Abstract
For a long time Th1 cells were considered the key players in the induction of inflammation and progression of disease in autoimmune diseases. With the discovery of IL-17-producing CD4+ T cells (Th17) being abundant at inflammation sites, this soon changed. Investigating this new T helper subset, it became clear that in comparison to Th1 and Th2 cells, Th17 cells have an increased tendency to change their phenotype and therefore become either more pathogenic or immunoregulatory. This makes them an attractive target for therapeutic interventions. As plasticity of Th17 cells differs between different autoimmune diseases, understanding its drivers is complex. This review focusses on the role of plasticity within Th17 cells in induction, aggravation and resolution of disease and points out IL-23 as a potential key player in controlling Th17 fate. Finally, current treatments targeting IL-23 and Th17 plasticity are highlighted and an outlook on future therapeutics is given.
Keywords
Th17, T cell adaptation, IL-23, IL-17, T cell subsets
Abbreviations
CD: Cluster of differentiation; cGN: Crescentic Glomerulonephritis; EAE: Exogene Autoimmune Encephalitis; GM-CSF: Granulocyte Macrophage Colony Stimulating Factor; HIES: Hyper IgE Syndrome; IBD: Inflammatory Bowel Disease; IFN-γ: Interferon-γ; IL: Interleukin; IL6R: Interleukin 6 Receptor; IL-23R: Interleukin 23 Receptor; NTN: Nephrotoxic Nephritis; SC: Single Cell; seq: Sequencing; Tfh: T follicular helper cell; TGF-β: Transforming Growth Factor β; Th1: T helper cell type 1; Th2: T helper cell type 2; Th9: T helper cell type 9; Th17: T helper cell type 17; Th22: T helper cell type 22
Introduction
Focusing on adaptive immunity there is a humoral and a cellular immune response. The humoral response is executed by antibodies acting via antigen specific binding. These antibodies are produced by activated B-lymphocytes in order to bind and neutralize antigens. The cellular response is performed via effector CD4+ and CD8+ T-lymphocytes. It was previously assumed that CD4+ T-lymphocytes only consist of Th1 and Th2 cells [1]. But over the last decades with the introduction of new T-helper subsets and more studies revealing grades of plasticity within CD4+ T-lymphocyte populations, this classic view was exposed to be lacking. Added subsets include regulatory T cells, Th17, Tfh, Th9 and Th22 cells [2]. Regarding these added subsets, Th17, Th9 and Tfh cells acquire their phenotype under the effect of master transcription factor RORγt, Etv5/PU.1 and Bcl6 respectively. The transcriptional program of Th1 and Th2 cells is also defined through a specific master transcription factor, the former by T-bet and the latter by GATA3 [3,4]. In order to polarize into specific effector subsets naive CD4+ T cells are dependent on cytokine signaling. These cytokines are produced by immune, epithelial and stromal cells in physiological conditions or as a response to pathogens. Therefore, the current composition of the cytokine milieu is what determines the fate of naïve CD4+ T cell. Furthermore, the different subsets of T cells are not only polarized through a certain cytokine environment but can also be defined through their cytokine output [1]. Differentiation of naive CD4+ cells is also impacted by commensals and pathogens within the intestinal microbiome. The pathogen Delta aroA Salmonella enterica triggers an IFN-γ dominated response, while Citrobacter rodentium leads to a Th17 answer [5]. The two helminths Heligmosomoides polygyrus and Nippostrongylus brasiliensis set off a Th2 response [6]. This work focuses on the recently discovered Th17 cells and the plasticity within this subset. In addition, the role of IL-23 in these processes will be highlighted.
