [1]. This latent phase allows HIV replication to rebound upon therapy interruption, rendering the disease incurable due to persistent viral particles throughout the patient's lifetime [1].
Numerous strategies for eliminating HIV latent reservoirs exist, with the "shock-and-kill" approach being extensively studied for functional cure [2]. This method involves activating viral reservoirs using latency-reversing agents (LRAs) to stimulate HIV RNA transcription and protein expression, leading to the elimination of activated infected cells by the immune system or ART [2,3].
To identify effective LRAs, we conducted a review of many molecular pathways involved in HIV-1 replication, elongation, and latency [4-6]. Latently infected cells, not constitutively producing viral particles, can be induced to do so through T-cell activation, often via T-cell receptor (TCR), CD3, CD28 [4,6], and/or PI3K/Akt [4]-related signaling pathways. These pathways activate downstream molecules like Zap-70, PLCγ1, and protein kinase C (PKC), sequentially activating transcription factors NFAT, NF-κB, Ap-1, and Sp-1, possessing consensus binding sequences in the HIV-1 LTR region crucial for replication [3,4].
During the elongation stage, recruitment of p-TEFb by Brd4 is vital, regulated by Tat protein, necessary for full-length HIV-1 mRNA synthesis. The CDK9/cyclin T1 complex within p-TEFb facilitates productive transcription elongation of HIV genomes [7]. Another significant pathway is Jak/Stat, where gp130 phosphorylation associates with Jak/Stat protein transcription factors, correlating with increased integrated viral HIV DNA and replication [8,9]. Jak/Stat pathways activated by p38α counteract AZT's inhibition of HIV-1 replication [10].
Various LRAs as shown in Schematic Figure, including PKC agonists, cytokines, Toll-like receptor agonists, histone deacetylase inhibitors, and bromodomain inhibitors, have been used to reactivate HIV-1 replication in vitro [11,12]. The activation of the NF-κB and AP-1 through PKC pathway stands out as a crucial route in the reactivation of HIV-1. There have been studies on PKC agonists, which are substances that activate PKC, as potential ways to eliminate hidden HIV-1 in the body. However, in clinical trials, some PKC agonists like bryostatin-1 and prostratin were found to increase a process called phosphorylation [13]. This process makes a protective protein called Bcl2 more effective in blocking cell death, and this happens in a way that depends on PKC. As a result, the cells become resistant to dying when exposed to various triggers [13]. Unfortunately, CD4 T cells (a type of immune cell) that produce HIV-1 were discovered to have higher levels of Bcl2 compared to uninfected cells, both in the body and when reactivated in a laboratory setting [13]. This suggests that the activation of certain protective components, like Bcl2, may play a role in why HIV-1 persists even after attempts to wake it up with PKC agonists.
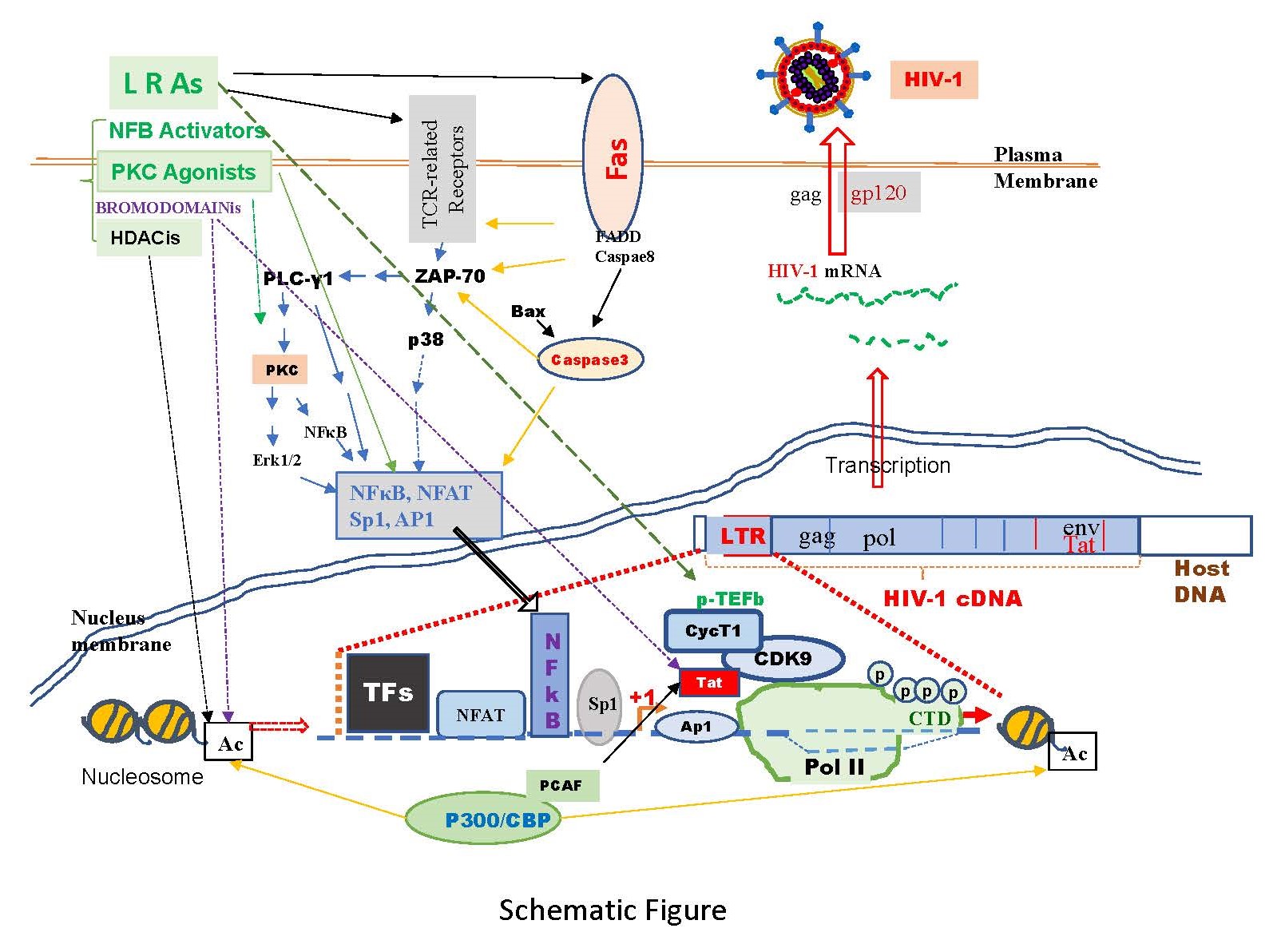
Jurkat cells, a human T lymphocyte (T cell) line, have gained extensive use as a model system for exploring different facets of T cell biology, immunology, and the expression of chemokine receptors vulnerable to viral entry, notably HIV. In this research, we employed Jurkat cells as a model to examine the impact of Phorbol-12-Myristate-13-Acetate (PMA) on the latency of HIV-1. PMA, a diester of phorbol known to activate protein kinase C (PKC) and stimulate cytokine production, has been previously documented to reactivate HIV-1 replication from latency [14]. Our findings indicate that PMA actively participates in the shock-and-kill strategy. It triggers the activation of T-cell receptor (TCR)-related pathways, p-TEFb (positive transcription elongation factor b), and Jak/Stat (Janus kinase/signal transducer and activator of transcription) pathways for the "shock" aspect. Simultaneously, PMA induces cell death through both apoptosis and autophagy, contributing to the "kill" aspect, particularly observed in HIV-1 infected Jurkat cells.
Materials and Methods
Chemicals and reagents
Phorbol-12-Myristate-13-Acetate (PMA) (Supplementary Figure 1A), Azidothymidine (AZT) (also known as Zidovudine (ZDV)) (Supplementary Figure 1B), along with all other chemicals were sourced from Sigma (St. Louis, MO).
