Abstract
As to better understand and characterize the dramatic differences in antioxidant response of human cells harboring mutations in the frataxin gene responsible for Friedreich's ataxia (FRDA), we studied primary cultures of skin fibroblasts derived from five FRDA patients with different major frataxin gene expansion sizes. Since oxidative stress has been previously established to play a critical role in FRDA, among the many enzymes that may modulate oxidative stress sensitivity, we selected some that have previously been shown to be critical in oxidative stress. However, we were unable to identify a consistent index to rank individual cultures based on these measures. We therefore focused our study on FRDA fibroblast cell death and its modulation by different antioxidants under culture conditions that exacerbate oxidative stress sensitivity. Under conditions that require cells to rely on mitochondrial activity, we observed significant but variable effect on FRDA cell proliferation. These conditions were then used to test the efficacy to prevent or slow down cell death, of a panel of antioxidant molecules targeting different steps of the pro-oxidant cascade previously documented in FRDA, namely uridine, pyruvate, and pioglitazone. We observed a surprising variability in the response of patient fibroblasts to antioxidant molecules even using similar culture conditions. We conclude that the individual-specific response already discernible at the cellular level may well play an important role in the frequent difficulties encountered in reaching definitive conclusions when testing the ability of antioxidants to counteract the consequences of frataxin depletion, a conclusion that may apply to clinical trials as well.
Keywords
Oxidative stress, Variability, Uridine, Pyruvate, Pioglitazone
Abbreviations
FRDA: Friedreich Ataxia; CM: Complete Medium; SC: Stressful Condition; SCU: Stressful Condition plus Uridine; SCP: Stressful Condition plus Pyruvate; SCUP: Stressful Condition plus Uridine and Pyruvate
Introduction
Friedreich ataxia (FRDA) is an autosomal recessive hereditary mitochondrial disease (1/30,000 births) ranked among neurological diseases [1,2]. Often detected during adolescence or young adulthood, the onset may also occur in early childhood and even in toddlerhood [3]. Conversely the disease may arise much latter, up to 50 years of age or over [4-6]. Some patients endure a rapid and severe progression while others experience a very slow, somewhat erratic course of the disease. Patients presenting with early-onset disease (before 15 years of age) generally worsen faster than others. Documented neurological features of FRDA include progressive trunk and limbs mixed cerebellar and sensory ataxia, associated with dysarthria, pyramidal signs. Besides neurological symptoms, skeletal deformities (scoliosis, foot deformities) and cardiomyopathy are observed in most patients, with an increased frequency of diabetes. Finally, visual and hearing impairment, dysphagia and bladder dysfunction can also occur. Neurological signs may be subtle at onset while hypertrophic cardiomyopathy may be the prominent symptom initially detected. Conversely, the heart can remain asymptomatic throughout life for other patients.
Most of the cases (>95%) result from abnormal GAA-triplet expansions of different lengths on both alleles of the first intron of the Frataxin gene located on chromosome 9 [7]. These expansions reduce gene transcription resulting in the lowering of Frataxin levels (30 to 2% of control), an essential mitochondrial protein involved in iron-sulfur cluster (ISC) biogenesis [8]. In the heart micro biopsies of a series of FRDA patients, a generalized deficiency of mitochondrial respiratory chain (RC) ISC-containing proteins (ISP) was evidenced together with defects of both cytosolic and mitochondrial ISC-containing aconitases [9]. Similarly, in a number of organisms studied in laboratories, Frataxin deficiency results in impaired synthesis of ISC in the mitochondria [8]. In cultured human skin fibroblasts grown under standard conditions, Frataxin depletion does not significantly affect the activity of ISC-containing enzymes, either involved in the RC or the Krebs cycle [9]. Yet, it often causes a slight overproduction of superoxides revealed by an elevation of the superoxide-inducible SOD activity [10] and a low content in reduced glutathione [11-13]. These cells simultaneously display a frequent hypersensitivity to external oxidizing agents [14] including oxygen itself [15]. This hypersensitivity is observed in a number of animal and human cell types where Frataxin gene is mutated causing poor handling of oxidizing pressure, a process presumably instrumental in disease progression [15-17]. The prominent role played by oxidant stress can be exacerbated when a long expansion of the Frataxin gene additionally to Frataxin gene extinction disturbs the transduction of the neighboring gene, PIP5K1β, coding the PIP5K protein [18,19]. Impairing the transcription on the PIP5K-encoding gene destabilizes cell actin-network and causes major disorganization of actin-bound Keap1/Nrf2 antioxidant signaling proteins ultimately resulting in impaired antioxidant cell defenses [10].
Thus, whether considering the genetic aspects (e.g., size of the expansion in the Frataxin gene or identity of the genes involved, i.e., Frataxin, PIP5K1β), the biochemical aspects (e.g., damage to soluble ISP, to RC components or to antioxidant systems), the clinical signs and the sequence of their apparition, a very contrasting picture between patients emerges. Indeed, this interindividual variability is at the forefront of the characteristics of the disease [3,20-31].
Considering the context of this rare, slowly progressive disease, possibly resulting from the combination of several mechanisms, with rather unpredictable steps, the almost insurmountable difficulty to find a drug or a procedure equally effective for all patients becomes understandable. For the same reasons, organizing, interpreting therapeutic trials, making predictions about chances of therapeutic success, all have turned out to be true nightmares. Thus, not surprisingly, 26 years after the discovery of the genetic bases of the disease in 1996 [7], therapeutic trials have proved to be essentially inconclusive. So far statistical power of trials remains insufficient to reach firm conclusion, despite the frequent occurrence of sub-groups of patients positively responding to the treatment when none of the individuals receiving a placebo did [32]. A much better identification and understanding of the mechanisms that could explain the great variability observed between individuals appears therefore essential.
In the present study, we focused on the oxidative stress and its management targeting few steps of the Nrf2 pathway in cultured skin fibroblasts. In 2009, we identified this pathway as defective in FRDA in association with abnormalities of fibroblasts actin network [10]. The defect of Nrf2 antioxidant pathway was found to be more severe in case of impaired expression of the Frataxin-adjacent PIP5k-encoding gene caused by particularly large expansion in the Frataxin-gene [19]. This pioneer work paved the way to identify molecules targeting this pathway, (pioglitazone, leriglitazone, omaveloxolone). Noticeably this latter drug, in the absence of any available therapy, is the only one so far approved by the FDA for FRDA (https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-first-treatment-friedreichs-ataxia), since we and other have documented its frequent occurrence and partly delineated the putative underlying mechanism. Using precisely the five cell lines derived from FRDA patients for which we previously gather a set of molecular and biochemical data, we here underlines the one-to-one response to three substances known to exert antioxidant effects by distinct mechanisms (Figure 1). Uridine, acting particularly on the antioxidant signaling pathway of SOD1 (cytosolic and mitochondrial intermembrane location) [33] through the glyoxalase DJ-1 [34-36]; pyruvate, a potent hydrogen peroxide detoxifying agent through a non- enzymatic reaction specific to dicarboxylic acids [37,38]; pioglitazone acting on antioxidant defenses signaling, in particular of mitochondrial SOD2, via the Nrf2 transcription factor to rescue cells mortality in FRDA [39,40].