Th17 Cells
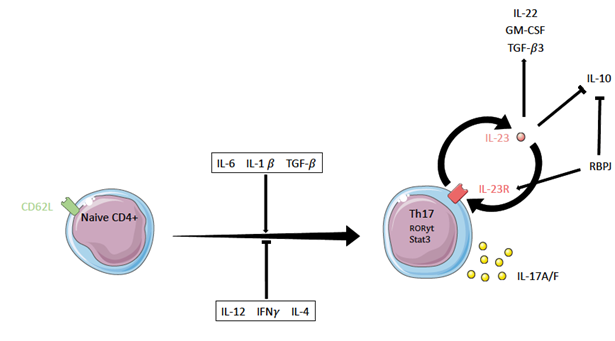
IFN-γ producing Th1 cells were considered to be the driver of inflammation within autoimmune disease. However, research revealed the substantial pathogenic role of a not yet described T cell subset within different types of autoimmune disease, which is now known as Th17 cells [7,8]. Th17 cells are crucial in eliciting inflammation in response to bacterial or fungal pathogens penetrating the mucosa [9,10]. In the multisystemic disorder HIES mutations in Stat3 occur [11]. Stat3 is an important transcription factor for the induction of Th17 cells. Patients suffering HIES have less Th17 cells and suffer mucocutaneous candidiasis [12]. Th17 cells are defined through their expression of the master transcription factor RORγt, the chemokine receptor CCR6 and production of the cytokines IL22 and IL17A-F. IL17A-F production can also be found in other cell types such as macrophages, γδT-cells and NK cells [13,14]. IL17A-F, RORγt, RORα, and IL-21 are expressed production of IFN-γ in Stat3 knockout mice [15,16]. For naive CD4+ T cells to become Th17 cells, the cytokines IL1-β, TGF-β1 in a Stat3 dependent manner, whileTh1 cells continue und IL-6 are necessary [17]. IL-23 was also believed to play a role in Th17 polarization, but as it only induces IL17 secretion in CD4+ CD62L- memory T cells and not in naïve CD4+ CD62L+ T cells its role is rather limited to maintaining Th17 fate. This is because naïve T cells do not express IL-23R [18]. IL6 and TGF-β1 also increase IL17 secretion in these naïve cells [15]. Once polarized Th17 cells do not show a stable IL-17-producing phenotype but rather display a certain degree of plasticity. Depending on the environment they can differentiate into regulatory T cells, Th1 cells or Tfh [19-21]. Apart from Stat3 another signal transducer and activator of transcription, Stat4, seems to affect Th17 cells. Stat4 knockout mice stimulated with IL-23 show decreased IL-17 output in comparison to WT mice, suggesting IL-23 exercises its Th17 stabilizing effect partly through Stat4. However, this is limited to IL17 output and no difference in RORγt expression could be found [15].
A more recent study came to the conclusion that IL6 acts upstream of IL-23 signaling in Th17 polarization. Only the IL6- IL6R interaction keeps a steady RORγt expression, which is required for IL-23R. Hence without IL6, IL-23 has no binding site [22]. This finding indicates that IL6 is not only crucial for induction of Stat3 and therefore inhibiting Foxp3 and a possible Treg fate, but is also a key regulator of stability of Th17 cells [22,23]. GM-CSF knockout mice did not develop severe EAE, underlining the importance of GM-CSF production by Th17 in order to elicit their pathogenic potential [24]. GM-CSF production in Th17 cells is dependent on RORγt, which in turn is negatively regulated by IFN-γ [24]. So called non-classical Th17 or Th1 like Th17 cells expressing IFN-γ are prevalent in active inflammation [25]. These proinflammatory cells express the master transcriptions factors of Th1 (T-bet) and Th17 (RORγt) cells at the same time and are believed to play a part in the induction of autoimmune disease [9].
IL-23 and Its Receptor
The cytokine IL-23 was first described by Oppmann et al. in the year 2000. Structural analysis revealed its composition of two different subunits p19 and p40. The latter also forms a part of interleukin 12, while the p19 subunit is unique for IL-23 [26]. The close structural relation of IL12 and IL-23 is reflected in their receptor affinity, as both cytokines bind to the receptor subunit IL12Rβ1 (Figure 1). Even though they have a mutual binding site, IL12 and IL-23 elicit their distinct effects in vivo by the means of a ligand specific subunit. For IL-23 the specific binding site is IL-23Rα [26]. A therapy targeting p19 or IL-23Rα can therefore specifically eliminate IL-23 signaling without interfering with the IL-12 signal [27]. Binding of IL-23 to IL- 23Rα induces signaling via receptor associated JAK2 and Tyk2. This leads to phosphorylation and dimerization of Stat3 and Stat4 [18].
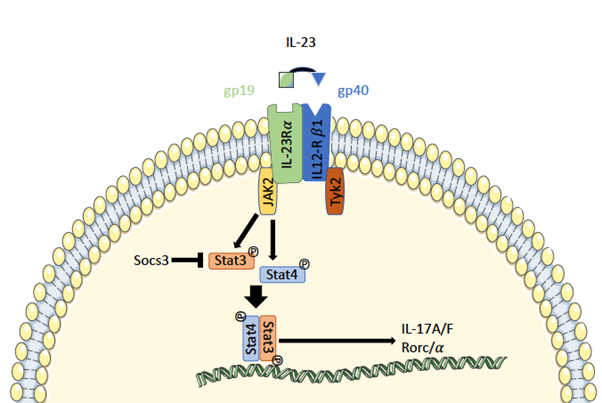
Figure 1: IL-23 signaling pathway in Th17 cells. The subunits p19 and p40 of IL-23 bind to their respective IL-23 receptor subunits IL-23Rα and IL12Rβ1. This induces activation of JAK2 and Tyk2 leading to phosphorylation and dimerization of Stat3 and Stat4. These can bind to promotor regions and induce transcription of various genes related to Th17 cells.