Cell culture and treatments
Jurkat, clone E6-1, cells were acquired from the National Institutes of Health AIDS Research Reference and Reagent Program (Germantown, MD). They were cultured at 37°C in 5% CO2 in RPMI 1640 medium containing 10% fetal calf serum, 2 mM glutamine, 50 μg/ml penicillin, and 50 μg/ml streptomycin.
To perform HIV-1 infection, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. The experimental setup is illustrated in Supplementary Figure 2: (A) Study Design I: On day 7, some HIV-1-infected cells were transferred to fresh medium and cultured for an additional 7 days (Treatment A), while others were treated with AZT (2 µm) for 7 days (Treatment B & C). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for another 7 days: Treatment A: Medium only; Treatment B: AZT (2 µm) continuously; Treatment C: AZT interruption. (B) Study Design II: On day 7, some HIV-1-infected cells were transferred to fresh medium for an additional 7 days (Treatment D & E), while others were treated with AZT (2 µm) for 7 days (Treatment F, G, H & I). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for an additional 24 hours: Treatment D: Medium only; Treatment E: PMA (10 ng/ml); Treatment F: AZT (2 µm) continuously; Treatment G: AZT (2 µm)/PMA (10 ng/ml); Treatment H: AZT interruption; Treatment I: AZT interruption with the presence of PMA (10 ng/ml).
RNA isolation and DNA isolation
Viral RNA was isolated from 140 µl of culture supernatant using the QIAamp Viral RNA Mini Kit (Valencia, CA 91355) according to the manufacturer’s protocol. Five (5) out of the 50 µl of the extracted RNA was used as templates for real-time RT-PCR.
Total DNA isolation of approximately 1 x 107 cells was performed with the QIAamp DNA Mini Kit (Valencia, CA 91355) according to the manufacturer’s protocol. Five (5) out of 100 µl of the extracted DNA was used as templates for real-time PCR.
Templates for quantitative PCR included known concentrations of HIV-1 (MN) viral RNA and viral DNA, serially diluted from 108 to 102 copies. A standard curve was generated through the quantitative PCR process. The values presented in the figures denote the average concentration derived from six reactions, with each reaction performed in triplicate and based on the established standard curve.
Real-time PCR
Primers and TaqMan probes for gag p24 were designed, and quantitative PCR was conducted using an automated TaqMan 7500 Analyzer according to our previous reports [10,15]. RT-PCR thermal steps included 55°C for 5 min, 50°C for 30 min, and 95°C for 10 min, followed by 45 cycles of two-step PCR at 95°C for 15 s and 60°C for 1 min.
Western blot analysis
Proteins were extracted from Jurkat cells using RIPA buffer, separated through SDS-PAGE, and transferred onto polyvinylidene difluoride membranes, as outlined in our prior publications [10,15]. The membranes were then incubated with primary antibodies, specifically rabbit polyclonal/mouse monoclonal antibodies targeting AKT, ATG5, Beclin1, Brd4, CD3ζ, CD4, CD28, CDK9, FADD, Fas, FLIP, Jak2, p53, PI3K, PLCγ1, SP1, ULK1, and GAPDH, which were sourced from Santa Cruz Biotechnology (Santa Cruz, CA). Additionally, rabbit polyclonal antibodies against AP-1, Bax, Bcl2, Caspase-3, Caspase-8, Cyclin T1, DRAM, Erk1/2, Grb2, IFI16, LAT, LC3, mTOR, NFAT, NF-kBp65, PKC, p38, p-Rpb-CTD, Ras, SQSTM1/p62, STAT3, TCR3, TCRβ, VPS34, and ZAP70 were obtained from Cell Signaling Technology, Inc (Danvers, MA).
Protein expression levels were comparatively assessed based on data derived from a minimum of two distinct and independent experiments. The quantification process utilized ImageJ (Image Processing and Analysis in Java) from NIH website (https://imagej.nih.gov/ij/).
Statistical analysis
The unpaired Student’s t-test was employed for data analyses. A p-value <0.05 (*) and p-value <0.01 (**) were considered significant and very significant, respectively.
Results
Antiretroviral therapy (ART) has been effective in reducing mortality and morbidity associated with HIV-1, inhibiting its replication and lowering viral RNA in plasma. In this study, Jurkat cells were treated with Azidothymidine (AZT) for 7 days post-HIV-1 infection exhibited a significant reduction in viral replication compared to untreated cells (sFig. 3).
AZT treatment interruption effects
Despite the critical importance of consistent ART usage, non-adherence is prevalent, especially within high-risk populations of individuals living with HIV [16]. To investigate the impact of interrupting AZT treatment on cell death and HIV-1 replication in vitro, Jurkat cells were employed in accordance with the methodology outlined in Study design I (Supplementary Figure 2A). As depicted in Figure 1A, a notable increase in cell death levels was observed following the interruption of AZT treatment (panel C) compared to the non-interruption or treatment continuation scenario (panel B). Furthermore, the interruption of AZT for 7 days led to a significant enhancement in HIV-1 replication (panel C) compared to the non-interruption condition (panel B), as illustrated in Figure 1B.
Recent reports from our studies highlight apoptotic cell death as a preferred mechanism for HIV-1 replication [15,17], with viral replication exhibiting an inverse correlation with HIV-1 latency [18]. Figure 1B underscores this correlation, showing that HIV-1 RNA load (indicative of replication) is indeed inversely proportional to DNA load (a marker of viral latency).
These findings collectively signify that the interruption of AZT treatment induces heightened cell death, increased HIV-1 replication, and decreased HIV-1 latency. Conversely, the sustained administration of AZT results in reduced cell death and HIV-1 replication, coupled with an increase in HIV-1 latency.
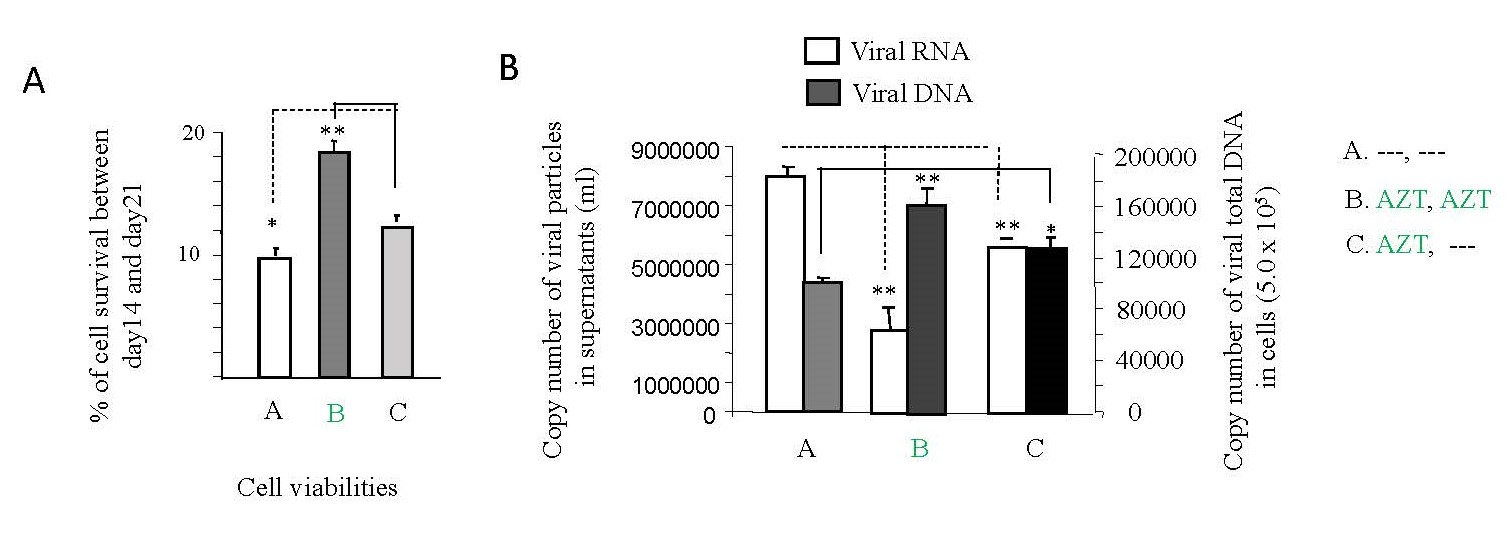
Figure 1. The effects of AZT interruption on HIV-1 replication and latency, cell viabilities. As illustrated in Study Design I, shown in Supplementary Figure 2A, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. On day 7, some HIV-1-infected cells were transferred to fresh medium and cultured for an additional 7 days (Treatment A), while others were treated with AZT (2 µm) for 7 days (Treatment B & C). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for another 7 days: Medium only (Treatment A); AZT (2 µm) continuously (Treatment B); AZT interruption (Treatment C). (A). The number of alive cells was determined by Trypan blue dye exclusion with using EVE™ Automatic Cell Counter, and the data represent the average of three different independent experiments. (B). 140 µl of culture supernatants containing HIV-1 particles and were used to isolate viral RNA for RT-PCR to test viral replication. 1 x 107 of live cells were used to isolate viral DNA for PCR to detect viral DNA levels.