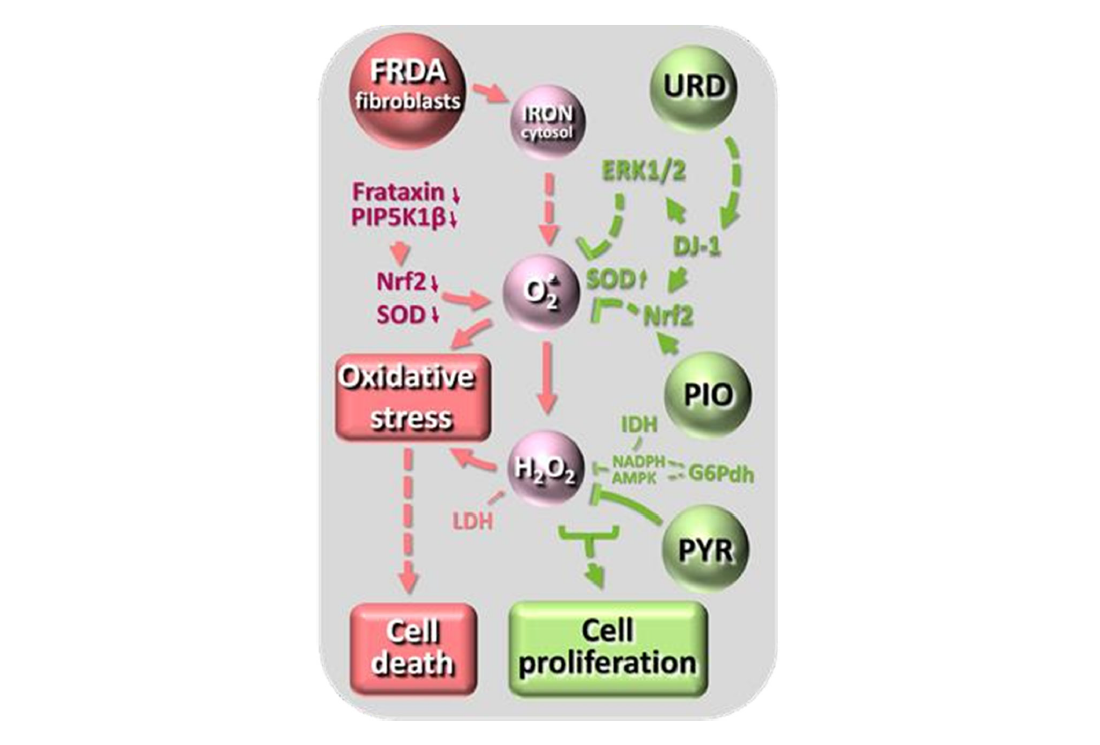
Figure 1. A very simplified diagram showing the action sites of the antioxidant molecules tested for their effect on fibroblast growth. On the left, the potential consequences of Frataxin gene expansion, in the middle the oxidative stress cascade, on the right, in green, the action sites of uridine (urd), pioglitazone (pio) and pyruvate (PYR) on the oxidative stress cascade. PIP5K1β, phosphatidylinositol-4-phosphate 5-kinase type 1 β; ERK1/2, extracellular signal- regulated kinase 1/2 cascade; DJ-1, a glyoxalase [34-35]) also known as Parkinson protein 7 (Park7); Nrf2, nuclear factor (erythroid-derived 2)-like 2; SOD, superoxide dismutase.
Materials and Methods
Human skin fibroblasts
Cultured fibroblasts were derived from skin biopsies obtained from five FRDA patients (FRDA1 to 5), and four anonymous healthy individuals, all from European ancestry. Written informed consent for research was obtained from patients and/or family members according to protocols in accordance with Robert Debré Hospital ethical committees (FRDA patient; 2011; Paris, France). The FRDA fibroblasts were from patients harboring a biallelic long GAA expansions (>2.6 kb) in the Frataxin (FXN) gene. These expansions were previously shown to impair the transcription of the adjacent PIP5K1β gene [18]. Compared to controls residual PIP5K1β mRNA was 58, 50, 43, 33 and 24% respectively [18] for patients noticed 1 to 5 in our study.
Cells were grown in T75 flasks in 10 ml Dulbecco’s modified Eagle's minimal essential medium (DMEM) containing 4.5 g/l glucose, 4 mM glutamine (Glutamax; Gibco Thermo Fisher Scientific), 2 mM pyruvate and 200 μM uridine (permissive complete medium, thereafter referred as CM) or lacking Glucose, Pyruvate and Uridine but containing 4 mM glutamine (non-permissive medium, thereafter referred as SC for stressful condition) or SC plus uridine (SCU), or SC plus pyruvate (SCP) or both uridine and pyruvate (SCUP). All media were supplemented with 10% fetal calf serum, 100 U/ml penicillin and streptomycin each. Cell pellets (1,500 g for 5 min) were kept frozen (-80°C) for ulterior biochemical enzyme assays studies.
Cells from a confluent culture (75-cm2 flask) were trypsinized and seeded in 4 × 75-cm2 flasks (20–25% confluency) containing permissive CM medium. After allowing the culture to stand for one night, the medium was changed to the desired conditions, i.e., CM medium, SC medium, or SCU, SCP, SCUP, SC or SCU plus DMSO (0.1 %) or 10 µM Pioglitazone in DMSO (pioglitazone hydrochloride, Sigma, France). We used control and patient fibroblasts with approximately the same number of passages, this by carrying out the manipulations with several different passages (from p3 to p14). The number of cells was estimated from random image sampling using the ImageJ processing program for 14 days without changing the medium. Noticeably, using brand new DMSO solutions is imperative as upon opening and several months of storage the solution becomes progressively pro-oxidative and deleterious for cultured cells. Due to its broad spectrum of solubility, DMSO still remains an often-irreplaceable solvent, e.g., for the development process of new antioxidants with neuroprotective properties [41,42].
Microscopy
Representative random phase-contrast photographs were taken on an LSM 5 Ex [1] citer optic microscope (Eclipse TE300 Nikon, France) (×4). The area of cells cultured in CM medium was estimated from random image sampling using the ImageJ processing program.
Enzyme measurements
Superoxide dismutase (total SOD) (EC 1.15.1.1) and catalase (EC 1.11.1.6) activities of cells cultured in CM and SC medium were measured either spectrophotometrically (SOD) or from oxygen release (catalase) at 37°C on frozen cell pellets according to previously described protocols [43].
The activity of various enzyme implicated in glucose metabolism and NADPH production: NADP+ glucose-6-phosphate dehydrogenase (G6PDH) (EC 1.1.1.49), NAD+ lactate dehydrogenase (LDH) (EC 1.1.1.27), NADP+- dependent isocitrate dehydrogenase (IDH1-2) (EC 1.1.1.42), NAD+ glutamate dehydrogenase (GDH) (EC 1.4.1.2) activities were measured in CM conditions. All activities were spectrophotometrically measured (Cary 60, Varian, Agilent Technologies) on frozen-thawed cell pellets, at 37°C according to published protocols [44]. Protein was estimated by the Bradford assay.
Statistics
Data are presented as mean ± SD for all experiments. Statistical significance was calculated by standard unpaired t-test or one-way ANOVA with Bonferroni post-test correction for more than two conditions. A p ≤ 0.05 was considered statistically significant (GraphPad Prism 5) and noticed with one asterisk, p values less than 0.01 with two asterisks, p values less than 0.001 with three asterisks.
Results
Stressful conditions for studying the effect of antioxidants
We first observed that, as previously reported [10], most FRDA fibroblasts cultured in medium containing high glucose and glutamine (providing substrates for both cell proliferation and antioxidant defenses), plus uridine and pyruvate increase their basal level of antioxidant defenses. This is reflected by a slight but significant increased level of basal SOD activity in the FRDA patient’s fibroblasts. This difference is abolished in SC medium containing only glutamine as carbon source (Table 1). No similar increase was observed for catalase activity whatever the culture condition used. Noticeably the fibroblasts from one of the FRDA patients displayed a particularly low catalase activity. Similarly, even when cells were cultured in CM condition, G6PDH, NADP+ IDH, NAD+ GDH and LDH activities were found unchanged (Table 1).