IL-23 is a proinflammatory cytokine of particular relevance for Th17 effector cells [7]. Different mouse models with an IL-23p19 knockout, a knockout particularly targeting IL-23 signaling, subline this finding. IL-23p19 knockout mice are protected from disease in models of EAE, collagen induced arthritis or IBD [7,28,29]. The protection from EAE was due to a lack of GM-CSF production in Th17 cells and not to a reduced IL17 expression [24]. GM-CSF production in Th17 cells is dependent on RORγt, which in turn is negatively regulated by IFN-γ [24]. In contrast IL-23 upregulates GM-CSF expression explaining the non-present inflammation in IL-23 knockoutmice [30]. In line with this finding stimulation with IL-23 resulted in exacerbated disease and increased IL17 production [31]. Transgene mice ubiquitously expressing p19 show elevated levels of inflammation in various organs and growth disturbances. Inflammation markers such as IL1 and TNFα were elevated in these animals [32]. These findings put targeting of IL-23 and its receptor on the map for the development of new pharmaceuticals for treatments of autoimmune diseases and potentially hyperinflammation in infection [33,34]. Genome wide association studies could associate mutations in components of the IL-23 signaling pathway with several immune mediated diseases [35-37]. A regulator of IL- 23 activity is the suppressor of cytokine signaling 3 (Socs3). At low Socs3 levels the phosphorylation frequency of Stat3, induced by IL-23, is amplified. Therefore, in the presence of Socs3 less Stat3 can bind to the promotor region of IL17A-F and when absent significantly more naïve CD4+ T-cells polarize into Th17 cells [38]. Th17 cells polarized under the influence of IL1β, IL6 and IL-23 expressed Tbx21 mRNA and T-bet protein, while cells stimulated with IL1β, IL6 and TGFβ1 did not [39]. Furthermore, polarization with IL-23 leads to significantly more IL22 production [39]. Elevated levels of IL22 can be found in psoriasis patient, correlating with the degree of inflammation. Furthermore, IL22 only affects digestive, epidermal and respiratory epithelial cells but not immune cells, lacking IL22R [40]. IL-23R is only expressed in memory T- cells therefore IL- 23 is unable to directly affect naïve CD4+ cells [15]. Cultures of Th17 cells either with or without IL-23 revealed a role for IL-23 in stabilizing the Th17 phenotype. Cells cultured with IL-23 kept their Th17 phenotype over time [20]. IL-6 knockout mice showed less IL-23R expression suggesting a role for IL-6 not only in inducing RORγt but also IL-23R [4]. IL-23 promotes a more pathogenic Th17 subset compared to IL6 and TGFβ1 primed more immunoregulatory Th17 cells (Figure 2). This is due to the inability of IL-23 primed cells to produce IL10, splitting Th17 cells into a rather pathogenic IL10- subset and a nonpathogenic IL10+ subset [41]. The nonpathogenic Th17 cells showed upregulation of transcription factors related to IL-10 production like MAF and the Aiolos encoder IKZF3 [25,42]. While IL10+ Th17 cells stimulate M2 macrophages, linked to immunoregulatory tasks, IL10- Th17 cells prime M1 macrophages driving inflammation [42].
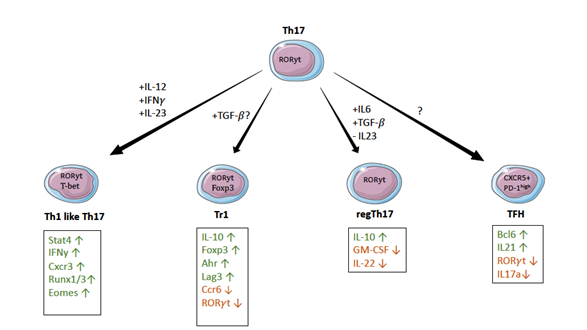
Th17 Plasticity (Figure 3)
In comparison to Th1 and Th2 cells which are rather stable Th17 cells display a high level of plasticity. Plasticity means that cells start to express markers (cytokines or transcription factors) that are known to define other T-helper subsets. With the finding that Th17 subsets can also secrete IFN-γ, the signature cytokine of Th1 cells, or IL-4, one of the signature cytokines of Th2 cells, it has been shown in vitro that definition of rigid subsets based on their cytokine expression may be lacking. In vitro studies with isolated IL-17 high cells from TGFβ+ IL6+ IL1β primed naïve CD4+ cells showed that adding IL-23 and IL1-β keeps IL17 production, while adding IL-12 or IL-4 reduced IL-17 output with cells starting to respectively produce IFN-γ and IL4 [20]. In order to further investigate the fate of Th17 cells in vivo a mouse model permanently marking IL17 producing cells has been developed. Cre-recombinase expression is linked to IL17 production and leads to a following permanent labeling of the cell with a fluorochrome. These cells can then be sorted based on their fluorochrome marking, making further investigation on their current cytokine output and their reaction to certain stimuli possible [43]. Possible subsets Th17 cells can transdifferentiate into are Tregs, Th1 cells, Tr1 and follicular T cells [19,43-45]. With this amount of possible differentiation and resulting effector functions encoding the drivers of Th17 plasticity is immensely important to understand disease and derive treatment options. An overlap of loci bound by master TF of Th17 cells RORγt and the targets of TF like STAT4, GATA3 and FOXP3 has been shown, which points at a central role of RORγt in influencing other subsets [46]. This overlap of targets further hints at a high grade of plasticity within the Th17 subset.
