Abstract
A role for oxidative stress in the etiology of myriad neuropathologies is well accepted. However, the specific effects of oxidative DNA damage in the onset or promotion of neuronal dysfunction have been less studied. In our recent publication by Behrouzi et al. (Oxidative DNA Damage and Cisplatin Neurotoxicity Is Exacerbated by Inhibition of OGG1 Glycosylase Activity and APE1 Endonuclease Activity in Sensory Neurons), inhibition of enzymes that play a role in repairing oxidative DNA damage exacerbated neurotoxic effects of the chemotherapeutic agent, cisplatin. In this Commentary, we aim to expand on the contribution of oxidative DNA damage to other neuropathologies within the peripheral and central nervous systems, including irritable bowel disease, aging and Alzheimer’s disease, amyotrophic lateral sclerosis, and other neurodegenerative diseases. Consistently, clinical neuropathology and disease progression correlates with increases in oxidative DNA damage within clinical biopsies. Progress in animal models of these diseases has elucidated a causative role for oxidative DNA damage in disease progression, as dampening the DNA repair response exacerbates disease, whereas promoting DNA repair mitigates disease. Overall, this Commentary highlights the importance of expanding our studies on oxidative DNA damage in the nervous system, as enhancing oxidative DNA repair might prove to be a potential therapeutic target for the mitigation of neurodegeneration.
Keywords
Oxidative stress, Oxidative DNA damage, Base excision repair, 8-oxoguanine DNA glycosylase-1, Apurinic/apyrimidinic endonuclease/redox effector factor 1, Chemotherapy-induced peripheral neuropathy, Inflammatory bowel disease, Alzheimer’s disease, Aging, Amyotrophic lateral sclerosis
Introduction
There are a wide range of neuropathologies in which oxidative and nitrooxidative stress play a predominant role in the etiology of the disease [1-3]. Extensive research efforts have focused on examining the effects of reactive oxygen and nitrogen species (ROS/RNS) on the production of oxidized proteins and lipid peroxidation within neurons [4-6]. Another byproduct of oxidative stress that has been understudied in neuroscience, however, is oxidative DNA damage and the role of that damage in affecting neuronal function. One potential reason for the paucity of neuroscience research in the area of oxidative DNA damage is the notion that DNA damage is less dangerous in fully differentiated neurons compared to other cell types, as neurons do not undergo cellular division. However, recent studies have highlighted the effects of oxidative DNA damage to increase transcription, decrease transcription, and alter signal transduction pathways [7-15].