AZT interruption and TCR-related pathways
Certain transcription factors, including NFAT, NF-κBp65, AP-1 and SP-1, whose consensus binding sites are located in the HIV-1 LTR region, play crucial roles in viral replication [3-6]. Previously, we verified that these host transcription factors were regulated by TCR-related pathways [10,15,19]. As shown in Figure 2A, AZT treatment inhibited the expression of these factors (panel B) relative to untreated cells (panel A) and after treatment interruption, their expressions (panel C) returned to levels comparable to those within untreated cells (panel A). Furthermore, examining the molecules downstream in the TCR pathways, AZT interruption increased TCRβ, Zap70, PLCγ1 and PKC (panel C) relative to the AZT treatment group (panel B) (Figure 2B).
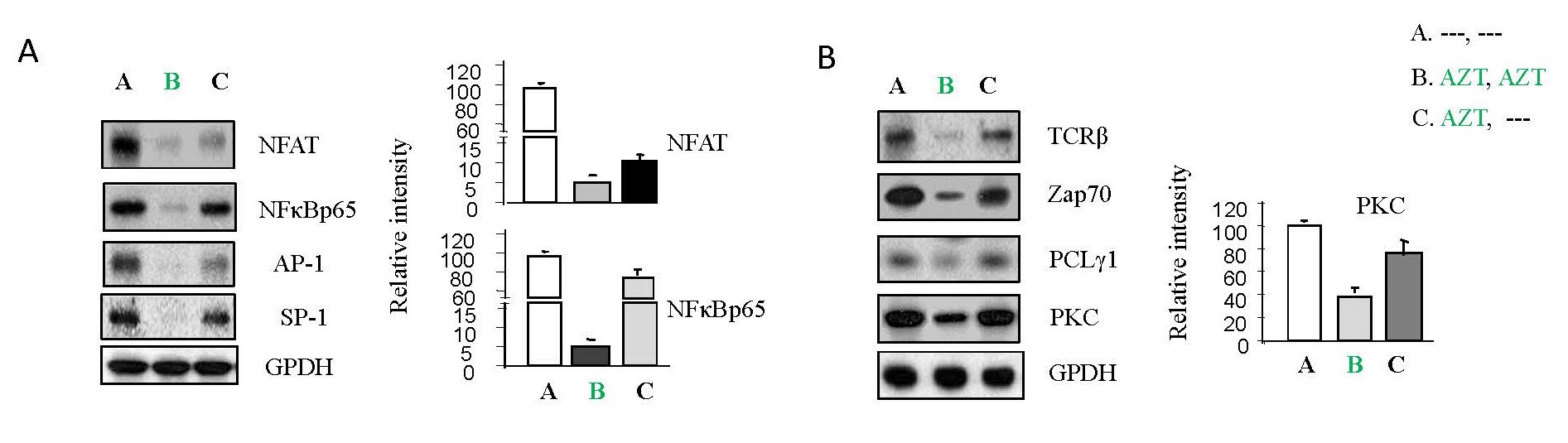
Figure 2. The effects of AZT interruption on host transcription factors and T-cell receptor (TCR)-related signaling pathways in HIV-1 infected Jurkat cells. As illustrated in Study Design I, shown in Supplementary Figure 2A, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. On day 7, some HIV-1-infected cells were transferred to fresh medium and cultured for an additional 7 days (Treatment A), while others were treated with AZT (2 µm) for 7 days (Treatment B & C). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for another 7 days: Medium only (Treatment A); AZT (2 µm) continuously (Treatment B); AZT interruption (Treatment C). To examine if PMA can activate TCR-related pathways, on day 21, total cell lysates were subjected to Western blot analysis to detect the expression of proteins that crucial components of the signaling pathways associated with TCR activation, (A) NFAT, NF-κB p65, Ap-1 and Sp-1; (B) TCRβ, ZAP 70, PLCγ1 and PKC.
AZT interruption and p-TEFb/Jak-Stat pathways
It has been noted that p-TEFb pathways, which activate viral transcription elongation, are also required for HIV-1 replication and reactivation of HIV-1 from latency [7]. As shown in Figure 3A and 3B, AZT interruption increased the expression of the proteins Brd4, CDK9 and Cyclin T1 (panel C) (Figure 3A), as well as the phosphorylation of RPb-CTD which is necessary for viral RNA elongation (panel C) (Figure 3B) relative to the sustained treatment group (panel B) (Figure 3A & 3B). This suggests that AZT treatment inhibits p-TEFb pathways while AZT interruption reactivates them.
We have reported that HIV-1 is able to activate Jak/Stat-related signaling pathways to counteract the AZT inhibition of HIV-1 replication [10]. AZT interruption increased protein expression of Jak2 and Stat3 (panel C) relative to the sustained treatment group (panel B) (Figure 3C).
Collectively, these results indicate that AZT inhibits viral replication through downregulation of TCR-related pathways, p-TEFb, and Jak/Stat pathways. Interruption of AZT reverses these effects, promoting viral replication.