Th17/Th1 axis Th-1 like Th17 cells co-express the master transcription factors of the Th17 and Th1 cell lineage and produce IFN-γ and IL17. These RORγt+ and T-bet+ double positive cells seem to display a link between what used to be considered terminally differentiated populations and therefore are the key player, when talking about the Th17/Th1 axis [44]. Th1 like Th17 cells can be found in several types of autoimmune disease like colitis [21,47], rheumatoid arthritis [48,49], multiple sclerosis [50] and EAE [43], where they have been linked to pathogenicity. Some Th17 cells, while starting to express IFN-γ, completely stop expression of IL17 and are therefore named Th1-like ex Th17 cells [44,51]. Stimulation of in vitro polarized Th17 cells with IL12 and IL-23 results in upregulation of T-bet and Stat4 and therefore arising IFN-γ expressing Th17 cells. When transferring polarized Th17 into Rag-/- mice, IFN-γ+ Th1 like Th17 cells emerged and induced colitis, proving that the in vitro findings translate to in vivo experiments [21]. Previous studies proved that transducing CD4+ T-cells with a retrovirus expressing RORγt lead to expansion of the Th17 population, while on the other hand transduction with T-bet blocked this expansion [52]. Yet combination of both failed to induce the “intermedium” Th1-like Th17 subset. Therefore, there may be inducers of IFN-γ apart from T-bet that lead to the formation of these IL-17A+ IFN-γ+ double positive cells [52]. Transcription factors upregulated in Th1-like Th17 cells stimulated with IL12 are T-bet, Runx1, Runx3 and Eomes. Especially Runx1 and Runx2 proved to be capable of inducing an IFN-γ and IL17A double positive population [53]. Chip analysis revealed an enrichment of Runx1 binding at the IFN-γ promotor in these IL12 treated Th17 cells [53]. In a T cell transfer model of colitis Harbour et al. showed that polarized Th17 cells transferred into Rag-/- mice, induce IBD. The majority of these Th17 cells became double positive for IL-17A and IFN-γ or acquired IFN-γ production combined with stagnation of IL17 expression. Since IFN-γ- Th17 cells did not induce wasting disease, these Th1 like Th17 cells and Th1 like ex Th17 cells appear to be crucial for disease induction [47]. The transformation of IFN-γ- Th17 to IFN-γ+ Th17 cells was STAT4 dependent, and it could be shown that the downstream effects of an IL12rb2- knockout in mice did not have a significant effect on this conversion. Due to the fact that this knockout targets only IL12 signaling, IL-23 which also is known to signal through STAT4, was proposed as the key player for Th17/Th1 plasticity [47]. However, there is a lack of direct evidence for this and further research is required. Th17 cells generated without TGF-β signal are RORγt+ T-bet+ double positive and express IL-23. They further expressed GMCSF and IFN-γ at the same time [43]. This hints at a possible generation of Th1 like Th17 cells in a cytokine environment lacking TGF-β. It has been shown that TGF-β inhibits T-bet and IFN-γ in Th1 cells, but further studies on its role in plasticity are required [54]. Considering the crucial role of Th1 like cells in autoimmune disease it is surprising that investigating mice with cGN, another immune disease model highly dependent on Th17 cells, this generation of Th1-like cells could not be observed. Although previous studies hinted at a possible generation of Th1-like cells more recent studies have proven that Th17 are quite stable within this model [43,55,56]. Th1- like Th17 cells could form a smaller population within cGN and be less relevant for disease than in IBD and EAE. Another explanation is, that the disease microenvironment in cGN lacks factors driving the transdifferentiation towards Th1 cells, which are present in other autoimmune models. This discrepancy between the models of EAE and colitis, with a lot of Th17 to Th1 plasticity and the missing one in cGN remains unexplained. Therefore, it will be interesting to further investigate the fate of exTh17 cells in cGN using the means of scRNA seq. IL-23/Th17axis Mice transferred with CD4+ T-cells stimulated with IL6, TGF-β and IL-23 or only IL6 and TGF-β did not suffer EAE, while IL-23 stimulated cells induced EAE in all specimens. This supports a role for IL-23 in producing a pathogenic Th17 phenotype while TGF-β and IL6 polarize an immunoregulatory subtype. Furthermore, it indicates that TGFβ and IL6 signaling is dominant to IL-23 [41]. Upregulation of IL-23R by IL-23 implies a positive feedback loop and further augments pathogenic potential of IL-23 [41]. IL22 is another Th17 effector interleukin driving inflammation, which is upregulated by IL-23 [41]. Further investigation on the influence of IL-23 on nonpathogenic Th17 cells revealed TGF-β3 to also be an IL-23 induced cytokine. Th17 cells induced by TGF-β3 and IL6 showed higher phosphorylation levels of Smad1 and Smad5, while polarization with TGF-β1 and IL6 lead to phosphorylation of Smad 2 and Smad3 [57]. Transfer of these TGF-β3 and IL6 polarized