Reactive oxygen and nitrogen species are produced by normal cellular metabolism and excessive production is ameliorated by the activity of endogenous antioxidant mechanisms, which are robust in neurons [16]. Oftentimes, however, the antioxidant mechanisms can be overwhelmed and unmitigated ROS/RNS can cause oxidative DNA damage. Oxidative DNA damage is induced by the interaction between a highly reactive oxygen or nitrogen species and a DNA base, resulting in production of various base lesions. The most abundant oxidative DNA lesion is called 7,8-dihydro-8-oxo-2'- deoxyguanosine (8oxo-dG), which is formed at the 8th carbon of the guanine base [17-19]. However, other lesions include 2,6-diamino-4-oxo-5-formamidopyrimidine (FapyG), which also forms on the guanine base; and 7,8-dihydro-8-oxoadenine (oxoA) on the adenine base. The relative abundance of these lesions highlights the importance of 8oxo-dG and FapyG; however, there is opportunity to explore less abundant lesions, such as oxoA and others, and to critically assess the relative incidence of these lesions in neurons relative to other cell types. As mentioned previously, oxidative lesions can cause altered DNA transcription and signal transduction [7-15] that can lead to functional impairment of cells. The base excision repair (BER) pathway is primarily responsible for the repair of oxidative DNA damage (See Figure 1 for a simplistic overview of the pathway). Cleavage of the damaged base is achieved by various glycosylases, depending on the lesion of interest. The primary glycosylase responsible for 8oxodG cleavage is 8-oxoguanine DNA glycosylase-1 (OGG1) [20]. There are other glycosylases that can recognize 8oxodG, including Nth Like DNA Glycosylase 1 (NTHL1) and Neilike DNA glycosylases 1 and 2 (NEIL1 and NEIL2), but they primarily recognize other pyrimidine lesions. Additional DNA glycosylases that recognize and cleave non-guanine lesions include Thymine-DNA glycosylase (TDG) and Uracil- DNA glycosylase (UNG). An abasic site is generated following removal of the base by the appropriate glycosylase, which is recognized and processed by AP endonuclease 1/Ref-1 (APE1/ Ref-1). APE1/Ref-1 processes the ends on the DNA backbone, which allows DNA polymerase β (Polβ) to recognize and fill the abasic site with the correct base through template-directed synthesis [21]. In addition to the endonuclease function of APE1/Ref-1, the enzyme also has a redox function, regulating transcriptional activity via the reduction of a number of transcriptional factors [22]. Another enzyme that minimizes oxidative DNA damage is Nudix hydrolase 1 (also known as MTH1), which hydrolyzes oxidized bases within the nucleoside triphosphate pools to avoid incorporation of a damaged base into a nucleic acid [23,24]. The relative contributions of these different components of the base excision repair pathway to ward off deleterious effects of oxidative DNA damage are now being explored in different neuropathologies. Here we will briefly discuss our recent findings in sensory neurons exposed to DNA-damaging chemotherapeutics and explore the role of oxidative DNA damage in other peripheral and central nervous system pathologies.
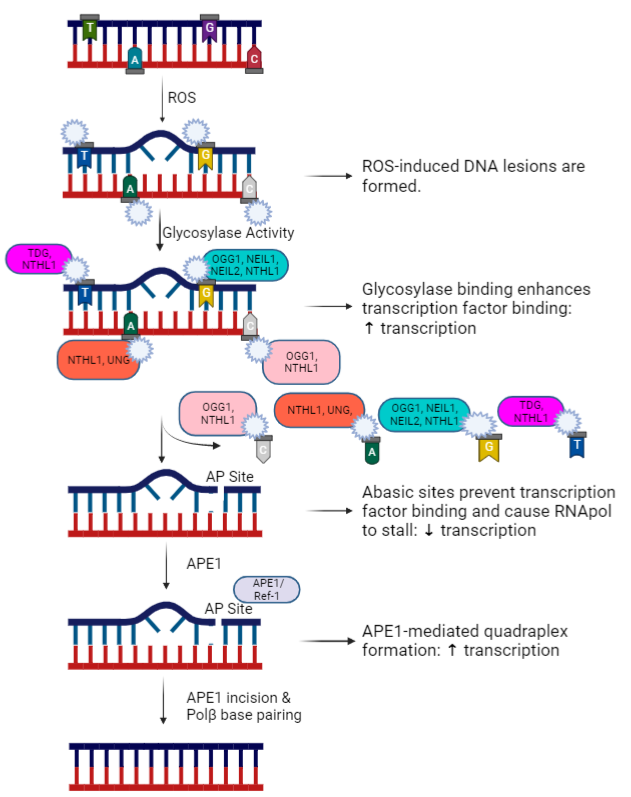
Figure 1: An overview of the base excision repair pathway with various base-specific glycosylases and stage-specific differential effects on transcription. Figure created in Biorender.com.
