Abstract
Interleukin-17 (IL-17) family proteins are involved in the control of infections. When unrestrained, these cytokines contribute to the development of chronic inflammatory diseases. The IL-17 family contains 6 members: IL-17A to F. In this review, we outline the current knowledge on the roles of each IL-17 member on breast cancer.
Keywords
IL-17, Breast tumor, Microenvironment, Cytokines, Stroma, Immune cells, Inflammation
Breast Cancer Generalities
Worldwide, breast cancer is the most-common invasive cancer in women. Commonly used treatments include surgery, hormonal therapy, radiotherapy, chemotherapy and targeted therapy. Failure of these treatments is often due to intrinsic or acquired resistances and is responsible for most relapses of cancer [1]. Heterogeneity among patients and tumors, together with the versatility of cancer make drug resistance more challenging to deal with. Drug combination is often proposed to overcome treatment resistance and novel targets are to be found to achieve better outcomes. According to the expression of molecular markers (estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2)), breast cancer can be divided into several subtypes: luminal A, luminal B, HER2-amplified, and triple-negative. The latter is defined by the lack of ER and PR expression, as well as the absence of HER2 amplification, which makes it unresponsive to hormonal therapy or HER2-targeted agents. Nevertheless, recent data highlighted the efficacy of therapeutic antibodies directed against immune checkpoint PD-1/PDL-1 in some breast cancer subtypes [2]. As immunotherapy enters clinical practice, it is important to identify new predictive biomarkers and potential targets.
Malignant Tumors are Wounds That Do Not Heal
Mediators that allow communication between tumor cells and cells of their microenvironment are potential targets for immunotherapy. Among them, cytokines control and shape the tumor microenvironment, in part through modulation of angiogenesis, cell migration and phenotype. In the tumor microenvironment, immune cells transitory fight both against and for tumor cell expansion, depending on tumor phases. Indeed, as proposed by Dunn et al. [3], during the elimination phase, immune surveillance successfully eradicates malignant cells; at equilibrium, the immune system exerts control over tumor cells; during the escape phase, tumor cells evade immune mechanisms and expand. Transition from one to the other phase may result from dysbalanced expression of mediators of inflammation and resolution of inflammation in response to environmental factors, to the tumor itself or even by anti-tumor therapies [4]. At the cellular level, inflammation and resolution of inflammation are finely driven by leukocyte migration, apoptosis and efferocytosis. Results linking efferocytosis to cancer progression are increasing. The milk fat globule-epidermal growth factor (EGF) factor 8 protein (MFG-E8), a key molecule of efferocytosis, is present at high levels in triple-negative breast cancers and its invalidation sensitizes triple-negative breast cancers to cisplatin treatment [5]. However, opposite results were found in ER(+) and HER2-amplified breast cancers [5]. Targeting of phosphatidyl serine, which is expressed at the cell membrane during cell apoptosis or in response to docetaxel, showed efficacy in breast cancer therapy [6,7]. New approaches in anticancer therapy propose to simultaneously block the proinflammatory response and activate endogenous resolution programs. For instance, preoperative stimulation of resolution and inflammation blockade with nonsteroidal anti-inflammatory drugs or resolvins, eradicated micrometastasis formation during tumor resection [8].
At the molecular level, chemokines, such as CCL2 and CCL5, and cytokines from the tumor microenvironment play crucial roles in leukocyte migration, phenotype and immune responses. Type 1 inflammation, characterized by production of interleukin-1β (IL-1β), IL-6, tumor necrosis factor–α (TNF-α), and interferon-γ (IFN-γ), is proinflammatory and antifibrogenic. Type 2 inflammation, characterized by production of IL-4, IL-5, IL-10, IL- 13, IL-25, and IL-33, is associated with reduced tissue inflammation but positively correlates with tissue fibrosis. Imbalance between type 1 and type 2 inflammation is the major driver of fibrosis. Type 3 inflammation is characterized by production of IL-17A, IL-17F, IL-22, and IL-26 produced by neutrophils, mast cells, group 3 innate lymphoid cells (ILC3), T helper 17 (Th17) and Th22 cells. Type 3 cytokines play an important physiological role in tissue homeostasis but can be pathogenic. Deregulation of type 3 immunity is associated with abnormal tissue repair, chronic inflammatory diseases, and some cancers. The cytokine equilibrium in the tumor microenvironment may then contribute to the dogma proposing that malignant tumors are described as wounds that do not heal or alternatively as wounds that overheal [9].