cells resulted in severe disease and even mortality [57]. With RBPJ (Recombining binding protein suppressor of hairless), Meyer zu Horste et al. proposed a key regulator of IL-23 signaling, both transactivating IL-23R and repressing IL10 in Th17 cells [58]. T-bet, the master TF of the Th1 program, is suspected to upregulate IL-23R and vice versa T-bet upregulation following IL-23 signaling has been shown [59]. The upregulation of IL-23R by T-bet is in line with the necessity of both Th17 and Th1 cells in order to elicit pathogenicity in autoimmune disease. IL-23 stimulation leads to more pathogenic IL10- Th17 cells while TGF-β and IL6 promotes immunoregulatory IL10+ Th17 cells [41]. Th17 to regulatory T cells When facing a pathogen, the induction of an immune response and the following inflammation are advantageous for the organism. Nevertheless, at some point the inflammation has to be opposed in order to restore homeostasis. Here two regulatory cell types, Foxp3 expressing regulatory T cells (T-regs) and the T regulatory type 1 cells (TR1), play a crucial role [19]. Th17 triggered inflammation within autoimmune disease is not beneficial for the host, yet the subsets opposing Th17 cells are comparable to the ones in acute infection [19]. While Th17 cells are considered a pathogenic T cell subset they still share TGF-β as a differentiation marker with the immune regulatory T-regs [60]. In 2008 Lochner et al. observed that Tregs can co-express RORγt and Foxp3 [61]. There are three hypotheses on the origin of these double positive cells, 1) they could form an independent cell type 2) they could be transdifferentiated from regulatory T cells 3) they have their origin in Th17 cells. The fate of Th17 cells can be investigated with IL17A-fate reporter mice, where cells expressing IL17A are permanently marked as YFP+. It could be shown that under homeostatic conditions almost 50% of YFP+ CD4+ lymphocytes of the colon stop IL17A expression [19]. Some of these so called exTh17 cells started to produce IL10 and express Foxp3 [19]. LAG3 a marker for Tr1 cells was also increased in exTh17 cells and similar to Tr1 cells, exTh17 cells displayed downregulated expression of CCR6 and RORγt. Th17 cells being able to downregulate Th17 markers and acquire regulatory markers, points at an axis between pathogenic Th17 cells and immunoregulatory T cells. Formation of exTh17 Tr1 cells correlates with the ongoing immune response [19]. A study following up on Th17 cell transdifferentiation in the kidney in cGN found no plasticity towards RORγt Foxp3 double positive cells. This led to the assumption that these double positive cells resemble an independent population [62]. Kannan et al. recently reported that IL-23 induced inflammation in psoriasis leads to higher amounts of IL17 producing Tregs and to more Tregs entering cell cycle [63]. Around 11% of these IL-23 induced IL17A+ Tregs where double positive for Foxp3 and RORγt [63]. These cells were most likely generated from IL17A- Foxp3+ Tregs suggesting a transdifferentiation from Treg to Th17 cells [63]. Kannan et al. therefore proposed IL-23 as a potential inducer of instability in the Treg lineage further exacerbating disease [63]. TH17/Tfh axis Compared to previously described axes there is not that much known about the Th17/Tfh axis. Tfh cells interact with B cells in the germinal center and are important for their differentiation. Sorting of Payer’s patch derived IL17 fate positive cells expressing Tfh markers (CXCR5+, PD-1high) revealed downregulation of RORc and IL-17a together with an upregulation of Tfh markers IL21 and Bcl6 [64]. IL-23 was not necessary for transdifferentiation of Th17 cells towards Tfh cells, as there were similar amounts of Th17 cells expressing Tfh markers in IL17 fate reporter mice and p19 deficient IL17 fate reporter mice [64].
Treatments Targeting IL-23
As previously described, IL-23 is critical for maintaining the Th17 phenotype and furthermore linked to plasticity towards Th1 cells. With Th1 and Th17 cells both being linked to pathogenicity in different autoimmune diseases, IL-23 appears to be a perfect target for a potential treatment. There are several Abs targeting IL-23 showing promising results. Guselkumab and Tildrakizumab are two of those candidates. Guselkumab targets the IL-23p19 subunit and showed to be viable as a new treatment option for psoriasis [65]. Recently tested in a phase 3 trial, it may be applicable for treatment of psoriatic arthritis in the future too [66]. Tildrakizumab is another humanized monoclonal antibody targeting the IL- 23/IL17 axis via the p19 subunit of IL-23. It is approved for the treatment of psoriasis [67]. Another recent approach is a gene-silencing Py-Im polyamide that can enter the nucleus and compete with c-Rel for binding at the IL-23p19 promotor, therefore limiting IL-23 expression on DNA level [68]. However, application of previously admitted drugs is still limited to few diseases and further clinical studies to expand treatment options are essential.