Peripheral Neuropathies
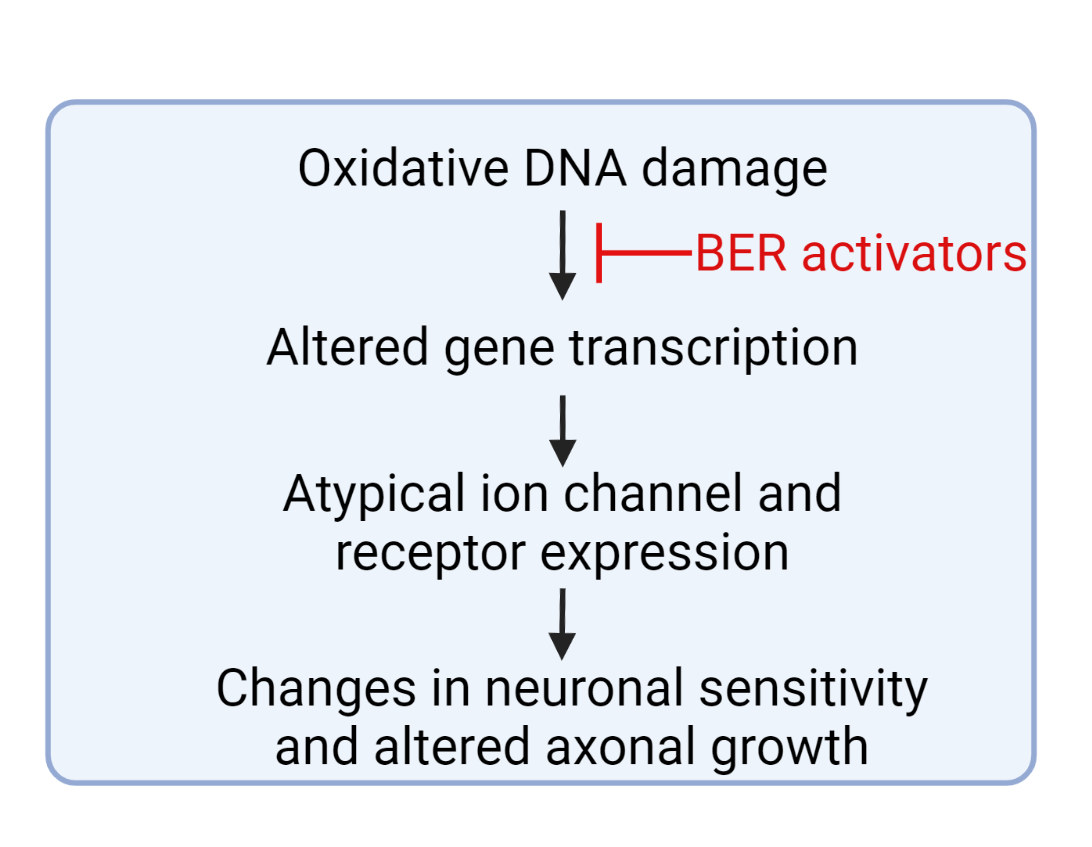
Figure 2: A proposed role for how enhancing DNA repair in sensory neurons could prevent or reverse chemotherapy-induced peripheral neuropathy. Figure created in Biorender.com.
Irritable Bowel Disease
Oxidative stress also contributes to tissue damage in patients with Irritable Bowel Disease (IBD). Mucosal biopsies from patients with IBD have enhanced reactive oxygen intermediates, diminished endogenous antioxidants, and increased oxidative DNA damage, as measured by 8oxodG [42]. The specific role of oxidative DNA damage in the pathophysiology of IBD was recently assessed in an animal model of spontaneous chronic colitis [43]. In this study, the investigators examined the effects of enhancing the DNA repair activity, while simultaneously diminishing the redox activity of APE1/Ref-1 via the pharmacological administration of APX3330. APX3330 administration reversed gross histological changes to the distal colon, diminished leukocyte infiltration into the colon, and restored gastrointestinal motility in the animal model of colitis. Upon further examination, reversal of these symptoms was associated with an increased survival of myenteric nerves and glial cells with diminished superoxide production and 8oxo-dG formation in the myenteric plexus [43]. These data suggest a prominent role for colonic oxidative stress to induce transcriptional activity and oxidative DNA damage within the enteric nervous system and highlights APE1/Ref-1 as a potential therapeutic target for patients with IBD.
While neuropathies and IBD are largely a result of dysfunction within peripheral sensory neurons, oxidative DNA damage may mediate pathology of many diseases and the following sections highlight research demonstrating a role for oxidative DNA damage in central nervous system diseases.