Understanding of the biological milieu associated with breast cancer has important implications for biobehavioral research. Circulating cytokines may image the temporal stage of cancer and can serve as biomarkers for prognosis and/or target for immunotherapy. Several years ago, proteomic analysis had showed that three cytokines, granulocyte colony-stimulating factor (G-CSF), IL-6, and IL-17, were markedly increased in women with breast cancer as compared with those in women who did not have breast cancer [10]. Very recently, a study showed that serum IL-17 was up-regulated in breast cancer patients and that high levels of IL-17 also correlated with poor clinical outcomes [11]. The association of serum IL- 17 levels with an increased level of both basic fibroblast growth factor (bFGF) and G-CSF was correlated with poor disease-free survival among breast cancer patients. The study suggested that systemic IL-17 levels correlated with pathological complete response rates in breast cancer patients [11]. IL-17-blocking monoclonal antibodies may then be interesting to use in breast cancer. Nevertheless, controversial experimental studies have pointed out both pro and anti-tumoral roles for IL-17. In this review, we will present the implication of IL-17 family members in the hallmarks of oncogenesis and in breast cancer development.
The IL-17 Family Members
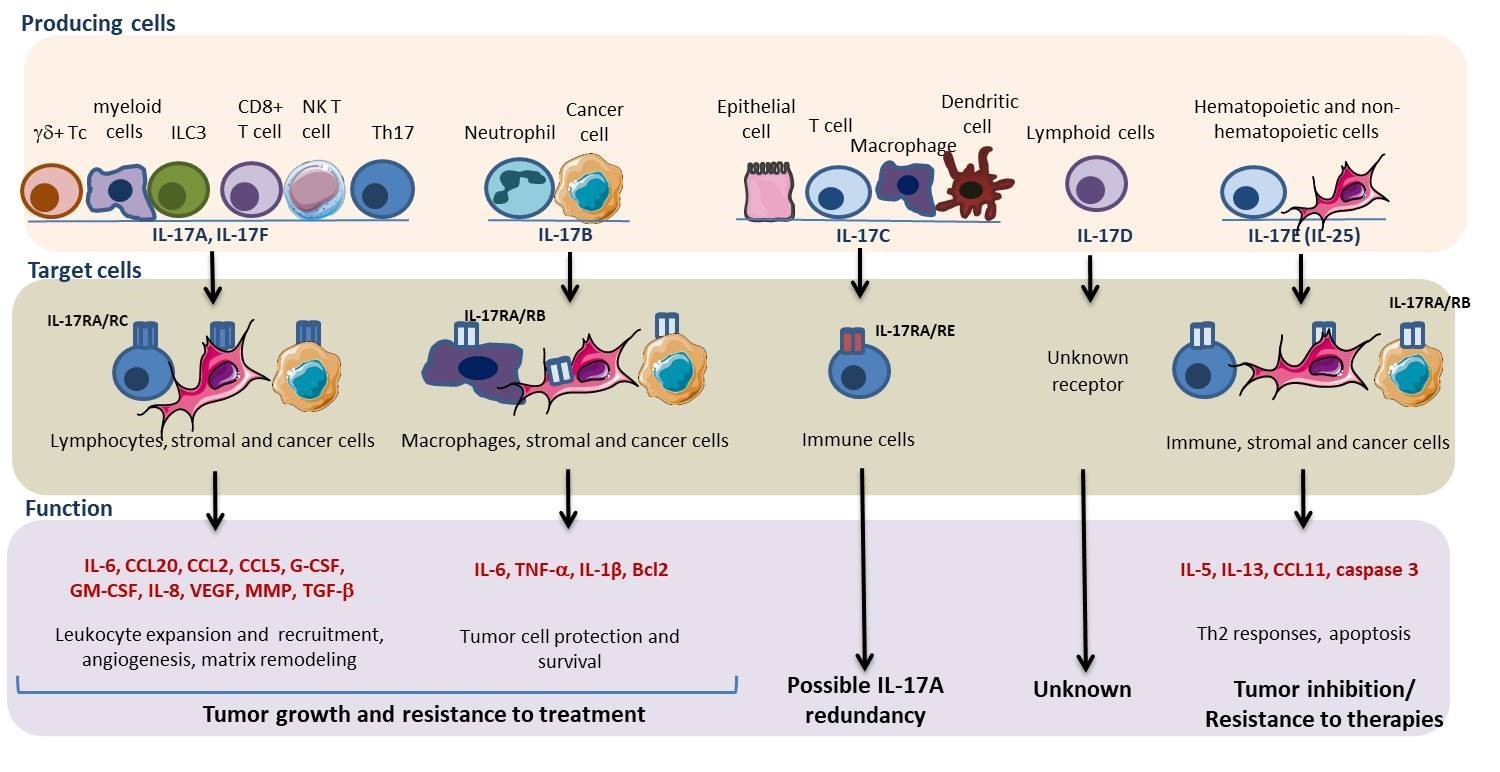
Originally thought to be produced by Th17 lymphocytes, IL-17 is now found to be secreted by many innate-like lymphocytes including, γδ T cells, invariant natural killer cells (iNKT), and ILC3, but also stromal cells, tumor cells and some myeloid cells. Many of these cell types are present in relatively high numbers at mucosal surfaces making IL-17 members important mediators of both host defense and homeostatic physiological processes at mucosal barrier sites. The wide cellular distribution of IL-17R contributes to the pleiotropic nature of the IL-17 family, allowing it to partake in a variety of physiological processes both beneficial and detrimental.