Discussion
In this review we presented the current understanding of Th17 cells and highlighted the role of IL-23 in polarization and plasticity. Understanding the factors that lead to the development of the Th17 cell subtype and decoding its interactions with tissue and other T helper subtypes remains crucial in order to understand autoimmune diseases. In summary, IL-23 is the key cytokine leading to pathogenicity in Th17 cells. Regulating the key effector molecules IL22 and GM-CSF, IL-23 drives inflammation in autoimmune disease (Codarri et al. 2011). With the findings from recent studies, factors that drive Th17 pathogenicity are better understood, but regarding Th17 plasticity, more studies are needed to illuminate the exact pathways and interactions of Th17 cells [24]. According to our current knowledge IL-23 is a key regulator of Th17 stability. However, there is evidence linking IL-23 to the Th17/Th1 axis as it can upregulate T-bet and Stat4 and therefore IFN-γ [21]. There could be other key regulators of Th17 fate yet to be discovered at this time that are important for stabilizing Th17 fate or pushing them towards another subset. Therefore, these contrary roles of IL-23 could be due to different cofactors. Future comprehensive studies focusing on these points will hopefully help us understand Th17 fate even better and translate findings into the clinic. A lot of the data on Th17 plasticity comes from only a few models of autoimmune disease, mainly EAE and colitis rheumatoid arthritis. Hence it is interesting to get more data on Th17 plasticity working with new autoimmune disease models in the future. There may be a bias in Th17 plasticity comparing different animal models, which could be addressed in the future and more studies on human samples are necessary due to the fact that findings in animal models seldom translate optimally to patients. A recent study investigating mice T cells in colitis facing different pathogens came to the conclusion that T cells cluster above all according to the pathogens they face and not to their cell type [6]. Therefore, getting more data on the behavior of T cells under changing microbiomes and pathogens may result in a better understanding of triggers of Th17 plasticity. Plasticity seems to not only be limited to pushing a phenotype towards another T helper subset, but may also play a role in fine-tuning within one subset. A more immunoregulatory Th17 subset producing IL10 and more pathogenic Th17 cells lacking IL10 production are reported [41]. These two subsets may merge. It will be interesting to test if it is more beneficial to push Th17 cells towards Tr1/Treg cells or if pushing them only towards a more IL10 producing Th17 subtype is advantageous. Remaining more Th17 like may combine the beneficial host defense mechanisms with immunoregulatory properties. There is a lot of evidence pointing towards an important role for IL-23 in Th17/Th1 plasticity, however direct evidence is necessary to further prove this [47]. Even though there is a lot of in vitro data and data from mouse models, the mechanisms behind Th17 plasticity in the actual in vivo situation in humans are still incompletely understood. The directions of plasticity in different diseases are still not clear and may differ between models. Th1-like Th17 cells in one disease may arise from the Th17 pool, while in the context of another disease have their origin in Th1 cells. Future studies addressing this question and the exact mechanisms driving Th17 plasticity are necessary. Investigation of Th17 cells with new technologies built on scRNAseq will help understand their properties and interactions [69]. Velocity indicates the direction of differentiation of a single cell within a scRNAseq dataset based on spliced and unspliced mRNA [70]. Using this package on Th17 scRNAseq data from different diseases may answer the questions regarding directions of plasticity. Analyzing the gene expression and all the proteins expressed by a single Th17 cell, will further reveal factors that drive transdifferentiation. This might also indicate the state of Th17 cells, when prone to transdifferentiation. With scRNAseq the future for plasticity research looks promising. Recently introduced bioinformatic evaluation packages, forming trajectories and illuminating interactions between T cell subsets, may fill the gaps in our current understanding of Th17 fate.
Acknowledgements
This study was supported by a grant from the Deutsche Forschungsgemeinschaft, DFG (SFB 1192 to C.F.K.) and a stipend from the SFB 1192 Research Training Group to J.K.
References
2. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. 2015 Jul 1;74(1):5-17.
3. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000 Mar 17;100(6):655-69.
4. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006 Sep 22;126(6):1121-33.
5. Krebs CF, Turner JE, Paust HJ, Kapffer S, Koyro T, Krohn S, et al. Plasticity of Th17 cells in autoimmune kidney diseases. The Journal of Immunology. 2016a Jul 15;197(2):449-57.
6. Kiner E, Willie E, Vijaykumar B, Chowdhary K, Schmutz H, Chandler J, et al. Gut CD4+ T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nature Immunology. 2021 Feb;22(2):216-228.
7. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. The Journal of Experimental Medicine. 2005 Jan 17;201(2):233-40.
8. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature Immunology. 2005a Nov;6(11):1133-41.
9. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17–producing T helper memory cells. Nature Immunology. 2007 Jun; 8(6):639-46.
10. Happel KI, Zheng M, Young E, Quinton LJ, Lockhart E, Ramsay AJ, et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL- 17 expression in response to Klebsiella pneumoniae infection. The Journal of Immunology. 2003 May 1;170(9):4432-6.
11. Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. The Journal of Experimental Medicine. 2008 Jul 7;205(7):1551-7.