Aging and Alzheimer’s Disease
Increasing levels of oxidative stress and subsequent accumulation of DNA damage is a hallmark of aging and has been shown to underlie age-related and Alzheimer’s Diseaseassociated cognitive impairment [44,45]. Further, exacerbation of DNA damage via suppression or dysfunction of DNA damage repair proteins can accelerate age-related cognitive decline in rodents and humans [46,47], supporting a specific role for oxidative DNA damage in the etiology of aging. These results have been replicated in an animal model, whereby the activity of OGG1 is experimentally reduced. Suppression of OGG1 activity results in an increase in age-induced 8oxo-dG levels in mouse brains with a concomitant decline in cognitive function [48]. Similarly, the same authors explored the effects of compromised OGG1 activity in a 5XFAD mouse model of Alzheimer’s disease (AD) and found an increase in 8oxo-dG in hippocampal neurons and further deficits in cognition compared to 5XFAD mice with normal OGG1 activity levels [48]. Similar findings were described by Oka et al., who demonstrated that knockout of OGG1 and the nucleoside hydrolase, MTH1, which work collectively to diminish the burden of 8oxo-dG, exacerbated the accumulation of 8oxo-dG in brain microglia of triple-transgenic AD model mice (3xTg- AD-H) and promoted degeneration of the hippocampus [49]. Furthermore, the loss of MTH1 and OGG1 activity resulted in an exacerbation of cognitive impairment in the AD mice [49]. Clinically, oxidative DNA damage is validated, as 8oxodG levels are more prevalent in AD patient brains [1,50] and serum [51] compared to age-matched controls. This increase in oxidative DNA damage suggests increased production of ROS in AD patients that overwhelm repair mechanisms, a diseaserelated dysfunction or diminution of endogenous antioxidant or DNA damage repair activity, or some combination of the two. Altogether, these publications support the notion that oxidative DNA damage plays a key role in the development or progression of AD, thus further research on ways to enhance the repair of oxidative DNA damage is warranted to identify novel putative therapeutics for AD.
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is a neurodegenerative disease of large motor neurons and oxidative stress is presumed to also contribute to the pathogenesis of the disease [52]. ALS patients exhibit elevated levels of 8oxo-dG in upper (motor cortex) and lower (spinal cord) motor neurons compared to agematched controls [53-56] and motor neurons with prominent nuclear 8oxo-dG staining exhibit markers of cellular death/ dying [56]. Interestingly, the genes for APE1/Ref-1 and OGG1 are hypomethylated in motor cortex and spinal cord motor neurons from ALS patients, suggesting continued expression of the repair proteins and the propensity for DNA repair, thus enhancing the activity of these proteins might be a viable therapeutic strategy for ALS. Indeed, in an animal model of spinal motor nerve degeneration via sciatic nerve avulsion where there is an increase in 8oxo-dG [57], overexpression of APE1/Ref-1 or OGG1 to enhance DNA repair and diminish the DNA damage response attenuates neuronal cell loss in the spinal cord [58].
Other Neurodegenerative Diseases
Correlations between an increase in oxidative DNA damage and clinical pathology have also been established for Parkinson’s disease (PD), with increases in 8oxo-dG levels and increased levels of DNA repair enzymes in the substantia nigra of PD patients [59,60]. Similar increases in oxidative DNA damage have been observed in Huntington’s Disease (HD) patient samples and in brain from mouse models of HD [61,62]. An elegant study has demonstrated that huntingtin plays a role in engaging the BER pathway in response to oxidative stress and is recruited to sites of oxidative DNA damage, but that mutant huntingtin introduces deficiencies in BER, leading to the accumulation of excess oxidative DNA damage [63].
Conclusions
The repair of oxidative DNA damage through engagement of the BER pathway in post-mitotic cells, such as neurons, is critical because the cells do not have robust alternative strategies to repair the damage, such as replication proofreading and mismatch repair [64]. As reviewed herein, there is strong evidence that oxidative DNA damage correlates with neuropathologies and moderate evidence that DNA damage contributes to the onset or progression of some diseases in both the peripheral and central nervous system. Behrouzi et al. (2022) built upon an existing body of work examining the role of oxidative DNA damage in CIPN; however, further work is needed to fully understand how oxidative DNA damage alters transcriptional profiles to alter neuronal function. Similarly, further work in other neurodegenerative diseases is necessary to identify a causal role for oxidative DNA damage and explore therapeutics that could promote DNA repair and potentially lead to breakthroughs in clinical patient care.