The IL-17 cytokine family of cytokines consists of 6 proteins (IL-17A to IL-17F) and 5 receptors (IL-17RA to IL-17RE). IL-17A, the founding and most studied member of the IL-17 family, was cloned in 1993 and initially named CTLA-8 [12]. IL-17F shares the highest degree of conservation to IL-17A (55%) and is commonly produced by the same cell types. IL-17E, also known as IL-25, displays the lowest degree of sequence conservation (16%). IL-17A, IL-17F, IL-17C and IL-17E function in host defense against pathogens and play various but not fully understood roles in mediating inflammation in autoimmune, allergic and chronic inflammatory conditions. Given the central role of IL-17A in autoimmunity, much effort has focused on the development of neutralizing antibodies for therapeutic use. IL-17A-blocking monoclonal antibodies secukinumab and ixekizumab recently received FDA-approval for the treatment of psoriasis, ankylosing spondylitis and psoriatic arthritis. Besides, it was also found that IL-17 cytokines bind IL-17 receptors on tumor cells, signaling the downstream activation of transcription factors (nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), signal transducer and activator of transcription (STAT), and activator protein 1 (AP-1)), kinases (mitogenactivated protein kinase (MAPK) and human epidermal growth factor receptor 1 (HER1)), tissue remodeling matrix metalloproteinases (MMPs), and anti-apoptotic proteins (Akt, Erk, mTOR, Bcl-2, and Bax) in a myriad of cancers [13,14].
IL-17A and IL-17F
Closely related molecules IL-17F and IL-17A are secreted as both homodimeric and heterodimeric forms, all of which are biologically active. The IL-17F homodimer is 100-fold less active than the IL-17A homodimer, but its expression was shown to be 10-fold higher than the IL-17A homodimer form, in the conditioned medium of human activated CD4+ T cells [15]. IL-17A is mostly secreted as a heterodimeric form with IL-17F, suggesting that the activity of IL-17A is, at least, partially attributed to the IL-17F/IL-17A heterodimeric cytokine. IL-17A and F were initially identified in T cells also expressing IL-21, IL-22 and granulocyte-macrophage colony stimulating factor (GM-CSF), which led to the denomination of a unique Th cell subset identified as Th17 [16]. The transition of naïve T cells to Th17 cells was due to a combination of IL-1β, IL-6, TGF-β and IL-21 for initial commitment, and IL-23 for full acquisition of their pathogenic capacity [17]. Later studies showed that IL-17A and F are also produced by CD8+ T cells, innate tissue-resident, Th17 and NKT cells. These cells share a common dependence on IL-23 and on the transcription factor ROR-γt for IL-17 production, and express the chemokine receptor CCR6, which binds C-C motif chemokine ligand 20 (CCL20) [18,19]. Recent reports have shown the expression of IL-17A by myeloid cells, including macrophages, neutrophils and mast cells, with possible artifacts due to their propensity for phagocytosis of Il-17A producing cells. IL-17A, IL- 17F and IL-17A/F signaling requires the formation of a heterotrimeric receptor complex comprising two IL-17RA and one IL-17RC [20]. The strength of IL-17 signaling varies widely among different cell types, in part due to differential expression profiles of IL-17RA and RC and binding affinities of IL-17A and IL-17F for these receptors [21,22]. Indeed, IL-17RA is highly expressed by leukocytes, whereas IL-17RC is preferentially, but not exclusively expressed in nonimmune cells [23]. Cells that express low levels or lack IL-17RC do not respond to IL-17A and F. Nevertheless, chronic inflammation can enhance the expression of IL-17RA and RC in some circulating leukocyte subsets, suggesting that the IL-17A/F signaling may be amplified in certain conditions. This is the case in bullous pemphigoid [24]. IL-17A and F signal leads to Act1-TRAF6-mediated activation of transcription factors NF-κB, MAPK-AP-1, and C/EBP. Downstream of IL- 17R, TRAF3 also associates with Act1 to inhibit TRAF6 signaling (reviewed in [25]).