12. Hanna S, Etzoni A. New host defense mechanisms against Candida species clarify the basis of clinical phenotypes. Journal of Allergy and Clinical Immunology. 2011 Jun 1;127(6):1433-7.
13. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nature Reviews Immunology. 2010 Jul;10(7):479-89.
14. Krebs CF, Reimers D, Zhao Y, Paust HJ, Bartsch P, Nuñez S, et al. Pathogen-induced tissue-resident memory TH17 (TRM17) cells amplify autoimmune kidney disease. Science Immunology. 2020 Aug 7;5(50):eaba4163.
15. Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. The Journal of Immunology. 2007 Apr 15;178(8):4901-7.
16. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010 May 28;32(5):605- 15.
17. Krebs CF, Schmidt T, Riedel JH, Panzer U. T helper type 17 cells in immune-mediated glomerular disease. Nature Reviews Nephrology. 2017 Oct;13(10):647-59.
18. Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL- 12Rβ1 and a novel cytokine receptor subunit, IL-23R. The Journal of Immunology. 2002 Jun 1;168(11):5699-708.
19. Gagliani N, Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015 Jul;523(7559):221-5.
20. Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. The Journal of Immunology. 2008 Nov 1;181(9):5948-55.
21. Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009 Jan 16;30(1):92-107.
22. Harbour SN, DiToro DF, Witte SJ, Zindl CL, Gao M, Schoeb TR, et al. TH17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Science Immunology. 2020 Jul 17;5(49):eaaw2262.
23. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006 May;441(7090):235- 8.
24. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nature Immunology. 2011 Jun;12(6):560-7.
25. Evans HG, Roostalu U, Walter GJ, Gullick NJ, Frederiksen KS, Roberts CA, et al. TNF-α blockade induces IL-10 expression in human CD4+ T cells. Nature Communications. 2014 Feb 4;5(1):1-2.
26. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000 Nov 1;13(5):715-25.
27. McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23–IL- 17 immune pathway. Trends in Immunology. 2006 Jan 1;27(1):17-23.
28. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17–producing effector T helper cells in vivo. Nature Immunology. 2009 Mar;10(3):314-24.
29. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro-and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. The Journal of Experimental Medicine. 2003 Dec 15;198(12):1951-7.
30. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, ET AL. The encephalitogenicity of TH17 cells is dependent on IL-1-and IL-23- induced production of the cytokine GM-CSF. Nature Immunology. 2011 Jun;12(6):568-75.
31. Raphael I, Forsthuber TG. Stability of T-cell lineages in autoimmune diseases. Expert Review of Clinical Immunology. 2012 May 1;8(4):299-301.
32. Wiekowski MT, Leach MW, Evans EW, Sullivan L, Chen SC, Vassileva G, et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. The Journal of Immunology. 2001 Jun 15;166(12):7563-70.
33. Tang C, Chen S, Qian H, Huang W. Interleukin‐23: as a drug target for autoimmune inflammatory diseases. Immunology. 2012 Feb;135(2):112-24.
34. Zhao Y, Kilian C, Turner JE, Bosurgi L, Roedl K, Bartsch P, et al. Clonal expansion and activation of tissue-resident memory-like TH17 cells expressing GM-CSF in the lungs of patients with severe COVID-19. Science Immunology. 2021 Feb 23;6(56):eabf6692.
35. Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. The American Journal of Human Genetics. 2012 Apr 6;90(4):636-47.
36. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nature genetics. 2012 Dec;44(12):1341-8.
37. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006 Dec 1;314(5804):1461-3.
38. Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, et al. Selective regulatory function of Socs3 in the formation of IL-17- secreting T cells. Proceedings of the National Academy of sciences. 2006 May 23;103(21):8137-42.
39. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature. 2010 Oct;467(7318):967-71.
40. Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, et al. IL‐22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. European Journal of Immunology. 2006 May;36(5):1309-23.
41. McGeachy MJ, Bak-Jensen KS, Chen YI, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-β and IL-6 drive the production of IL 17 and IL-10 by T cells and restrain TH-17 cell–mediated pathology. Nature Immunology. 2007 Dec;8(12):1390-7.
42. Aschenbrenner D, Foglierini M, Jarrossay D, Hu D, Weiner HL, Kuchroo VK, et al. An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nature Immunology. 2018 Oct;19(10):1126-36.
43. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nature Immunology. 2011 Mar;12(3):255-63.
44. Kamali AN, Noorbakhsh SM, Hamedifar H, Jadidi-Niaragh F, Yazdani R, Bautista JM, et al. A role for Th1-like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Molecular Immunology. 2019 Jan 1;105:107-15.
45. Bartsch P, Kilian C, Hellmig M, Paust HJ, Borchers A, Sivayoganathan A, et al. Th17 cell plasticity towards a T-bet-dependent Th1 phenotype is required for bacterial control in Staphylococcus aureus infection. PLoS Pathogens. 2022 Apr 21;18(4):e1010430.