|
Enzyme |
Control fibroblasts (n=4; 3 assays each) |
FRDA fibroblasts (n=5; 3 assays each) |
FRDA1 |
FRDA2 |
FRDA3 |
FRDA4 |
FRDA5 |
|
Total SOD (U/mg/prot) |
|||||||
|
3.7 ± 0.5 |
4.9 ± 0.5* |
4.4 ± 1.8 (n=3) |
4.4 ± 1.4 (n=4) |
5.3 ± 1.5 (n=3) |
5.2 ± 2.7 (n=3) |
5.2 ± 1.7 (n=3) |
|
3.7 ± 1.0 |
3.8 ± 0.4 |
4.2 ± 0.4 (n=4) |
4.0 ± 0.5 (n=3) |
4.1 ± 0.8 (n=3) |
3.6 ± 1.0 (n=3) |
3.2 ± 0.3 (n=4) |
|
Catalase (nmol /mg/prot) |
|||||||
|
377 ± 118 |
434 ± 215 |
550 ± 137 (n=3) |
534 ± 136 (n=3) |
411 ± 150 (n=3) |
71 ± 24* (n=3) |
603 ± 112 (n=4) |
|
485 ± 90 |
584 ± 73 |
627 ± 105 (n=3) |
623 ± 152 (n=4) |
649 ± 113 (n=3) |
172 ± 67* (n=3) |
550 ± 168 (n=3) |
|
Glucose-6-phoshate dehydrogenase (nmol/min/mg prot) |
117 ± 15 |
96 ± 20 |
125 ± 30 (n=9) |
78 ± 11 (n=8) |
99 ± 23 (n=7) |
74 ± 23 (n=7) |
100 ± 30 (n=7) |
|
NADP+ Isocitrate dehydrogenase (nmol/min/mg prot) |
16.3 ± 2.2 |
15.7 ± 3.5 |
18.9 ± 6.6 (n=7) |
13.6 ± 4.3 (n=7) |
12.8 ± 5.6 (n=7) |
13.3 ± 5.2 (n=7) |
22.3 ± 3.0 (n=7) |
|
NAD+ Glutamate dehydrogenase (nmol/min/mg prot) |
9.8 ± 4.1 |
8.9 ± 3.1 |
7.7 ± 0.2 (n=3) |
11 ± 0.1 (n=3) |
9.8 ± 1.0 (n=3) |
5 ± 0.8 (n=3) |
8.7 ± 2.9 (n=3) |
|
Lactate dehydrogenase (nmol/min/mg prot) |
2306 ± 750 |
3103 ± 527 |
2470 ± 264 (n=3) |
3793 ± 504 (n=3) |
2600 ± 656 (n=3) |
3234 ± 168 (n=3) |
2726 ± 605 (n=3) |
Finally, we noticed that the area of FRDA patient’s cells was significantly lower than that of controls cells which can be attributed to previously described cytoskeletal structure disorganization possibly resulting from inconspicuous oxidative stress (Figure 2).
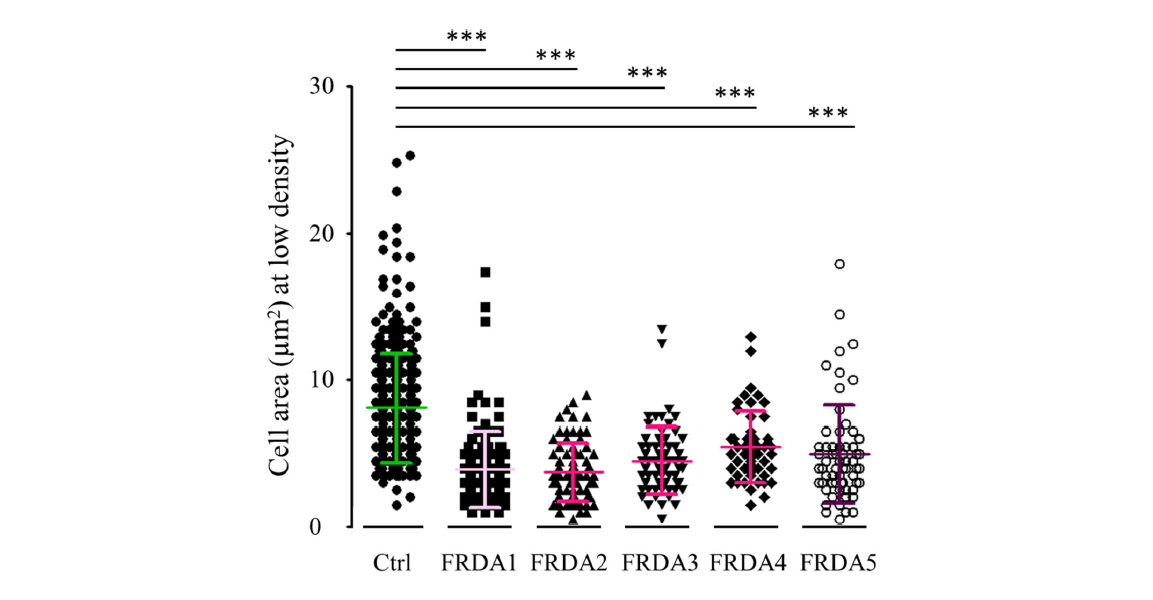
Figure 2. Comparison of control and FRDA fibroblast cell areas. Cell area was estimated from random image sampling and found much reduced for FRDA fibroblasts.
To select SC culture, we first evaluate cell proliferation in rich (CM) versus stringent (SC) culture medium (2 FRDA patients and 2 controls). Of note, FRDA fibroblasts actively grow under standard conditions, echoing the absence of significant enzyme defect in these cells [9]. Thereafter referred as the complete culture medium, CM consisted of a glucose-rich DMEM medium added with glutamine, pyruvate, and uridine (CM). In contrast, SC medium was limited to glutamine as a carbon source, and devoid of any molecule possibly providing an indirect antioxidant reservoir. After 14 days of culture, cell counting revealed a significant difference between control and FRDA fibroblasts: while the growth of control cells in SC medium (Figure 3) was not significantly affected, the FRDA fibroblasts hardly survived 14 days of culture (at most 25% compared to CM condition).
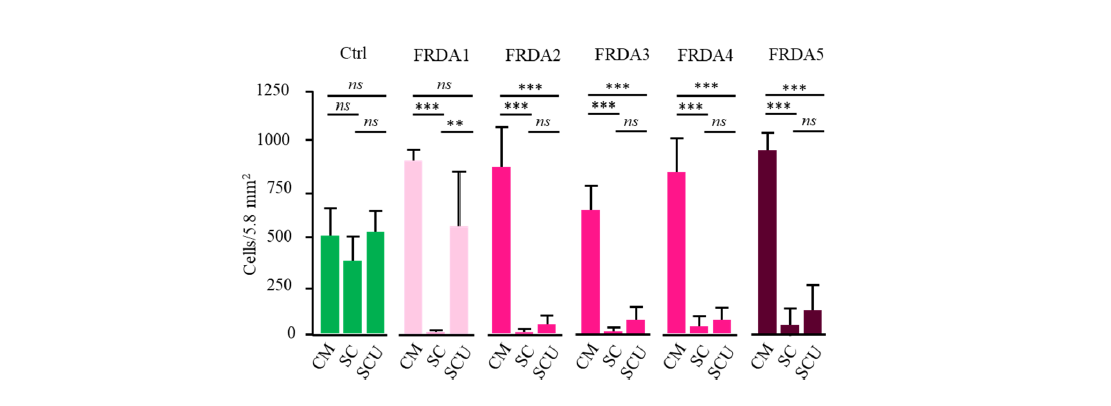
Figure 3. Proliferation of control (n=4; green) and FRDA fibroblast cells (FRDA1 to 5; red) in complete medium (CM), under stressful conditions (SC) and under SC in the presence of uridine (SCU). Red intensity of the graphs for FRDA cells corresponds to the PIP5K1β RNA decrease inversely proportional to the extent of the expansions in the Frataxin gene (see Table 2). ns: non-significant.
Noticeably, a limited, yet significant, cell mortality (50%) could be observed in one control under these quite harsh culture conditions. Addition of uridine fully preserved cell growth in all control cells. We therefore used SCU medium for studying the effect of pioglitazone in the next experiments (Figure 5). We also verified that none of these cells endured impaired use of pyruvate. such impairment would result in lactate accumulation in case of a RC dysfunction as it has been reported in several human tissues with Frataxin mutation. After 14 days of culture the color of the medium remained unchanged indicating that no significant medium acidification and lactic acid accumulation (not shown).