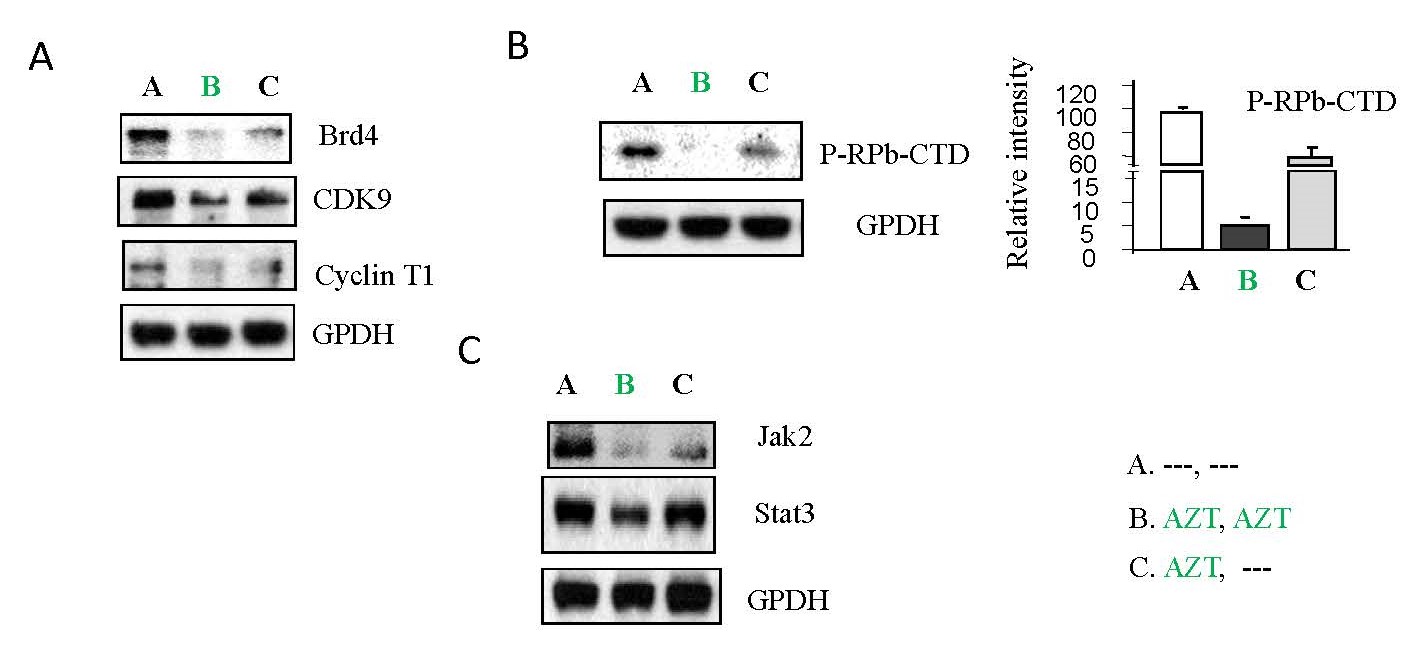
Figure 3. The effects of AZT interruption on P-TEFb pathways and Jak/Stat pathways in HIV-1 infected Jurkat cells. As illustrated in Study Design I, shown in Supplementary Figure 2A, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. On day 7, some HIV-1-infected cells were transferred to fresh medium and cultured for an additional 7 days (Treatment A), while others were treated with AZT (2 µm) for 7 days (Treatment B & C). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for another 7 days: Medium only (Treatment A); AZT (2 µm) continuously (Treatment B); AZT interruption (Treatment C). To examine if PMA can activate p-TEFb pathways and Jak/Stat pathways, on day 21, total cell lysates were subjected to Western blot analysis to detect the expression of proteins that crucial components of the signaling pathways associated with p-TEFb activation, (A) Brd4, CDK9 and Cyclin T1; (B) P-RPb-CTD; and (C) Jak2 and Atat3.
PMA effects
In a previous investigation, we discovered that PMA could induce a substantial reactivation of HIV-1 replication from latency, a condition induced by the transient expression of FLIP or XIAP [18]. To assess the impact of PMA on AZT treatments and AZT interruption, HIV-1 infected Jurkat cells were cultured in the presence of AZT for 7 days and subsequently treated with PMA for 24 hours, following the experimental design outlined in Study design II (Supplementary Figure 2B). As illustrated in Figure 4A, PMA treatment led to a significant upregulation of host transcription factors, including NF-κBp65, NFAT, Ap-1, and Sp1 (panel E, G, I), in comparison to untreated cells (panel D), medium-only conditions, AZT treatment-only conditions (panel F), or AZT interruption-only conditions (panel H). These results demonstrate that PMA has the potential to notably activate or reactivate HIV-1 replication in HIV-1 infected Jurkat cells.
PMA and TCR-related pathways
PMA exhibits the potential to activate TCR-related pathways, as demonstrated in Jurkat cells treated according to the outlined Study design II (Supplementary Figure 2B). Initial findings revealed an upregulation of host transcription factors associated with TCR signaling, including NFAT, NF-κBp65, AP-1, and SP-1, following PMA treatment (panel E, G, I), compared to non-PMA treatment groups (Panel D, F, H) (Figure 4A). Furthermore, PMA significantly increased the protein expression of receptors such as TCRα, TCRβ, CD3ζ, and CD28 (panel E, G, I) relative to the non-PMA treatment groups (Panel D, F, H) (Figure 4B), suggesting that PMA can activate or reactivate HIV-1 replication through TCR-related receptors in HIV-1 infected Jurkat cells.
The crucial role of the cellular surface receptor CD4 in HIV infection is well-known [15]. PMA treatment resulted in an increase in CD4 protein expression (panel E, G, I) compared to non-PMA treatment groups (Panel D, F, H) (Figure 4C), potentially indicating that PMA can enhance the entry of viral particles into host cells through the upregulation of CD4 [18].
Several other host molecules, including Zap-70, PLCγ1, and PKCζ, known to act as connectors between TCR-related receptors and host transcription factors essential for HIV-1 replication [3-6], exhibited increased protein expression in Jurkat cells treated with PMA (panel E, G, I) relative to non-PMA treatment groups (Panel D, F, H) (Figure 4D).
The transmembrane adapter Linker for activation of T-cells (LAT), which propagates antigen receptor signaling pathways following TCR signaling activation [20,21], displayed a substantial upregulation induced by PMA. The phospho-LAT, bridged by Grb2 family adaptors, activated Ras signaling downstream, contributing to the production of HIV-1 RNA (Figure 4E).
Recent findings suggest that HIV-1 replication or reactivation from latency can be influenced by PI3K/Akt signaling pathways [10,22]. PMA treatment increased the protein expression of PI3K/Akt (panel E, G, I) relative to non-PMA treatment groups (Panel D, F, H) in Jurkat cells (Figure 4F).