46. Xiao S, Yosef N, Yang J, Wang Y, Zhou L, Zhu C, et al. Smallmolecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity. 2014 Apr 17;40(4):477- 89.
47. Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proceedings of the National Academy of Sciences of the United States of America. 2015 Jun 2;112(22):7061-6.
48. Hickman-Brecks CL, Racz JL, Meyer DM, LaBranche TP, Allen PM. Th17 cells can provide B cell help in autoantibody induced arthritis. Journal of Autoimmunity. 2011 Feb 1;36(1):65-75.
49. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proceedings of the National Academy of Sciences of the United States of America. 2010 Aug 17;107(33):14751- 6.
50. Fletcher JM, Lalor SJ, Sweeney CM, Tubridy N, Mills KH. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clinical & Experimental Immunology. 2010 Oct;162(1):1-11.
51. Brucklacher-Waldert V, Ferreira C, Innocentin S, Kamdar S, Withers DR, Kullberg MC, et al. Tbet or continued RORγt expression is not required for Th17-associated immunopathology. The Journal of Immunology. 2016 Jun 15;196(12):4893-904.
52. Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, et al. T-bet represses TH17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nature Immunology. 2011 Jan;12(1):96-104.
53. Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, et al. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity. 2014 Mar 20;40(3):355-66.
54. Park IK, Shultz LD, Letterio JJ, Gorham JD. TGF-β1 inhibits T-bet induction by IFN-γ in murine CD4+ T cells through the protein tyrosine phosphatase Src homology region 2 domaincontaining phosphatase-1. The Journal of Immunology. 2005b Nov 1;175(9):5666-74.
55. Krebs CF, Paust HJ, Krohn S, Koyro T, Brix SR, Riedel JH, et al. Autoimmune renal disease is exacerbated by S1P-receptor-1- dependent intestinal Th17 cell migration to the kidney. Immunity. 2016b Nov 15;45(5):1078-92.
56. Summers SA, Steinmetz OM, Li M, Kausman JY, Semple T, Edgtton KL, et al. Th1 and Th17 cells induce proliferative glomerulonephritis. Journal of the American Society of Nephrology. 2009 Dec 1;20(12):2518-24.
57. Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, et al. Induction and molecular signature of pathogenic TH17 cells. Nature Immunology. 2012 Oct;13(10):991-9.
58. Meyer Zu Horste G, Wu C, Wang C, Cong L, Pawlak M, Lee Y, et al. RBPJ Controls Development of Pathogenic Th17 Cells by Regulating IL-23 Receptor Expression. Cell Reports. 2016 Jul 12;16(2):392-404.
59. Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, Lovett-Racke AE. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. Journal of Immunology. 2007 Feb 1;178(3):1341-8.
60. Hatton RD. TGF-β in Th17 cell development: the truth is out there. Immunity. 2011 Mar 25;34(3):288-90.
61. Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORγt+ T cells. The Journal of Experimental Medicine. 2008 Jun 9;205(6):1381-93.
62. Kluger MA, Meyer MC, Nosko A, Goerke B, Luig M, Wegscheid C, et al. RORγt+ Foxp3+ cells are an independent bifunctional regulatory T cell lineage and mediate crescentic GN. Journal of the American Society of Nephrology. 2016 Feb 1;27(2):454-65.
63. Kannan AK, Su Z, Gauvin DM, Paulsboe SE, Duggan R, Lasko LM, et al. IL-23 induces regulatory T cell plasticity with implications for inflammatory skin diseases. Scientific Reports. 2019 Nov 27;9(1):1-8.
64. Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, et al. Plasticity of TH17 cells in Peyer’s patches is responsible for the induction of T cell–dependent IgA responses. Nature Immunology. 2013 Apr;14(4):372-9.
65. Reich K, Armstrong AW, Foley P, Song M, Wasfi Y, Randazzo B, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo-and active comparator–controlled voyage 2 trial. Journal of the American Academy of Dermatology. 2017 Mar 1;76(3):418-31.
66. Deodhar A, Helliwell PS, Boehncke WH, Kollmeier AP, Hsia EC, Subramanian RA, et al. Guselkumab in patients with active psoriatic arthritis who were biologic-naive or had previously received TNFα inhibitor treatment (DISCOVER-1): a double-blind, randomised, placebo-controlled phase 3 trial. The Lancet. 2020 Apr 4;395(10230):1115-25.
67. Frampton JE. Tildrakizumab: a review in moderate-to-severe plaque psoriasis. American Journal of Clinical Dermatology. 2019 Apr;20(2):295-306.
68. He X, Liu R, Fan T, Huang X, Wu C, Su W, et al. Treating Autoimmune Diseases by Targeting IL-23 with Gene-Silencing Pyrrole–Imidazole Polyamide. The Journal of Immunology. 2020 Apr 15;204(8):2053-63.
69. Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Experimental & Molecular Medicine. 2018 Aug;50(8):1-4.
70. La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells. Nature. 2018 Aug;560(7719):494-8.