Antioxidant effect of uridine
SOD being a key limiting enzyme controlling oxidative stress in FRDA, we first selected uridine as an antioxidant shown to positively act on SOD enzyme expression. Both the manganese SOD2- and the copper-zinc SOD1-induction by uridine occurs through the Nrf2- signaling pathway [45,46]. Moreover, SOD1-induction by uridine also occurs through the Elk/Erk pathway. Noticeably the SOD1 isoform is present in both the cytosol and the mitochondrial inter-membrane while SOD2 is confined to the mitochondrial matrix space. In SCU medium, control fibroblasts readily and similarly proliferate. In contrast, SCU medium did not provide protection against cell death in 4 of the 5 FRDA fibroblasts (Figure 3). Nevertheless, this medium still allowed a significant growth for cells from patient 1 (FRDA1) (35 % of cell death). FRDA1 patient is distinguished from the other four FRDA patients by harboring the smallest expansion in the Frataxin gene with a limited effect on the transcription of neighboring genes (Table 2). We previously showed that very large expansion caused silencing of both the FXN and PIP5K1β genes [18]. Loss of PIP5K function results in destabilization of the actin network and impaired Keap1-Nrf2 signaling of superoxide dismutase (SOD). This resulted in cells becoming highly sensitive to superoxides [10]. Accordingly, these cells (FRDA2) were previously shown to be rapidly killed by exposure to even low doses of distal Q-binding site inhibitors of complex II (SDHI fungicide) due to slight overproduction of superoxides [47].
Antioxidant effect of pyruvate
We next tested the effect of pyruvate, known as other keto acids, to promptly react with hydrogen peroxides [48]. Compared to uridine (SCU; Figure 3), pyruvate alone provides a significantly better protective effect for FRDA fibroblasts (SCP; Figure 4). However, a significant mortality was still observed, especially substantial for FRDA5. Patient 5 distinguished from the other four FRDA patients by the largest expansion in the Frataxin gene (Table 2), previously shown to impair PIP5K1β transcription. This indicated that impaired SOD signaling in this patient’s cells could not be fully overcome by peroxide trapping by pyruvate.
Protecting effect provided by the simultaneous presence of both uridine and pyruvate
The combination of the two antioxidants, uridine and pyruvate provides a quite significant protection against cell death for four of the five FRDA fibroblasts (SCUP; Figure 4). Noticeably, the fibroblasts of patient 5, whose growth remained severely affected despite the presence of pyruvate alone, benefited from the simultaneous presence of pyruvate and uridine.
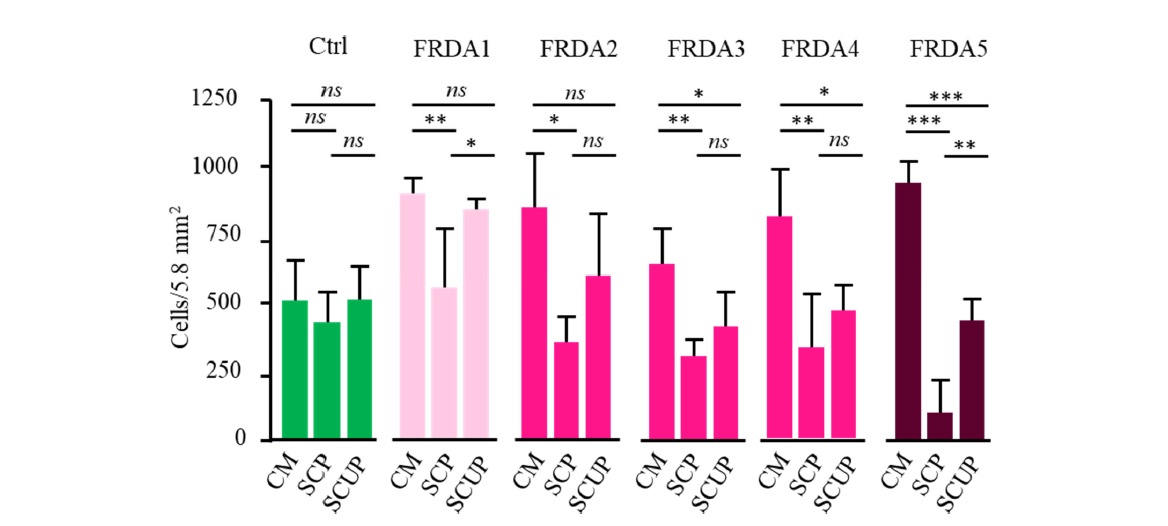
Figure 4. Proliferation of control (n=4; green) and FRDA fibroblast cells (FRDA1 to 5; red) in complete medium (CM), under stressful conditions in the presence of pyruvate (SCP) or in the presence of uridine plus pyruvate. Color code as described in Figure 3 legend. ns: non-significant.
Probing the antioxidant effect of pioglitazone
The peroxisome proliferator-activated receptor γ (PPARγ) agonist pioglitazone was selected because it has been shown to possibly enhance the transcription of Nrf2, and several target genes involved in cellular antioxidant response. Noticeably, pioglitazone has been reported to reduce disease progression in a subset of the neurologically affected mice, the so-called Harlequin mice. As patients with FRDA disease, this mouse possesses a heterogeneous genetic background resulting from a non-intended proviral insertion in the X-linked Aifm1 locus [49]. This causes a 90 to 35% reduction of Aifm1 protein according to tissues [50]. This leads to a time- and tissue-dependent respiratory chain complex I deficiency which classified this affection as a mitochondrial disease similar to FRDA. Similarity with FRDA, Harlequin mouse phenotype is also marked by the spectacularly inconsistent nature and severity of symptoms, including muscle, neurological, ocular, and cardiac symptoms [51]. Individual responders to pioglitazone were noticed in the Harlequin mice [52].
As shown in Figure 5, pioglitazone afforded no protection against cell death when cells were grown under stressful conditions even when uridine was added to lighten the stress pressure. This lack of effect indicates that in the fibroblasts from this set of patients, the antioxidant defenses pathway targeted by pioglitazone is profoundly affected. Noteworthy, this pathway is functional and was shown to respond to pioglitazone under similar condition [52] in fibroblasts from humans suffering from other pathological conditions such as mitochondrial infantile encephalomyopathy linked to Aifm1 mutation [53].
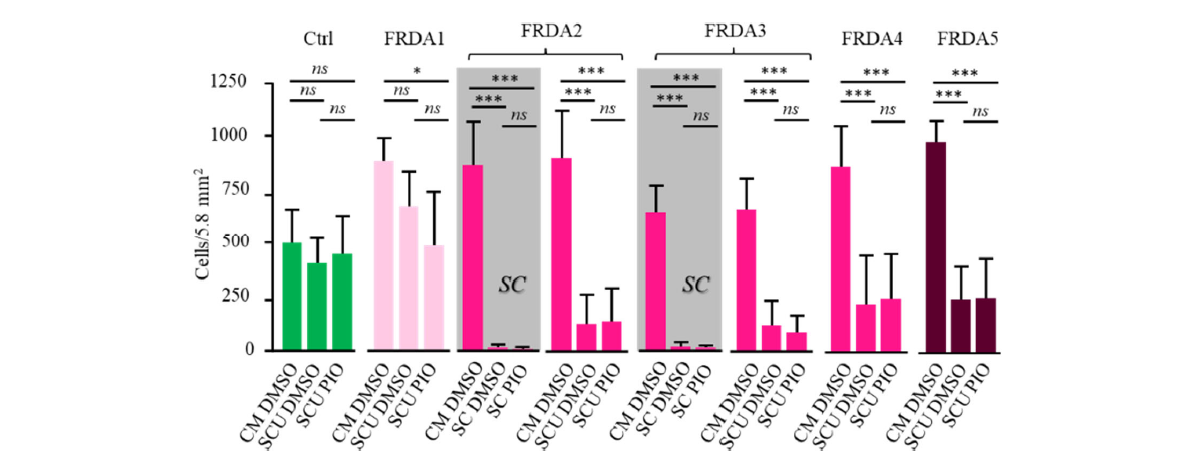
Figure 5. Proliferation of control (n=4; green) and FRDA fibroblast cells (FRDA1 to 5; red) in complete medium in the presence of DMSO (CM DMSO), under stressful conditions in the presence of DMSO and uridine (SCU DMSO), or under stressful condition in the presence of uridine and pioglitazone (SCU PIO). In the two grey boxes, representative sets of data (obtained with FRDA2 and 3 fibroblasts) illustrate the total inefficiency of pioglitazone to rescue growth of FRDA fibroblasts. Similar inefficiency was observed in the presence of uridine for the five FRDA cells (FRDA1-5). Notice that added pioglitazone was resuspended in DMSO. DMSO (0.001% final) was similar under the three conditions compared. Color code as described in Figure 3 legend. ns: non-significant.