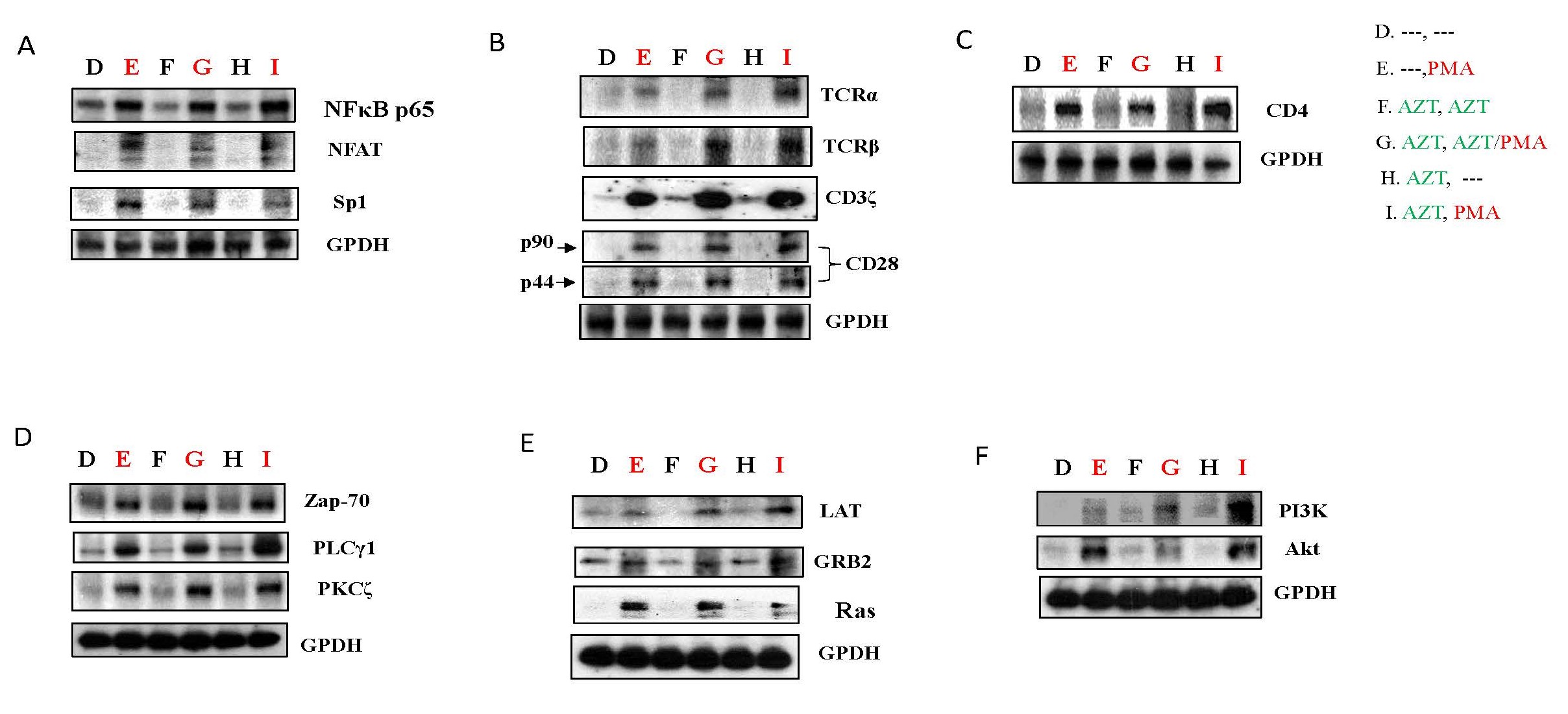
Figure 4. PMA could activate TCR-related pathways in HIV-1 infected Jurkat cells. As depicted in Study Design II, shown in Supplementary Figure 2B, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. On day 7, some HIV-1-infected cells were transferred to fresh medium for an additional 7 days (Treatment D & E), while others were treated with AZT (2 µm) for 7 days (Treatment F, G, H & I). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for an additional 24 hours: Medium only (Treatment D); PMA (10 ng/ml) (Treatment E); AZT (2 µm) continuously (Treatment F); AZT (2 µm)/PMA (10 ng/ml) (Treatment G); AZT interruption (Treatment H); and AZT interruption with the presence of PMA (10 ng/ml) (Treatment I). To examine if PMA can activate TCR-related pathways, on day 15 the cells were harvested and total cell lysates were subjected to Western blot analysis to detect the expression of proteins that crucial components of the signaling pathways associated with TCR activation, (A) NF-κB p65, NFAT and Sp-1; (B) TCRα, TCRβ, CD3ζ and CD28; (C) CD4; (D) ZAP 70, PLCγ1 and PKCζ; (E) LAT, Grb2 and Ras; and (F) PI3K and Akt.
PMA and MAPK pathways
Moreover, PMA exhibited the capacity to activate MAPK pathways, a type of signaling pathway involved in the activation of host transcription factors such as Ap-1 and NF-κB [3,4,23]. Analysis of total cell lysates by Western blot revealed that PMA increased the expression of Erk1/2, p38, and JNK (panel E, G, I) compared to non-PMA treatment groups (Panel D, F, H) (Figure 5).
These findings collectively suggest that PMA can enhance HIV-1 replication, rescue HIV-1 from latency induced by AZT, and contribute to the recovery of HIV viral load following AZT interruption at the viral transcriptional levels. This effect is achieved through the activation of TCR-related/PI3K/MAPK signaling pathways.
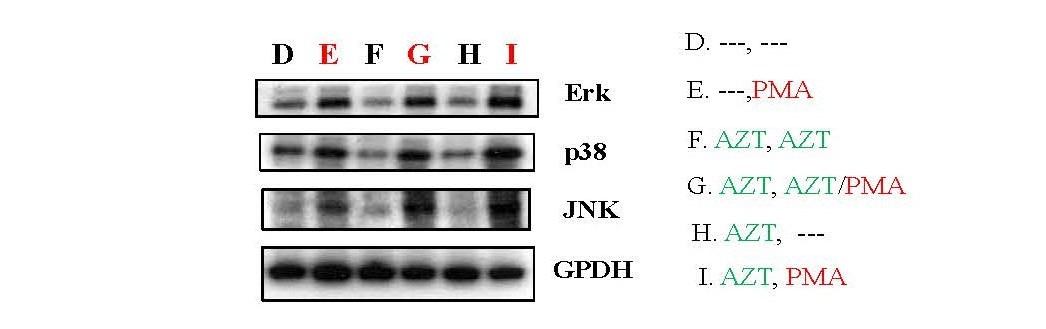
Figure 5. PMA could activate MAPK pathways in HIV-1 infected Jurkat cells. As depicted in Study Design II, shown in Supplementary Figure 2B, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. On day 7, some HIV-1-infected cells were transferred to fresh medium for an additional 7 days (Treatment D & E), while others were treated with AZT (2 µm) for 7 days (Treatment F, G, H & I). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for an additional 24 hours: Medium only (Treatment D); PMA (10 ng/ml) (Treatment E); AZT (2 µm) continuously (Treatment F); AZT (2 µm)/PMA (10 ng/ml) (Treatment G); AZT interruption (Treatment H); and AZT interruption with the presence of PMA (10 ng/ml) (Treatment I). To examine if PMA can activate MAPK pathways, on day 15 the cells were harvested and total cell lysates were subjected to Western blot analysis to detect the expression of proteins that crucial components of the signaling pathways associated with the activation of MAPKs, Erk1/2, p38 and JNK.
PMA-induced apoptosis
Given PMA's ability to activate/reactivate HIV-1 replication in Jurkat cells independently of AZT treatment, we explored whether the elevated viral loads induced by PMA could lead to cell death [24,25]. In a study design consistent with Study design II (Supplementary Figure 2B), we investigated the impact of PMA on the death of viral-infected cells. In Figure 6A, it is evident that a noteworthy increase in cell death, particularly in panels E, G, and I, following the 7-day PMA treatment, in contrast to the untreated cells depicted in panels D, F, and H. This experimental sequence suggests that PMA treatment may play a substantial role in influencing cellular outcomes, leading to a significant induction of cell death in the context of HIV-1 infection and treatment.
Caspase-3, a well-established indicator of apoptotic death [26], showed a significant increase in activation with PMA treatment (panel E, G, I), particularly in the AZT interruption group (panel I), compared to non-PMA treatment groups (panel D, F, H) (Figure 6B). This suggests that PMA dominantly induces apoptotic death in HIV-1 infected Jurkat cells.
Two major apoptotic signaling pathways associated with HIV-1 infection were examined: extrinsic (Fas-mediated) and intrinsic (Bax/mitochondria-mediated) [24-26]. PMA upregulated the protein expression of Fas and FADD (panel E, G, I) (Figure 6C), leading to the formation of the death-induced signaling complex (DISC) and subsequent caspase-8 activation. This cascade contributes to caspase-3 activation in the Fas-mediated apoptotic pathway [24,25]. Additionally, PMA increased Bax protein levels through its regulator, p53 (panel E, G, I), compared to non-PMA treatment groups (Panel D, F, H) (Figure 6D). Activated Bax proteins translocate from the cytosol to mitochondria, inducing caspase-3 activity through cytochrome c/caspase-9 [24,25].
IFI16 (interferon-γ-inducible protein 16), capable of detecting cDNA produced during HIV-1 replication, exhibited increased protein levels with PMA treatment (panel E, G, I) relative to non-PMA treatment groups (Panel D, F, H) (Figure 6D). This increase in IFI16 suggests heightened T-cell death through the intrinsic apoptotic pathway [27].