Acknowledgements
A. Behrouzi and J.C. Fehrenbacher wrote the Commentary, with M.R. Kelley providing edits and suggestions.
Financial support for this work was provided by the National Institutes of Health by the National Cancer Institute CA205166 (M.R. Kelley and J.C. Fehrenbacher) and CA231267 (J.C. Fehrenbacher and M.R. Kelley). Additional financial support was provided by the Earl and Betty Herr Professor in Pediatric Oncology Research, the Jeff Gordon Children’s Foundation, and the Riley Children’s Foundation (M.R. Kelley).
References
2. Zhou Y, Zhen Y, Wang G, Liu B. Deconvoluting the Complexity of Reactive Oxygen Species (ROS) in Neurodegenerative Diseases. Front Neuroanat. 2022;16:910427.
3. Fukae J, Mizuno Y, Hattori N. Mitochondrial dysfunction in Parkinson's disease. Mitochondrion. 2007;7(1-2):58-62.
4. Angelova PR, Esteras N, Abramov AY. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: Finding ways for prevention. Med Res Rev. 2021;41(2):770-84.
5. Hajieva P, Bayatti N, Granold M, Behl C, Moosmann B. Membrane protein oxidation determines neuronal degeneration. J Neurochem. 2015;133(3):352-67.
6. Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. 2012;2012:428010. Epub 2012/06/12.
7. Hailer-Morrison MK, Kotler JM, Martin BD, Sugden KD. Oxidized guanine lesions as modulators of gene transcription. Altered p50 binding affinity and repair shielding by 7,8-dihydro-8-oxo-2'- deoxyguanosine lesions in the NF-kappaB promoter element. Biochemistry. 2003;42(32):9761-70.
8. Ramon O, Sauvaigo S, Gasparutto D, Faure P, Favier A, Cadet J. Effects of 8-oxo-7,8-dihydro-2'-deoxyguanosine on the binding of the transcription factor Sp1 to its cognate target DNA sequence (GC box). Free Radic Res. 1999;31(3):217-29.
9. Moore SP, Toomire KJ, Strauss PR. DNA modifications repaired by base excision repair are epigenetic. DNA Repair (Amst). 2013;12(12):1152-8.
10. Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst). 2004;3(5):483-94.
11. Allgayer J, Kitsera N, von der Lippen C, Epe B, Khobta A. Modulation of base excision repair of 8-oxoguanine by the nucleotide sequence. Nucleic Acids Res. 2013;41(18):8559-71.
12. Visnes T, Cazares-Korner A, Hao W, Wallner O, Masuyer G, Loseva O, et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science. 2018;362(6416):834-9.
13. Pan L, Zhu B, Hao W, Zeng X, Vlahopoulos SA, Hazra TK, et al. Oxidized Guanine Base Lesions Function in 8-Oxoguanine DNA Glycosylase-1-mediated Epigenetic Regulation of Nuclear Factor kappaB-driven Gene Expression. J Biol Chem. 2016;291(49):25553- 66.
14. Boldogh I, Hajas G, Aguilera-Aguirre L, Hegde ML, Radak Z, Bacsi A, et al. Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J Biol Chem. 2012;287(25):20769-73.
15. Luo J, Hosoki K, Bacsi A, Radak Z, Hegde ML, Sur S, et al. 8-Oxoguanine DNA glycosylase-1-mediated DNA repair is associated with Rho GTPase activation and alpha-smooth muscle actin polymerization. Free Radic Biol Med. 2014;73:430-8.
16. Lee KH, Cha M, Lee BH. Neuroprotective Effect of Antioxidants in the Brain. Int J Mol Sci. 2020; 21(19):7152.
17. Cadet J, Douki T, Ravanat JL. One-electron oxidation of DNA and inflammation processes. Nat Chem Biol. 2006;2(7):348-9.
18. Margolin Y, Cloutier JF, Shafirovich V, Geacintov NE, Dedon PC. Paradoxical hotspots for guanine oxidation by a chemical mediator of inflammation. Nat Chem Biol. 2006;2(7):365-6.
19. Steenken S, Jovanovic SV. How Easily Oxidizable Is DNA? One-Electron Reduction Potentials of Adenosine and Guanosine Radicals in Aqueous Solution. Journal of the American Chemical Society. 1997;119(3):617-8.