Correlation between sensitivity to oxidative stress and PIP5K1β mRNA expression
We next examined the potential correlation between mortality rate in FRDA fibroblast cultures grown in different media and the residual PIP5K1β mRNA in these cells (Table 2). Using cell survival as a criterion, 3 distinct types of behavior emerged. The first observations for patient 1 (FRDA 1) characterize a significant rescue by the addition of uridine alone. Fibroblasts from this patient have been also the least impacted by stressful conditions. Interestingly enough, they have the highest Frataxin level and PIP5K1β mRNA expression. At the other end of the spectrum, fibroblast cultures from patient 5 were the most impacted by the stress conditions with less than 5% of the cell survival. The fibroblast culture from this patient was rescued neither by uridine nor by pyruvate alone. A partial survival could only be observed when both uridine and pyruvate were added (SCUP). Noticeably, patient 5 fibroblasts had the lowest PIP5K1β mRNA expression (Table 2). The growth of the three other patients could not be rescued by uridine alone, while it was significantly improved by pyruvate alone. Considering the recognized mechanism of pyruvate acting through peroxide chelation, this suggests that impaired peroxide elimination, previously reported in fibroblasts from FRDA patients [11] was the likely cause of the poor survival of patient 2-5 fibroblasts under stressful conditions.
|
Medium |
SC |
SCU |
SCP |
SCUP |
CM |
PIP5K1β mRNA (% of CTRL) |
| Cells | ||||||
|
FRDA1 |
- |
+ |
± |
+ |
+ |
58 |
|
FRDA4 |
- |
- |
± |
+ |
+ |
50 |
|
FRDA5 |
- |
- |
± |
± |
+ |
43 |
|
FRDA2 |
- |
- |
± |
± |
+ |
33 |
|
FRDA3 |
- |
- |
- |
± |
+ |
24 |
Quite noticeably, if the effect of uridine, or uridine plus pyruvate on cell survival is judged by averaging the results obtained on cells from the five patients, it turns out to be statistically different (Figure 6). This was also observed in the case of fibroblasts from one FRDA patient (FRDA3) with an unexplained low catalase (Table 1). Conversely, if repeated sets of data obtained for each patient are analyzed independently, it can be concluded that uridine (FRDA1), or the simultaneous presence of uridine and pyruvate (FRDA1 to 4) exerts a statistically confirmed effect for 4 of the 5 patients (Figures 3 and 4). This clearly highlights the inter- individual variability and the impasse with statistics based on means.
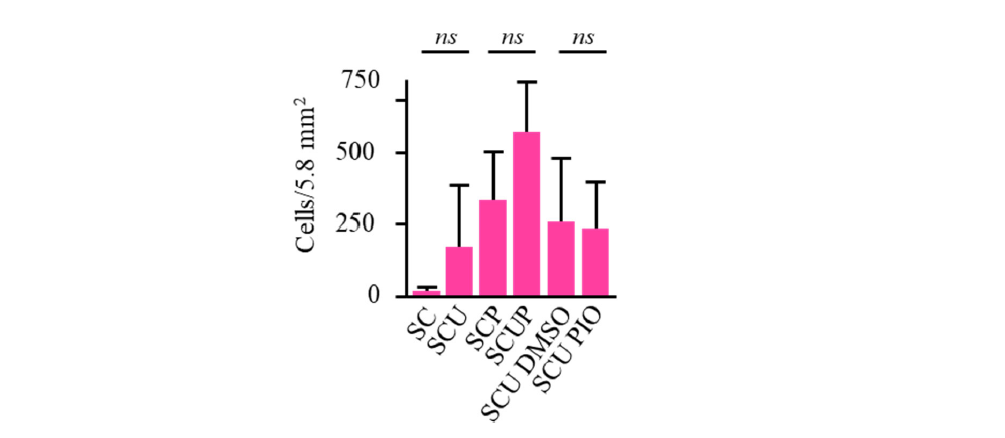
Figure 6. Averaging growth data for the five FRDA fibroblasts fully masks the protecting effect of uridine (SC/SCU), or uridine plus pyruvate (SCP/SCUP), previously characterized for FRDA cells individually considered. When necessary (SCU DMSO/SCU PIO), DMSO (0.001% final) was similar. Ns: non-significant.
Discussion
This study reveals an unpredicted variability of cultured skin fibroblasts from patients harboring expansion in the frataxin gene. It is of course not envisaged here that cell cultures could reflect in any way the complexity of living organisms, especially humans. As a result, their responses to the antioxidant agents cannot prefigure the results that might be subsequently observed on clinical trials. On the other hand, as is the case here, such studies can early point to inherent difficulties linked to the eminently variable consequences of even neighboring mutations, i.e. expansion in the Frataxin gene, when the effects of antioxidant agents are studied. They thus make it possible to discuss the difficulties encountered for more than 30 years in therapeutic trials using antioxidants to counter Friedreich's Ataxia. The study has been carried out under strictly controlled laboratory conditions and on a limited set of cells which would be usually described as homogeneous (all with large expansion, our initial selection criterion and PIP5K1β mRNA decrease). Yet, it reveals quite variable responses of cells to antioxidants. Most importantly, it first evokes the extreme variability of biochemical phenotypes, even when studying a single cell type (here, patient’s fibroblasts) from just five patients. Then it points to the place held by oxidative stress in this variability as seen through the response to three substances interfering with the oxidative stress. Indeed, this variability represents a hardly solvable difficulty in the management of clinical trials, as illustrated by the trial of pioglitazone (Figure 7).
The extreme variability of biochemical phenotypes
Most research aiming at counteracting FRDA, including ours, attempts to identify efficient approaches or molecules to counterbalance the Frataxin mutation. For 30 years, several supposedly effective molecules, selected based on their indisputable effects in vitro or using so-called “models” of the disease (yeasts, C. elegans, Drosophila, or mice) have been discarded because the real-life tests in human turned out to be disappointing, as none of these effects reached sufficient statistical significance.
Several points must be considered when interpreting results from studies of models. First, as pointed out repeatedly in our study, variability is a fundamental characteristic of FRDA which undoubtedly stands, at least in part, from the genetic heterogeneity of patients. In contrast, most of the research models are constructed by introducing targeted genetic modifications which results in clonal populations thereby eliminating the essential genetic variability. These transgenic animals or cell lines are undoubtedly very valuable and irreplaceable tools for studying molecular mechanisms, but they are poor predictors of the efficacy or inefficacy of treatment, and, in a sense, poor models. Indeed, to evaluate any therapeutic approach, one can hardly extrapolate the results obtained on clonal animal population as opposed to the genetically heterogeneous human population, or when bearing in mind the short lifespan of animal models while it takes several years, or even dozens for a disease to set in this for a same mechanism at work, or when taking into account the highly controlled and homogenous conditions in the animal facilities which cannot reflect a real life of patients exposed to variable and ever changing external factors. These factual concerns, which constitute real obstacles in carrying out therapeutic trials in humans, allows understanding the reason of the failures of numerous trials using substances deemed promising in the initial phases carried out on so-called models.
As mentioned above, a major limitation of these models results from the shortness of their life (at best 4-5 years) compared to the very progressive development of Friedreich's disease. The reasons for this progressive character, observed in most mitochondrial diseases, are essentially unknown, but could turn out to be decisive in bringing about the eventual success or failure of any therapy. This point is far from being specific to Friedreich's ataxia, having all its value for most mitochondrial diseases whose evolution is so poorly understood, and stands true for many complex human diseases.