IL-17A and F induce a proinflammatory milieu with enhanced cytokine, chemokine, growth factor and MMP production. The biological effects of IL-17A and F include thrombosis, inflammation and bone degradation. IL- 17A indirectly induces neutrophil expansion, migration and survival through synthesis of G-CSF, GM-CSF and IL-8 [26]. IL-17-A also stimulates the apoptosis and efferocytosis of neutrophils by macrophages in response to IL-10 and glucocorticoids [27,28]. These results contrast with the study by Hou et al, who showed that during the acute phase of inflammation, in experimental autoimmune myocarditis, IL-17A attenuates efferocytosis by monocytes [29]. IL-17A increases fibrosis through increased TGF-β expression and signaling pathways. Indeed, IL-17A enhanced responses to TGF-β by increasing cell surface expression of its receptor on stellate cells and allowing the activation of SMAD2/3 transcription factors in response to suboptimal doses of TGF-β [30]. IL-17A also dramatically increases the release of angiogenic factors, such as IL-8 [31] and vascular endothelial growth factor (VEGF) [32]. IL-17A, therefore potentiates both induction and resolution of inflammation, which might explain the controversy around its implication in several inflammatory diseases [33].
In breast cancers, studies have shown higher expression of IL-17A on tumor infiltrating lymphocytes and macrophages when compared to normal breast tissue [34,35]. Positive correlations were found between the number of IL-17-expressing cells and the histological grade of the tumors, aggressiveness of cancers and shorter disease-free survival [36-38]. Polymorphisms in the IL- 17A gene are associated with increased breast cancer risk in a Chinese patient population [39]. All together, these studies suggest that IL-17A exerts pro-tumoral activities in breast cancer. On the other hand, several papers suggest a possible anti-tumoral effect of IL-17A. Horlock et al. found that patients with HER2-positive breast carcinomas have reduced numbers of circulating Th17 cells and that treatment with anti-HER2 trastuzumab increased Th17 frequencies [40]. Yang et al. found that Th17 cells and expression of IL-17, IL-1β, and IL-6 cytokines were higher in breast cancer tissue compared to normal breast tissue but found a negative correlation between the numbers of Th17 cells and the stage of cancer [34]. Although these studies are mostly based on correlations and associations, they challenge the pro-tumoral effect of IL-17A.
Experimental models of breast cancers mainly support the pro-tumoral role of IL-17A. IL-17R knockdown or TGF-β neutralization, upstream of IL-17 synthesis, decreased tumor growth in vivo in 4T1 mouse mammary carcinoma model [41]. Also, IL-17A neutralization inhibited tumor growth and prevented the migration of tumorigenic neutrophils in various murine mammary tumor models [42,43]. In the study of Novitskiy et al., results supported that in human breast cancer, as well as in mouse models, decreased TGF-β signaling is correlated with increased CXCL1/5 genes and Th17 response [43].