20. Monden Y, Arai T, Asano M, Ohtsuka E, Aburatani H, Nishimura S. Human MMH (OGG1) type 1a protein is a major enzyme for repair of 8-hydroxyguanine lesions in human cells. Biochem Biophys Res Commun. 1999;258(3):605-10.
21. Singhal RK, Prasad R, Wilson SH. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J Biol Chem. 1995;270(2):949-57.
22. Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid Redox Signal. 2009;11(3):601-20.
23. Fujikawa K, Kamiya H, Yakushiji H, Fujii Y, Nakabeppu Y, Kasai H. The oxidized forms of dATP are substrates for the human MutT homologue, the hMTH1 protein. J Biol Chem. 1999;274(26):18201- 5.
24. Fujikawa K, Kamiya H, Yakushiji H, Nakabeppu Y, Kasai H. Human MTH1 protein hydrolyzes the oxidized ribonucleotide, 2-hydroxy-ATP. Nucleic Acids Res. 2001;29(2):449-54.
25. Loprinzi CL, Lacchetti C, Bleeker J, Cavaletti G, Chauhan C, Hertz DL, et al. Prevention and Management of Chemotherapy- Induced Peripheral Neuropathy in Survivors of Adult Cancers: ASCO Guideline Update. J Clin Oncol. 2020;38(28):3325-48.
26. Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS One. 2014;9(9):e106485.
27. Vasko MR, Guo C, Thompson EL, Kelley MR. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA Repair (Amst). 2011;10(9):942-52.
28. Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/ apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68(15):6425-34.
29. Ta LE, Espeset L, Podratz J, Windebank AJ. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. NeuroToxicology. 2006;27(6):992- 1002.
30. Dzagnidze A, Katsarava Z, Makhalova J, Liedert B, Yoon MS, Kaube H, et al. Repair capacity for platinum-DNA adducts determines the severity of cisplatin-induced peripheral neuropathy. J Neurosci. 2007;27(35):9451-7.
31. Sawant A, Floyd AM, Dangeti M, Lei W, Sobol RW, Patrick SM. Differential role of base excision repair proteins in mediating cisplatin cytotoxicity. DNA Repair (Amst). 2017;51:46-59.
32. Kim HS, Guo C, Thompson EL, Jiang Y, Kelley MR, Vasko MR, et al. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat Res. 2015;779:96-104.
33. Duggett NA, Griffiths LA, McKenna OE, de Santis V, Yongsanguanchai N, Mokori EB, et al. Oxidative stress in the development, maintenance and resolution of paclitaxel-induced painful neuropathy. Neuroscience. 2016;333:13-26.
34. Wani TH, Chakrabarty A, Shibata N, Yamazaki H, Guengerich FP, Chowdhury G. The Dihydroxy Metabolite of the Teratogen Thalidomide Causes Oxidative DNA Damage. Chem Res Toxicol. 2017;30(8):1622-8.
35. Lee CJ, Goncalves LL, Wells PG. Embryopathic effects of thalidomide and its hydrolysis products in rabbit embryo culture: evidence for a prostaglandin H synthase (PHS)-dependent, reactive oxygen species (ROS)-mediated mechanism. FASEB J. 2011;25(7):2468-83.
36. Manguinhas R, Fernandes AS, Costa JG, Saraiva N, Camoes SP, Gil N, et al. Impact of the APE1 Redox Function Inhibitor E3330 in Non-small Cell Lung Cancer Cells Exposed to Cisplatin: Increased Cytotoxicity and Impairment of Cell Migration and Invasion. Antioxidants (Basel). 2020;9(6):550.
37. Fishel ML, Xia H, McGeown J, McIlwain DW, Elbanna M, Craft AA, et al. Antitumor Activity and Mechanistic Characterization of APE1/Ref-1 Inhibitors in Bladder Cancer. Mol Cancer Ther. 2019;18(11):1947-60.
38. Pang L, Lian X, Liu H, Zhang Y, Li Q, Cai Y, et al. Understanding Diabetic Neuropathy: Focus on Oxidative Stress. Oxid Med Cell Longev. 2020;2020:9524635.