Oxidative stress can trigger death in FRDA skin fibroblasts cultures
Many biochemical abnormalities have been reported as the consequence of mutations in the Frataxin gene. They combine, depending on the tissue and possibly on individual, the deficit of iron-sulfur proteins [9], damage to the respiratory chain [9], impairment of oxidation of fatty acids [55], dysfunction of pyruvate dehydrogenase [56], the abnormal levels of oxidative stress [10], the disorganization of cytoskeletal structures [12], glycolytic [57] or signaling pathways for antioxidant defenses [14]. By selecting fibroblasts for this study and selecting controlled stressful conditions resulting from strongly limited glucose availability and subsequent, limited source of reducing power (NADPH) for glutathione known to control oxidative stress in FRDA cells [11,12], we can assess the cell lethality in response to oxidative stress and to compare the effect of compounds acting such stress and likely balancing these stressful conditions.
Although using one type of cells from only five patients, we observed a strong disparity between culture behavior and cells response to antioxidants. Stressful conditions had a devastating effect on all five patient’s cultures, but it could be significantly counterbalanced by uridine only for patient 1 fibroblasts. In the presence of pyruvate, the deleterious effect of stressful conditions was much less pronounced, being roughly annihilated and strongly reduced by uridine for 4 of the 5 patients. Uridine and pyruvate have been described previously as neuroprotective compounds [33], our results confirmed their protective effects against oxidative stress in FRDA fibroblasts. Throughout this study, we were unable to evidence any positive effect of the glitazone (pioglitazone) on the small number of FRDA fibroblasts tested.
Overall, it appears that the deleterious effects of oxidative stress and the protective effect of uridine and/or pyruvate on fibroblasts depend on each patient from which the culture originates.
From culture flasks to clinical trials
Among the molecules tested here to illustrate the individual response to the oxidative stress of fibroblasts cultured under stressful conditions, pioglitazone is worth discussing despite the lack of effect on cultured fibroblasts we have studied. Indeed, using this molecule to counteract Friedreich ataxia in human patients perfectly illustrates the difficulty in conducting human trials, when considering the variability between individuals, in the context of a rare disease such as FRDA.
Due to its ability to activate PPARγ and cellular antioxidant defenses, this molecule was proposed for a proof of concept (Phase II) study in 2009. All the information on the procedures used in this trial is freely available (https://www.orpha.net/data/eth/GB/ID50165GB.pdf). The results of this trial have been made public, and it was stated that firm conclusion using the planned statistical analyses is impossible to reach [54]. However, three lessons can be drawn from this trial now completed in France and which correspond to a recurrent problem in clinical trials in this disease. The first is related to the extreme heterogeneity among patients affected by this rare disease which makes it almost impossible to constitute sufficiently large cohorts allowing valid comparisons in studies carried out against placebo. A second finding relates to the duration of placebo effects. The careful management during the trials of patients who otherwise might feel to be left alone, without sufficient attention and hope for long period, results in some improvement in patients of the placebo group lasting even longer than a year. Importantly, the duration of many trials does not exceed such a long period of time. Finally, for the pioglitazone trial, processing the data using a chosen Bayesian approach proved ineffective to obtain any convincing conclusion only allowing to state that the conditions were not met to seriously carry out this type of analysis. Indeed, this approach utilizes predictions made for the foreseeable outcome of the treatment for each patient which in the case of this disease, are based on unverified assumptions. Neither the profile of a patient at time t, nor his history prior to the start of the trial, sufficiently predicts the evolution of the disease with or without a treatment.
Summarized very briefly in (Figure 7), the results indicate that on the basis of a composite score adding results from three scales for estimating the progression of the disease (ICARS [58], FARS [59] and SARA [60]) the score of a minority group of patients receiving pioglitazone improved during the period of the test. In comparison, all patients receiving the placebo saw their composite score deteriorate or see inconsistent changes according to scale considered.
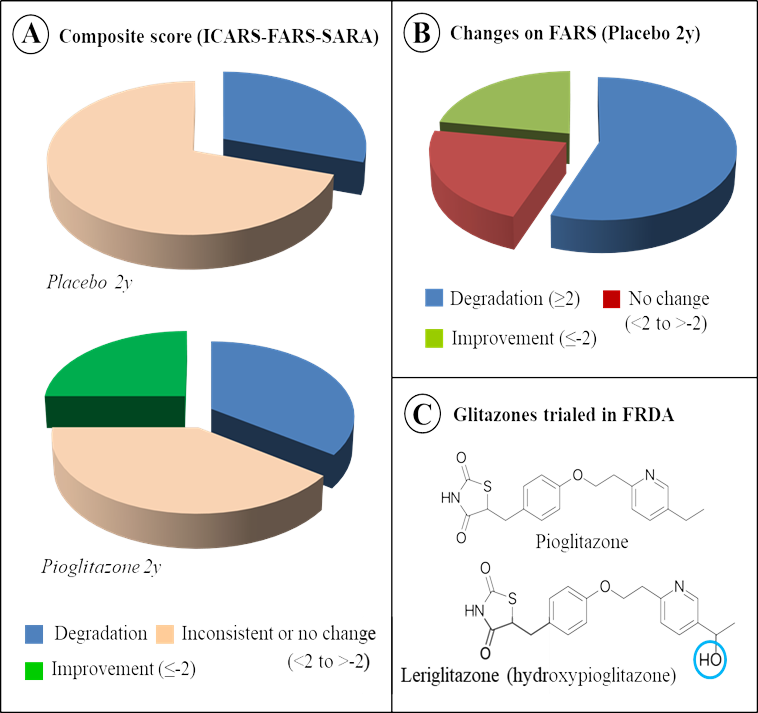
Figure 7. State of art for the use of glitazone to counteract Friedreich ataxia. A. The effect of Pioglitazone versus placebo on the composite score of FRDA patients in the clinical trial carried in France in starting 2008. Details of the trial conditions are available at https://www.orpha.net/data/eth/GB/ID50165GB.pdf, while conclusions tentatively drawn from the trial have been previously published [54]. B. The spectacular duration (2 years) of the placebo effect, traced by the significant number of patients displaying an improvement when considering the FARS (Friedreich Ataxia Rating Scale) scale. C. Another member of the glitazone family, Leriglitazone (hydroxypioglitazone), has been reported to be beneficial against FRDA in a phase 2 proof of concept (and a phase 2/3 clinical trial in another central nervous system disorder, adrenomyeloneuropathy) in US.
Thus, similarly to Idebenone, an antioxidant tested in rare mitochondrial diseases (FRDA and Leber's hereditary optic neuropathy), the power of the trials was not, and probably will hardly be, sufficient to reach a confident statistical value [61]. In the case of Idebenone, several clinical trials have shown a delay of neurological deterioration in FRDA disease in about 50% of the patients. Noticeably, the first publications focused on cardiac issues of a series of young patients report a reduction of cardiac hypertrophy and an improvement of cardiac functions resulting from the administration of Idebenone correlated with a recovery of enzymatic activities of iron- sulfur proteins, but did not at that time mention positive effect on neurological symptoms [62- 67].
Conclusion
To get out of this tricky situation, the way might be to better stratify the patients before performing trials and analyze their results. In the absence of reliable biomarkers, we need to find consensual criteria not reduced to a clinical picture at a given time. In that regard, distributing a few dozen people in placebo and treated groups has shown its limits in the case of FRDA over the past 30 years. The reality is that in the case of a rare, slowly progressive, and so heterogeneous disease like Friedreich's ataxia, the group size, added to the stratification into subgroups, will always be insufficient to allow standard tests to reach credible statistical values. As discussed above, even the Bayesian approach, as used for the pioglitazone trial in France, has shown its limits. These latter could be due to our incapacity based on pre-trial progression of the disease to produce credible predictions for each individual disease evolution.
Defining the criteria is a risky business. These criteria should include a maximum of parameters likely to determine the expression of the disease. Beside detailed clinical parameters, molecular and biochemical parameters might/should include size of the Frataxin expansion, respiratory chain and Krebs cycle activities, oxidative stress intensity, extent of PIP5K1β silencing, etc.
These are all related to the primary FRDA mutation and supposedly determining for clinical expression of the disease. As illustrated for skin fibroblasts in our study, the impairment of the expression of the PIP5K1β expression might be one of these criteria and failure to consider this parameter can be sufficient to mask the positive effect of a cell treatment. As for the last decades, the distribution of quite limited number of patients affected by this rare disease between treated- and placebo-groups will likely lead to inconclusive data or in conclusions biased by a given sampling. In particular, this greatly increases the risk of being unable to conclude on the usefulness of any molecule or approach trialing and to throw the baby (promising molecules or strategies) out with the bath water.