Mechanistically, IL-17A both plays on tumor cells and cells in microenvironment (Figures 1 and 2). IL-17A was shown to promote metalloproteinase (MMP)-dependent invasion of breast cancer cell lines MDA-435 and MDAMB231 [44], to inhibit cell death in 4T1, MDA-MB-231 and EM6 BC cell lines in a TGF-β dependent manner [41], to stimulate cell proliferation in MCF-7, through tyrosine kinase activation [45] and in MDA-MB231 through signal transducer and activator of transcription 3 (STAT3) activation [46]. IL-17A was also shown to stimulate the release of IL6 and C-C motif chemokine ligand 20 (CCL20) by 4T-1 tumor cells [42]. Interestingly, IL-17A was found to promote resistance to docetaxel-based chemotherapy in various cell lines, through activation of the extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) pathway [35]. In the tumor microenvironment, IL-17A recruits and drives myeloid cells toward suppressive and angiogenic [47] phenotype, through G-CSF and proinflammatory cytokines (such as IL-6 and TNF-α) release, which is reversed by antagonism of IL-17A [48-50]. On the other hand, IL-17A was also found to inhibit myeloid-derived suppressor cell (MDSC) proliferation and to trigger their apoptosis, which suggests that other cytokines take over IL-17A to maintain MDSCs in the tumor microenvironment [51].
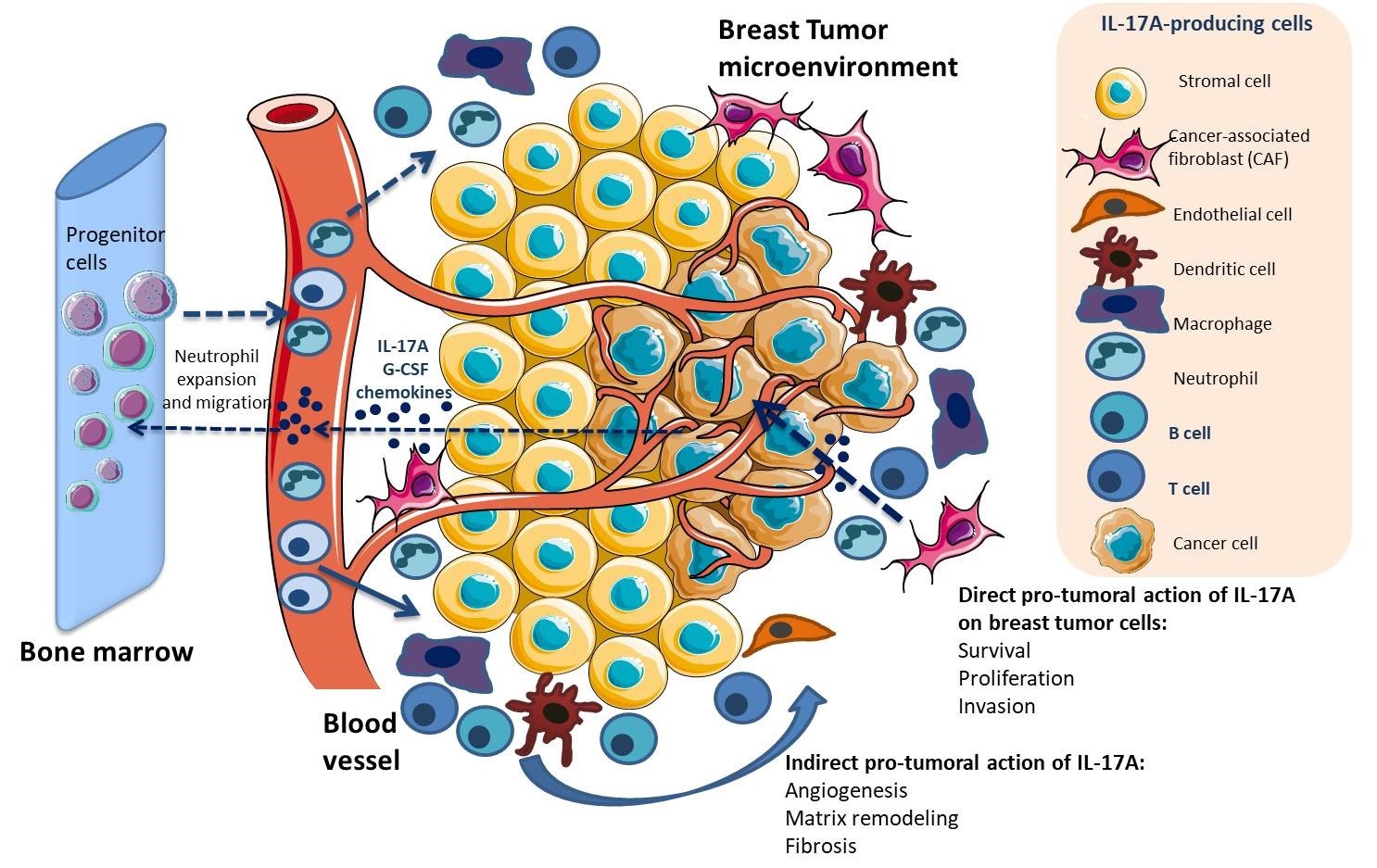
The role of IL-17F in breast tumor development has not been well described. Considering the high homology of IL-17F to –A, it is expected that many of the biological effects of IL-17F are similar to those of IL-17A, although differential function of IL-17F versus A was recently suggested [52].