Bcl2, which negatively influences Bax activity by disrupting Bax translocation through binding, showed decreased protein levels with PMA treatment (panel E, G, I) relative to non-PMA treatment groups (Panel D, F, H) (Figure 6E). It has been reported that Bcl2 inhibition can reduce HIV-1 latent reservoirs, leading to the selective death of HIV-infected cells after viral reactivation from latency [13,28].
FLIP, an inhibitor of caspase-8 that decreases HIV-1 replication and increases HIV-1 latency [17-19], displayed reduced production in Jurkat cells treated according to Study design II (Supplementary Figure 2B) (panel E, G, I) compared to non-PMA treatment groups (Panel D, F, H) (Figure 6E).
Collectively, these findings suggest that, in addition to directly activating/reactivating HIV replication, PMA can induce apoptotic death through the upregulation of proapoptotic molecules and downregulation of antiapoptotic proteins in HIV-1 infected Jurkat cells. Such patterns promote HIV-1 replication [17,18,20] and inhibit viral latency [18,22].
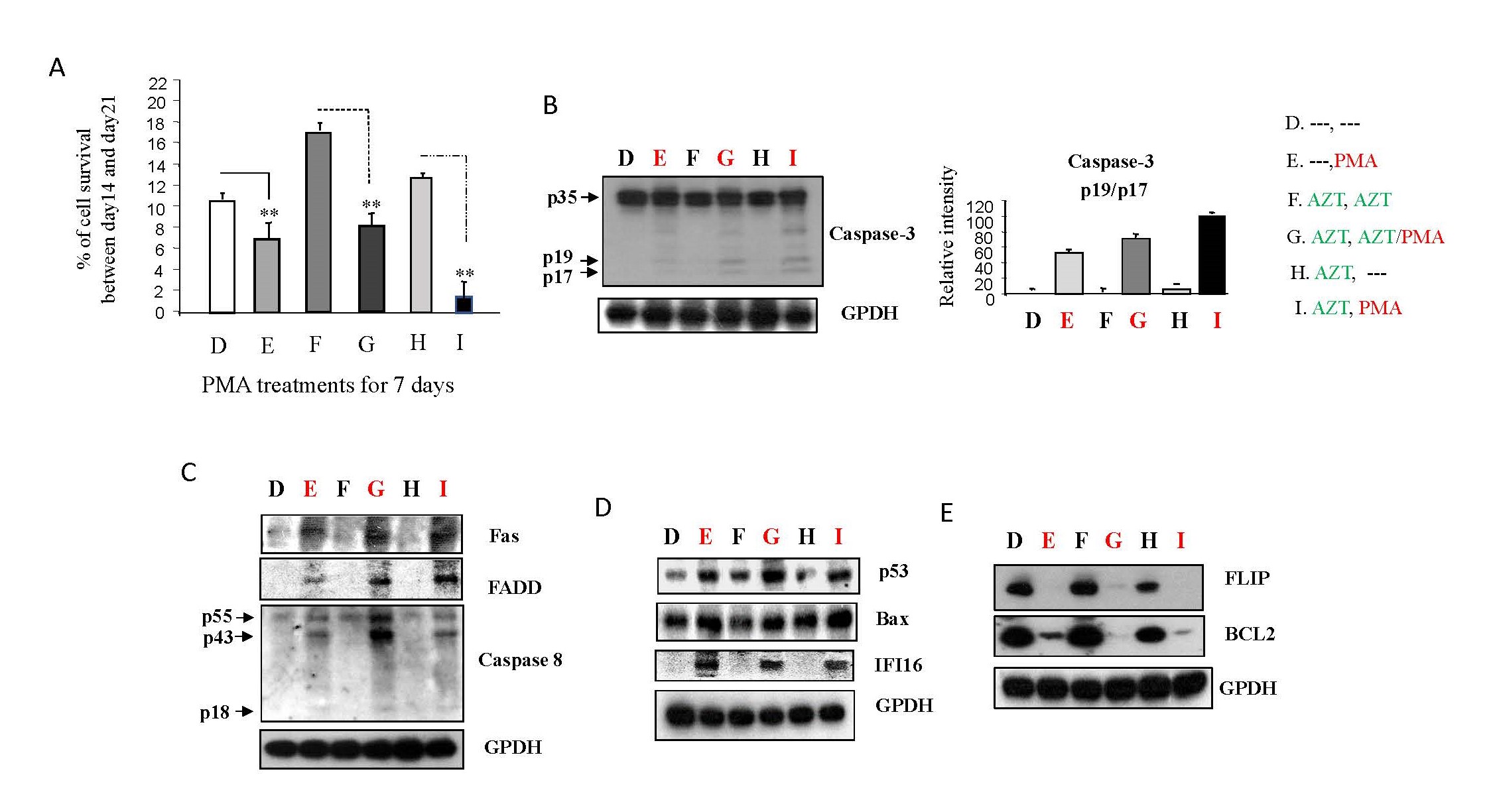
Figure 6. PMA could activate apoptotic pathways in HIV-1 infected Jurkat cells. As depicted in Study Design II, shown in Supplementary Figure 2B, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. On day 7, some HIV-1-infected cells were transferred to fresh medium for an additional 7 days (Treatment D & E), while others were treated with AZT (2 µm) for 7 days (Treatment F, G, H & I). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for an additional 7 days or 24 hours: Medium only (Treatment D); PMA (10 ng/ml) (Treatment E); AZT (2 µm) continuously (Treatment F); AZT (2 µm)/PMA (10 ng/ml) (Treatment G); AZT interruption (Treatment H); and AZT interruption with the presence of PMA (10 ng/ml) (Treatment I). (A). On day 21 the number of live cells was determined by Trypan blue dye exclusion with using EVE™ Automatic Cell Counter, and the data represent the average of three different independent experiments. To examine if PMA can affect apoptotic pathways, on day 15 the cells were harvested and total cell lysates were subjected to Western blot analysis to detect the expression of proteins that crucial components of the signaling pathways associated with apoptotic cell death, (B) caspas-3 activation; (C) Fas, FADD and caspase-8 activation; (D) IFI 16, p53 and Bax; and (E) FLIP and Bcl2.
PMA and autophagy
DRAM (damage-regulated autophagy modulator) helps control cell damage and is linked to a process called macroautophagy, as per reports [29]. Vps34, a type of enzyme, is involved in macroautophagy by changing a substance called phosphatidylinositol into phosphatidylinositol 3-phosphate [PI(3)P] [30]. In Figure 7A, it is evident that PMA can elevate the protein expression of DRAM and Vps34 in HIV-1 infected Jurkat cells (panel E, G, I) compared to non-PMA treatment groups (Panel D, F, H).
ULK1, or the Unc-51 like autophagy-activating kinase 1, plays a crucial role at the beginning of autophagy. It kickstarts the process by modifying a part of the PI3KC3 complex called Beclin1 through a process called phosphorylation. This modification activates PI3KC3 [31]. PMA upregulated the expression of ULK1 and Beclin1 (panel E, G, I) relative to non-PMA treatment groups (Panel D, F, H) in HIV-1 infected Jurkat cells (Figure 7B).
Other important markers in the autophagy pathway include ATG5, which starts making the autophagosome membrane and helps it fuse with lysosomes [32]. SQSTM1/p62 is a crucial scaffold protein in the macroautophagy process [33]. Protein light chain 3 (LC3) is found in autophagosomes, helping choose what to target for autophagy and participating in creating autophagosomes [34]. HIV-1 infected Jurkat cells treated with PMA exhibited higher expression levels of ATG5 and SQSTM1/p62, along with the cleavage of LC3 protein (panel E, G, I), compared to non-PMA treatment groups (Panel D, F, H) (Figure 7C).
These findings suggest that PMA can induce cell death mediated through autophagy in HIV-1 infected Jurkat cells.