Here, the study of FRDA fibroblasts was useful to stress the major inter individual differences between patient’s cells toward oxidative stress and their responses to antioxidants, presumably associated with different genetic background. In the future, the use of the powerful tools now available for omics analyses (genomics, transcriptomics, proteomics, metabolomics) might disclose one or more rational that could account for the observed differences between FRDA individuals at cellular and organismal levels. At cellular level, these rather expensive investigations should be better undertaken using more appropriated cell types (neurons, cardiomyocytes) known to be involved in the disease. This panel of omics which should be completed by past and present “environmental omics” when dealing with patients. So far, omics approaches did not provided markers strong enough to be used in clinical trials.
Funding
General expenses were covered by grants from AAJI (Association pour l'Aide aux Jeunes Infirmes & aux Personnes Handicapées), AFAF (Association Française de l'Ataxie de Friedreich) and Association OLY (Ouvrir Les Yeux).
Author Contributions
Conceptualization: Paule Bénit. Funding acquisition: Paule Bénit, Pierre Rustin. Investigation: Paule Bénit. Methodology: Paule Bénit. Supervision: Pierre Rustin, Malgorzata Rak. Writing – original draft: Paule Bénit, reviewing & editing: Pierre Rustin, Malgorzata Rak, Paule Bénit.
Competing Interests
The authors have declared that there are no competing interests.
References
2. Harding AE. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981 Sep;104(3):589-620.
3. Baban A, Cicenia M, Travaglini L, Calì F, Vasco G, Francalanci P, et al. Remember Friedreich ataxia even in a toddler with apparently isolated dilated (not hypertrophic!) cardiomyopathy. Revisited. Minerva Pediatr (Torino). 2023 Feb;75(1):117-23.
4. Rota S, Marchina E, Todeschini A, Nanetti L, Rinaldi F, Vanotti A, et al. Very late-onset friedreich ataxia with laryngeal dystonia. Case Rep Neurol. 2014 Dec 12;6(3):287-90.
5. Silvers DS, Felice KJ. Late-Onset Friedreich's Ataxia Presenting as a Spastic Paraparesis. J Clin Neuromuscul Dis. 2000 Sep;2(1):27-8.
6. Bidichandani SI, Garcia CA, Patel PI, Dimachkie MM. Very late-onset Friedreich ataxia despite large GAA triplet repeat expansions. Arch Neurol. 2000 Feb;57(2):246-51.
7. Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996 Mar 8;271(5254):1423-7.
8. Lill R, Mühlenhoff U. Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669-700.
9. Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997 Oct;17(2):215-7.
10. aupe V, Dassa EP, Goncalves S, Auchère F, Lönn M, Holmgren A, et al. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One. 2009;4(1):e4253.
11. Piemonte F, Pastore A, Tozzi G, Tagliacozzi D, Santorelli FM, Carrozzo R, et al. Glutathione in blood of patients with Friedreich's ataxia. Eur J Clin Invest. 2001 Nov;31(11):1007-11.
12. Pastore A, Tozzi G, Gaeta LM, Bertini E, Serafini V, Di Cesare S, et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich's ataxia: a potential role in the pathogenesis of the disease. J Biol Chem. 2003 Oct 24;278(43):42588-95.
13. Auchère F, Santos R, Planamente S, Lesuisse E, Camadro JM. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich's ataxia. Hum Mol Genet. 2008 Sep 15;17(18):2790-802.
14. Chantrel-Groussard K, Geromel V, Puccio H, Koenig M, Munnich A, Rötig A, et al. Disabled early recruitment of antioxidant defenses in Friedreich's ataxia. Hum Mol Genet. 2001 Sep 15;10(19):2061-7.
15. Ast T, Meisel JD, Patra S, Wang H, Grange RMH, Kim SH, et al. Hypoxia Rescues Frataxin Loss by Restoring Iron Sulfur Cluster Biogenesis. Cell. 2019 May 30;177(6):1507-1521.e16.
16. Lupoli F, Vannocci T, Longo G, Niccolai N, Pastore A. The role of oxidative stress in Friedreich's ataxia. FEBS Lett. 2018 Mar;592(5):718-27.
17. Schulz JB, Dehmer T, Schöls L, Mende H, Hardt C, Vorgerd M, et al. Oxidative stress in patients with Friedreich ataxia. Neurology. 2000 Dec 12;55(11):1719-21.
18. Bayot A, Reichman S, Lebon S, Csaba Z, Aubry L, Sterkers G, et al. Cis-silencing of PIP5K1B evidenced in Friedreich's ataxia patient cells results in cytoskeleton anomalies. Hum Mol Genet. 2013 Jul 15;22(14):2894-904.
19. Bayot A, Rustin P. Friedreich's ataxia, frataxin, PIP5K1B: echo of a distant fracas. Oxid Med Cell Longev. 2013;2013:725635.
20. Koohi N, Thomas-Black G, Giunti P, Bamiou DE. Auditory Phenotypic Variability in Friedreich's Ataxia Patients. Cerebellum. 2021 Aug;20(4):497-508.
21. Vogel AP, Wardrop MI, Folker JE, Synofzik M, Corben LA, Delatycki MB, et al. Voice in Friedreich Ataxia. J Voice. 2017 Mar;31(2):243.e9-243.e19.
22. Poole ML, Wee JS, Folker JE, Corben LA, Delatycki MB, Vogel AP. Nasality in Friedreich ataxia. Clin Linguist Phon. 2015 Jan;29(1):46-58.
23. Reetz K, Dogan I, Hohenfeld C, Didszun C, Giunti P, Mariotti C, et al. Nonataxia symptoms in Friedreich Ataxia: Report from the Registry of the European Friedreich's Ataxia Consortium for Translational Studies (EFACTS). Neurology. 2018 Sep 4;91(10):e917-e930.
24. Seco-Cervera M, González-Rodríguez D, Ibáñez-Cabellos JS, Peiró-Chova L, González-Cabo P, García-López E, et al. Circulating miR-323-3p is a biomarker for cardiomyopathy and an indicator of phenotypic variability in Friedreich's ataxia patients. Sci Rep. 2017 Jul 12;7(1):5237.
25. Legrand L, Weinsaft JW, Pousset F, Ewenczyk C, Charles P, Hatem S, et al. Characterizing cardiac phenotype in Friedreich's ataxia: The CARFA study. Arch Cardiovasc Dis. 2022 Jan;115(1):17-28.
26. Harding IH, Chopra S, Arrigoni F, Boesch S, Brunetti A, Cocozza S, et al. Brain Structure and Degeneration Staging in Friedreich Ataxia: Magnetic Resonance Imaging Volumetrics from the ENIGMA-Ataxia Working Group. Ann Neurol. 2021 Oct;90(4):570-83.
27. Montermini L, Richter A, Morgan K, Justice CM, Julien D, Castellotti B, et al. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997 May;41(5):675-82.
28. Rummey C, Corben LA, Delatycki M, Wilmot G, Subramony SH, Corti M, et al. Natural History of Friedreich Ataxia: Heterogeneity of Neurologic Progression and Consequences for Clinical Trial Design. Neurology. 2022 Oct 3;99(14):e1499-e1510.
29. Dutka DP, Donnelly JE, Nihoyannopoulos P, Oakley CM, Nunez DJ. Marked variation in the cardiomyopathy associated with Friedreich's ataxia. Heart. 1999 Feb;81(2):141-7.
30. Badhwar A, Jansen A, Andermann F, Pandolfo M, Andermann E. Striking intrafamilial phenotypic variability and spastic paraplegia in the presence of similar homozygous expansions of the FRDA1 gene. Mov Disord. 2004 Dec;19(12):1424-31.
31. Klopstock T, Chahrokh-Zadeh S, Holinski-Feder E, Meindl A, Gasser T, Pongratz D, et al. Markedly different course of Friedreich's ataxia in sib pairs with similar GAA repeat expansions in the frataxin gene. Acta Neuropathol. 1999 Feb;97(2):139-42.