39. Shakeel M. Recent advances in understanding the role of oxidative stress in diabetic neuropathy. Diabetes Metab Syndr. 2015;9(4):373-8.
40. Brinkman K, ter Hofstede HJM, Burger DM, Smeitink JAM, Koopmans PP. Adverse effects of reverse transcriptase inhibitors: mitochondrial toxicity as common pathway. AIDS. 1998;12(14):1735-44.
41. Fehrenbacher JC, Guo C, Kelley MR, Vasko MR. DNA damage mediates changes in neuronal sensitivity induced by the inflammatory mediators, MCP-1 and LPS, and can be reversed by enhancing the DNA repair function of APE1. Neuroscience. 2017;366:23-35.
42. Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci. 1996;41(10):2078-86.
43. Sahakian L, Filippone RT, Stavely R, Robinson AM, Yan XS, Abalo R, et al. Inhibition of APE1/Ref-1 Redox Signaling Alleviates Intestinal Dysfunction and Damage to Myenteric Neurons in a Mouse Model of Spontaneous Chronic Colitis. Inflamm Bowel Dis. 2021;27(3):388-406.
44. Moneim AE. Oxidant/Antioxidant imbalance and the risk of Alzheimer's disease. Curr Alzheimer Res. 2015;12(4):335-49.
45. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475-85.
46. de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, et al. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296(5571):1276-9.
47. Borgesius NZ, de Waard MC, van der Pluijm I, Omrani A, Zondag GC, van der Horst GT, et al. Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J Neurosci. 2011;31(35):12543-53.
48. Pao PC, Patnaik D, Watson LA, Gao F, Pan L, Wang J, et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer's disease. Nat Commun. 2020;11(1):2484.
49. Oka S, Leon J, Sakumi K, Abolhassani N, Sheng Z, Tsuchimoto D, et al. MTH1 and OGG1 maintain a low level of 8-oxoguanine in Alzheimer's brain, and prevent the progression of Alzheimer's pathogenesis. Sci Rep. 2021;11(1):5819.
50. de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer's disease. Lab Invest. 2000;80(8):1323-35.
51. Sliwinska A, Kwiatkowski D, Czarny P, Toma M, Wigner P, Drzewoski J, et al. The levels of 7,8-dihydrodeoxyguanosine (8- oxoG) and 8-oxoguanine DNA glycosylase 1 (OGG1) - A potential diagnostic biomarkers of Alzheimer's disease. J Neurol Sci. 2016;368:155-9.
52. Cookson MR, Shaw PJ. Oxidative stress and motor neurone disease. Brain Pathol. 1999;9(1):165-86.
53. Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69(5):2064-74.
54. Kikuchi H, Furuta A, Nishioka K, Suzuki SO, Nakabeppu Y, Iwaki T. Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol. 2002;103(4):408-14.
55. Shibata N, Nagai R, Miyata S, Jono T, Horiuchi S, Hirano A, et al. Nonoxidative protein glycation is implicated in familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Acta Neuropathol. 2000;100(3):275-84.
56. Kim BW, Jeong YE, Wong M, Martin LJ. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSCderived motor neurons with SOD1 mutations. Acta Neuropathol Commun. 2020;8(1):7.
57. Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci. 2005;25(27):6449-59.
58. Martin LJ, Wong M. Enforced DNA repair enzymes rescue neurons from apoptosis induced by target deprivation and axotomy in mouse models of neurodegeneration. Mech Ageing Dev. 2017;161(Pt A):149-62.
59. Fukae J, Takanashi M, Kubo S, Nishioka K, Nakabeppu Y, Mori H, et al. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson's disease and related neurodegenerative disorders. Acta Neuropathol. 2005;109(3):256-62.
60. Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y. Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson's disease. Ann Neurol. 1999;46(6):920-4.
61. Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington's disease. J Neurochem. 2001;79(6):1246-9.
62. Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, et al. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41(5):646-53.
63. Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WM, Truant R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. 2017;26(2):395-406.
64. Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don't divide what's to repair? Mutat Res. 2007;614(1-2):24-36.