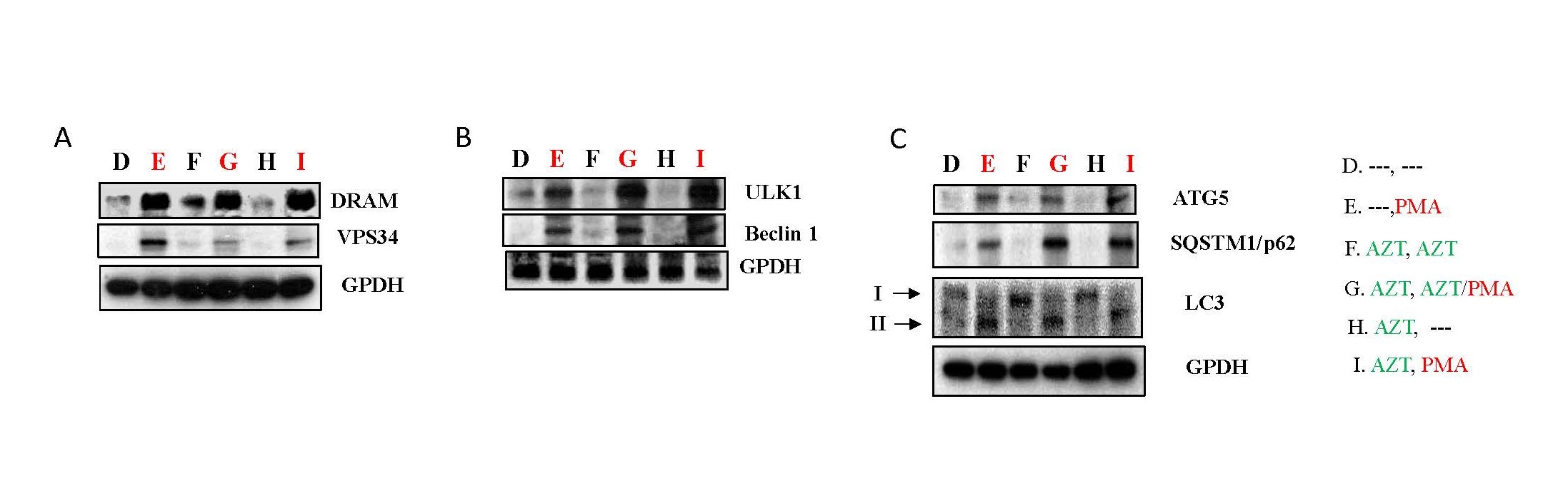
Figure 7. PMA could activate autophagy pathways in HIV-1 infected Jurkat cells. As depicted in Study Design II, shown in Supplementary Figure 2B, Jurkat cells were exposed to HIV-1 (MN stained) for 2 hours. Subsequently, the cells were washed twice with PBS and cultured at 37°C for 7 days. On day 7, some HIV-1-infected cells were transferred to fresh medium for an additional 7 days (Treatment D & E), while others were treated with AZT (2 µm) for 7 days (Treatment F, G, H & I). On day 14, equal amounts of live cells were transferred to fresh medium and cultured for an additional 24 hours: Medium only (Treatment D); PMA (10 ng/ml) (Treatment E); AZT (2 µm) continuously (Treatment F); AZT (2 µm)/PMA (10 ng/ml) (Treatment G); AZT interruption (Treatment H); and AZT interruption with the presence of PMA (10 ng/ml) (Treatment I). To examine if PMA can activate autophagy pathways, on day 15 the cells were harvested and total cell lysates were subjected to Western blot analysis to detect the expression of proteins that crucial components of the signaling pathways associated with cell autophagy, (A) DRAM and VPS34; (B) ULK1 and Beclin1; and (C) ATG5, SQSTM1/p62 and LC3.
Discussion
The study emphasizes the challenges associated with life-long antiretroviral therapy (ART) for HIV-1 management due to the persistence of viral latency and the common issue of non-adherence in high-risk populations [15]. Interruptions in ART lead to viral load rebounds, and despite the ongoing efforts in the "shock and kill" approach with latency-reversing agents (LRAs), current data show limitations in reducing the size of the HIV-1 reservoir.
Despite progress, LRAs face challenges in achieving the desired "shock and kill" effect. While LRAs can activate viral transcription, their inability to induce effective cell death limits their efficacy as stand-alone therapies [2,36]. Viral latency involves complex molecular mechanisms, including downregulation of host transcriptional factors, epigenetic modifications, and interference with transcriptional efficiency and elongation in host cells. Resting CD4 T-cells in tissues pose additional obstacles to the shock-and-kill strategy [37-39]. Research on LRAs for HIV-1 in vitro using a single type of cell faces challenges. Studies using cell lines to simulate HIV-1 latency may not fully represent the complexity seen in actual human cells. Reactivated cells may not be efficiently cleared by the immune system. Patient responses to LRAs vary, complicating the development of universally effective treatments.Top of Form
The study highlights the inverse correlation between anti-apoptotic molecules (Bcl-XL/Bcl2, FLIP, XIAP) and HIV-1 replication [16-18]. Bcl2, through its interaction with Casp8p41, facilitates an anti-apoptotic state [40], promoting HIV survival and latent reservoir establishment [27]. Conversely, Bcl2 inhibition decreases virally infected cells and latent reservoir size. Pro-apoptotic molecules (Fas, FADD, Bax, p53) positively correlate with TCR activation, HIV-1 replication, and cell death, reducing viral latent reservoirs [16,17].
PKC activators, known as robust LRAs, are proposed as potential alternatives. The study introduces PMA as a model for PKC activators [41]. PMA effectively reactivates HIV-1 from latency independently of AZT treatment status. Moreover, PMA demonstrates a dual role in the "shock and kill" approach by inducing apoptotic cell death through both extrinsic and intrinsic pathways. The study suggests that the downregulation/inactivation of anti-apoptotic molecules, such as Bcl2 or FLIP, by LRAs could activate TCR-related pathways, inducing apoptotic cell death for HIV-1 latency cure.
The findings suggest that PMA, and potentially other PKC activators, could enhance the effectiveness of existing therapies by reactivating HIV-1 from latency and inducing cell death. The study underscores the need for further investigations into the effects of different PKC activators in various cell types and conditions. The identified molecular mechanisms may guide the exploration of new LRAs and other therapeutic strategies for HIV-1 treatment. Additionally, the potential clinical applications of these findings, such as in developing HIV therapies, would require further research and validation. Overall, the study contributes valuable insights into potential avenues for improving HIV-1 management beyond conventional ART.
In conclusion, AZT, the conventional HIV-1 therapy, inhibits HIV-1 infection of Jurkat cells and its treatment interruption in cell culture reactivates viral replication via the upregulation of host transcription factors involved in the TCR-related, p-TEFb and Jak/Stat pathways. We have explored the effectiveness of an alternative treatment approach through a distinct mechanism (i.e. LRA) using PMA, which has been found to increase these transcription factors independent of AZT treatment status. Furthermore, PMA was also able to induce cell death in combination treatment conditions through both extrinsic and intrinsic apoptotic signaling pathways as well as via autophagy pathways. These results demonstrate that PMA, and possibly other PKC activators, could reactivate HIV-1 from latency and induce cell death, which may have the potential to enhance the effectiveness of existing therapies. Finally, the various molecular mechanisms we have identified here may be useful in the exploration of other strategies for new LRAs and other therapies that could be studied in the clinical setting for the treatment of HIV-1.
Author Contributions
XW and IH conceived the project. XW and JZ performed the experiments. XW formatted datasets and analyzed data. XW and IH participated in manuscript writing, engaged in data interpretation, and contributed to the intellectual content of this work.