32. Schiff M, Rustin P. Idebenone in Friedreich ataxia and Leber's hereditary optic neuropathy: close mechanisms, similar therapy? Brain. 2016 Jul;139(Pt 7):e39.
33. Al N, Çakir A, Koç C, Cansev M, Alkan T. Antioxidative effects of uridine in a neonatal rat model of hyperoxic brain injury. Turk J Med Sci. 2020 Dec 17;50(8):2059-66.
34. Mazza MC, Shuck SC, Lin J, Moxley MA, Termini J, Cookson MR, et al. DJ-1 is not a deglycase and makes a modest contribution to cellular defense against methylglyoxal damage in neurons. J Neurochem. 2022 Aug;162(3):245-61.
35. Choi J, Tak S, Jung HM, Cha S, Hwang E, Lee D, et al. Kinetic evidence in favor of glyoxalase III and against deglycase activity of DJ-1. Protein Sci. 2023 May;32(5):e4641.
36. Stewart RE, Deeb GM, Kalus ME Jr, Wahl RL. 'Double right atrium' demonstrated by radionuclide ventriculography in cardiac transplantation. Clin Nucl Med. 1990 Feb;15(2):122-3.
37. Zhou FQ. Advantages of pyruvate over lactate in peritoneal dialysis solutions. Acta Pharmacol Sin. 2001 May;22(5):385-92.
38. Ramos-Ibeas P, Barandalla M, Colleoni S, Lazzari G. Pyruvate antioxidant roles in human fibroblasts and embryonic stem cells. Mol Cell Biochem. 2017 May;429(1-2):137-50.
39. Marmolino D, Manto M, Acquaviva F, Vergara P, Ravella A, Monticelli A, et al. PGC-1alpha down-regulation affects the antioxidant response in Friedreich's ataxia. PLoS One. 2010 Apr 7;5(4):e10025.
40. Al-Muzafar HM, Alshehri FS, Amin KA. The role of pioglitazone in antioxidant, anti-inflammatory, and insulin sensitivity in a high fat-carbohydrate diet-induced rat model of insulin resistance. Braz J Med Biol Res. 2021 May 24;54(8):e10782.
41. Galvao J, Davis B, Tilley M, Normando E, Duchen MR, Cordeiro MF. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2014 Mar;28(3):1317-30
42. Sanmartín-Suárez C, Soto-Otero R, Sánchez-Sellero I, Méndez-Álvarez E. Antioxidant properties of dimethyl sulfoxide and its viability as a solvent in the evaluation of neuroprotective antioxidants. J Pharmacol Toxicol Methods. 2011 Mar-Apr;63(2):209-15.
43. Marklund S, Marklund G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem. 1974 Sep 16;47(3):469-74.
44. Bergmeyer HU. Methods of Enzymatic Analysis. New York: Academic Press; 1965. pp. 1-1088.
45. Wasik U, Milkiewicz M, Kempinska-Podhorodecka A, Milkiewicz P. Protection against oxidative stress mediated by the Nrf2/Keap1 axis is impaired in Primary Biliary Cholangitis. Sci Rep. 2017 Mar 23;7:44769.
46. Kasai S, Shimizu S, Tatara Y, Mimura J, Itoh K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules. 2020 Feb 17;10(2):320.
47. Bénit P, Kahn A, Chretien D, Bortoli S, Huc L, Schiff M, et al. Evolutionarily conserved susceptibility of the mitochondrial respiratory chain to SDHI pesticides and its consequence on the impact of SDHIs on human cultured cells. PLoS One. 2019 Nov 7;14(11):e0224132.
48. Guarino VA, Oldham WM, Loscalzo J, Zhang YY. Reaction rate of pyruvate and hydrogen peroxide: assessing antioxidant capacity of pyruvate under biological conditions. Sci Rep. 2019 Dec 20;9(1):19568.
49. Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, et al. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002 Sep 26;419(6905):367-74.
50. Bénit P, Goncalves S, Dassa EP, Brière JJ, Rustin P. The variability of the harlequin mouse phenotype resembles that of human mitochondrial-complex I-deficiency syndromes. PLoS One. 2008 Sep 15;3(9):e3208.
51. van Empel VP, Bertrand AT, van Oort RJ, van der Nagel R, Engelen M, van Rijen HV, et al. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol. 2006 Aug 15;48(4):824-32.
52. Bénit P, Pelhaître A, Saunier E, Bortoli S, Coulibaly A, Rak M, et al. Paradoxical Inhibition of Glycolysis by Pioglitazone Opposes the Mitochondriopathy Caused by AIF Deficiency. EBioMedicine. 2017 Mar;17:75-87.
53. Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D'Adamo P, et al. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010 Apr 9;86(4):639-49.
54. Andriss B, Husson I, Crepon SG, Jacqz-Aigrain E, Zohar S, Alberti C. Application de la méthodologie bayésienne a un essai clinique de petit effectif. Exemple de l’essai ACTFRIE: étude de l’effet de la pioglitazone dans l’ataxie de Friedreich. Revue d'Épidémiologie et de Santé Publique. 2015 May 1;63:S63.
55. Wang D, Ho ES, Cotticelli MG, Xu P, Napierala JS, Hauser LA, et al. Skin fibroblast metabolomic profiling reveals that lipid dysfunction predicts the severity of Friedreich's ataxia. J Lipid Res. 2022 Sep;63(9):100255
56. Barbeau A, Butterworth RF, Ngo T, Breton G, Melançon S, Shapcott D, Geoffroy G, Lemieux B. Pyruvate metabolism in Friedreich's ataxia. Can J Neurol Sci. 1976 Nov;3(4):379-88.
57. Angulo MB, Bertalovitz A, Argenziano MA, Yang J, Patel A, Zesiewicz T, et al. Frataxin deficiency alters gene expression in Friedreich ataxia derived IPSC-neurons and cardiomyocytes. Mol Genet Genomic Med. 2023 Jan;11(1):e2093.
58. Trouillas P, Takayanagi T, Hallett M, Currier RD, Subramony SH, Wessel K, et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J Neurol Sci. 1997 Feb 12;145(2):205-11.
59. Lynch DR, Farmer JM, Tsou AY, Perlman S, Subramony SH, Gomez CM, et al. Measuring Friedreich ataxia: complementary features of examination and performance measures. Neurology. 2006 Jun 13;66(11):1711-6.
60. Schmitz-Hübsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006 Jun 13;66(11):1717-20.
61. Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007 May;39(2):175-91.
62. Rustin P, Bonnet D, Rötig A, Munnich A, Sidi D. Idebenone treatment in Friedreich patients: one-year-long randomized placebo-controlled trial. Neurology. 2004 Feb 10;62(3):524-5; author reply 525; discussion 525.
63. Rustin P, von Kleist-Retzow JC, Chantrel-Groussard K, Sidi D, Munnich A, Rötig A. Effect of idebenone on cardiomyopathy in Friedreich's ataxia: a preliminary study. Lancet. 1999 Aug 7;354(9177):477-9.
64. Hausse AO, Aggoun Y, Bonnet D, Sidi D, Munnich A, Rötig A, et al. Idebenone and reduced cardiac hypertrophy in Friedreich's ataxia. Heart. 2002 Apr;87(4):346-9.
65. Mariotti C, Solari A, Torta D, Marano L, Fiorentini C, Di Donato S. Idebenone treatment in Friedreich patients: one-year-long randomized placebo-controlled trial. Neurology. 2003 May 27;60(10):1676-9.
66. Buyse G, Mertens L, Di Salvo G, Matthijs I, Weidemann F, Eyskens B, et al. Idebenone treatment in Friedreich's ataxia: neurological, cardiac, and biochemical monitoring. Neurology. 2003 May 27;60(10):1679-81.
67. Geromel V, Darin N, Chrétien D, Bénit P, DeLonlay P, Rötig A, Munnich A, Rustin P. Coenzyme Q(10) and idebenone in the therapy of respiratory chain diseases: rationale and comparative benefits. Mol Genet Metab. 2002 Sep-Oct;77(1-2):21-30.