Acknowledgments
The authors wish to acknowledge Dr. Viswanath Ragupathy and Dr. Mohan Haleyurgirisetty, for their critical review of this manuscript. The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Competing Interests
The authors declare no competing interests.
References
2. Campbell GR, Spector SA. Current strategies to induce selective killing of HIV-1-infected cells. J Leukoc Biol. 2022;112:1273-84.
3. Al-Harthi L, Roebuck KA. Human immunodeficiency virus type-1 transcription: role of the 5'-untranslated leader region (review). Int J Mol Med. 1998;1:875-81.
4. Pluta A, Jaworski JP, Cortés-Rubio CN. Balance between retroviral latency and transcription: based on HIV Model. Pathogens. 2020;10:16-41.
5. Khanal S, Schank M, El Gazzar M, Moorman JP, Yao ZQ. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells. 2021;10:475-97.
6. Carlin E, Greer B, Lowman K, Duverger A, Wagner F, Moylan D, et al. Extensive proteomic and transcriptomic changes quench the TCR/CD3 activation signal of latently HIV-1 infected T-cells. PLoS Pathog. 2021;17:e1008748.
7. Fujinaga K. P-TEFb as A Promising Therapeutic Target. Molecules. 2020;25:838-365.
8. Schumertl T, Lokau J, Rose-John S, Garbers C. Function and proteolytic generation of the soluble interleukin-6 receptor in health and disease. Biochim Biophys Acta Mol Cell Res. 2021;1869:119143.
9. Gavegnano C, Brehm JH, Dupuy FP, Talla A, Ribeiro SP, Kulpa DA, et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017;13:e1006740.
10. Wang X, Zhao J, Ragupathy V, Hewlett I. The effects of MAPK p38alpha on AZT resistance against reactivating HIV-1 replication in ACH2 cells. Mol Cell Biochem. 2019;462:41-50.
11. Lopes JR, Chiba DE, Dos Santos JL. HIV latency reversal agents: A potential path for functional cure? Eur J Med Chem. 2021;213:113213.
12. Kula-Pacurar A, Rodari A, Darcis G, Van Lint C. Shocking HIV-1 with immunomodulatory latency reversing agents Semin Immunol. 2021;51:101478.
13. French AJ, Natesampillai S, Krogman A, Correia C, Peterson KL, Alto A, et al. Reactivating latent HIV with PKC agonists induces resistance to apoptosis and is associated with phosphorylation and activation of BCL2. PLoS Pathog. 2020;16:e1008906.
14. Kim CH, Gollapudi S, Kim A, Lee T, Gupta S. Role of protein kinase C-beta isozyme in activation of latent human immunodeficiency virus type 1 in promonocytic U1 cells by phorbol-12-myristate acetate AIDS Res Hum Retroviruses. 1996;12:1361-6.
15. Wang X, Tan J, Zhao J, Ragupathy V, Haleyurgirisetty M, Hewlett I. Some findings of FADD knockdown in inhibition of HIV-1 replication in Jurkat cells and PBMCs Mol Cell Biochem. 2014;393:181-90.
16. Gwadz M, Cleland CM, Freeman R, Wilton L, Collins LM, L Hawkins R, et al. Stopping, starting, and sustaining HIV antiretroviral therapy: a mixed-methods exploration among African American/Black and Latino long-term survivors of HIV in an urban context. BMC Public Health. 2021;21:419.
17. Wang X, Ragupathy V, Zhao J, Hewlett I. Molecules from apoptotic pathways modulate HIV-1 replication in Jurkat cells. Biochem Biophys Res Commun. 2011;414:20-4.
18. Wang X, Zhao J, Biswas S, Devadas K, Hewlett I. Components of apoptotic pathways modulate HIV-1 latency in Jurkat cells Microbes Infect. 2022;24:104912.
19. Tan J, Wang X, Devadas K, Zhao J, Zhang P, Hewlett I. Some mechanisms of FLIP expression in inhibition of HIV-1 replication in Jurkat cells, CD4+ T-cells and PBMCs. J Cell Physiol. 2013;228:2305-13.
20. Yablonski D. Bridging the Gap: Modulatory Roles of the Grb2-Family Adaptor, Gads, in Cellular and Allergic Immune Responses Front Immunol. 2019;10:1704.
21. Rom S, Pacifici M, Passiatore G, Aprea S, Waligorska A, Del Valle L, et al. HIV-1 Tat binds to SH3 domains: cellular and viral outcome of Tat/Grb2 interaction. Biochim Biophys Acta. 2011;1813:1836-44.
22. Wang X, Zhao J, Mbondji C, Hewlett I. p53 Expression Activation of HIV-1 Latency in U1 Cells. Int J Virol AIDS. 2017;3:036.
23. Wang X, Sun B, Mbondji C, Biswas S, Zhao J, Hewlett I. Differences in Activation of HIV-1 Replication by Superinfection With HIV-1 and HIV-2 in U1 Cells. J Cell Physiol 2017;232:1746-53.
24. Wang X, Viswanath R, Zhao J, Tang S, Hewlett I. Changes in the level of apoptosis-related proteins in Jurkat cells infected with HIV-1 versus HIV-2. Mol Cell Biochem. 2010;337:175-83.
25. Wang X, Zhao J, Tang S, Lee S, Glazer RI, Hewlett I. c-FLIPL regulates PKC via AP-2 to inhibit Bax-mediated apoptosis induced by HIV-1 gp120 in Jurkat cells. Mol Cell Biochem. 2009;330:23-9.
26. Iwata M, Ohoka Y, Kuwata T, Asada A. Regulation of T-cell apoptosis via T-cell receptors and steroid receptors Stem Cells. 1996;14:632-41.
27. Hurst TP, Aswad A, Karamitros T, Katzourakis A, Smith AL, Magiorkinis G. Interferon-Inducible Protein 16 (IFI16) Has a Broad-Spectrum Binding Ability Against ssDNA Targets: An Evolutionary Hypothesis for Antiretroviral Checkpoint. Front Microbiol. 2019;10:1426.
28. Chandrasekar AP, Badley AD. Prime, shock and kill: BCL-2 inhibition for HIV cure. Front Immunol. 2022;13:1033609.
29. Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121-34.
30. Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol. 2008;181:655-66.
31. Wang C, Wang H, Zhang D, Luo W, Liu R, Xu D, et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat Commun. 2018;9:3492.
32. Codogno P, Meijer AJ. Atg5: more than an autophagy factor. Nat Cell Biol. 2006;8:1045-7.
33. Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431-41.
34. Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011;14:2201-14.
35. Rogov V, Dötsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167-78.
36. Kim Y, Anderson JL, Lewin SR. Getting the "Kill" into "Shock and Kill": Strategies to Eliminate Latent HIV. Cell Host Microbe. 2018;23:14-26.
37. Battistini A, Sgarbanti M. HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses. 2014;6:1715-58.
38. Rodari A, Darcis G, Van Lint CM. The Current Status of Latency Reversing Agents for HIV-1 Remission. Annu Rev Virol. 2021;8:491-514.
39. Khoury G, Darcis G, Lee MY, Bouchat S, Van Driessche B, Purcell DFJ, et al. The molecular biology of HIV latency. Adv Exp Med Biol. 2018;1075:187-212.
40. Cummins NW, Sainski-Nguyen AM, Natesampillai S, Aboulnasr F, Kaufmann S, Badley AD. Maintenance of the HIV Reservoir Is Antagonized by Selective BCL2 Inhibition. J Virol. 2017;91:e00012-17.
41. Washizaki A, Murata M, Seki Y, Kikumori M, Tang Y, Tan W, et al. The Novel PKC Activator 10-Methyl-Aplog-1 Combined with JQ1 Induced Strong and Synergistic HIV Reactivation with Tolerable Global T-cell Activation. Viruses. 2021;13:2037.