IL-17B
IL-17B is expressed in many cell types including tumor cells and neutrophils [53], but not T cells [54]. IL-17B binds to IL-17RA/IL-17RB. IL-17B induces an increased production of TNF-α and IL-1β in THP-1 cell line, and synergizes the effect of TNF-α on G-CSF and IL-6 expression by fibroblasts. IL-17B was shown to induce the migration of a B cell subset [55] and neutrophil in vivo [56]. IL-17B can oppose IL-17E-driven inflammation and has been shown to play an antagonistic role [57]. Several studies suggest that IL-17B plays important roles in embryonic organogenesis and tissue regeneration (reviewed in [58]).
In clinical and experimental breast cancers, IL-17B is found to be pro-tumoral. IL-17B and IL-17RB are associated with poor prognosis in patients and IL-17B expression upregulation is specifically associated with poorer survival [59,60]. Further, two separate studies revealed that IL-17B/IL-17RB upregulates anti-apoptotic proteins of the BCL-2 family, which induced resistance to etoposide and paclitaxel [59,60] (Figure 2).
IL-17C
IL-17C is secreted by epithelial cells in response to Toll-like receptor stimulation, which facilitates bacterial clearance through the recruitment of neutrophils [61]. Its expression has also been reported in T cells, dendritic cells and macrophages in inflamed tissues [62]. IL-17C acts via IL-17RA and IL-17RE and activates NF-κB, mediating a seemingly overlapping function to that of IL-17A and IL-17F. Its role in psoriasis is clear, but its implication in inflammation is beginning to be unraveled. IL-17C induces a similar pattern of gene expression to IL-17A, which poses the question of functional redundancy. IL- 17C is pro-tumoral in lung cancer [63]; its role in breast cancer is unknown.
IL-17D
IL-17D mRNA is detected in various tissues, including brain, heart, lung, pancreas, skeletal muscle and adipose tissue. In the immune system, expression seems to be restricted to naïve CD4+ T cells and B cells [64]. IL-17D most closely resembles IL-17B, with which it shares 27% homology. To date, its receptor remains unknown [57]. IL-17D has been shown to stimulate monocyte chemoattractant protein-1 (MCP-1) production from tumor endothelial cells, which leads to the recruitment of natural killer (NK) cells [65]. The action of IL-17D remains entirely obscure. Its expression in breast cancer is weak1, suggesting little impact on the disease progression.
IL-17E
IL-17E (IL-25) derives from both hematopoietic and non-hematopoietic cells and shares little homology with the other IL-17 members. IL-17E is expressed by innate immune cells such as mast cells and alveolar macrophages in response to allergic stimuli. Like IL-17B, IL-17E can bind to IL-17RA/IL-17RB, but with a higher affinity [66]. IL-17E is a pleiotropic cytokine, acting on stromal, innate immune and adaptive immune cells to orchestrate Th2- type inflammation [67], as mentioned above. Consistent with the association of dysregulated Th2 responses with the development of allergy, IL-17E production is linked to the severity of chronic allergic conditions. Thus, IL- 17E-induced inflammation can be distinguished from IL-17A- and IL-17F-induced inflammation through the nature of the immune infiltrate, which mostly consists of eosinophils for the former and neutrophils for the latter. However, IL-17E can inhibit Th17 development mainly by skewing the immune system toward a Th2 response, leading to the regulation of automimmune diseases [68] (Figure 2).
IL-17E receptor subunits IL-17RA/IL-17RB are upregulated in breast cancers versus normal samples. IL-17E, similarly to EGF, activates the epidermal growth factor receptor (EGFR) in triple-negative breast cancer (TNBC) cells that are resistant to EGFR inhibitors. It also activates the PYK-2, Src and STAT3 kinases, which are essential for EGFR activation and nuclear translocation [69]. Accordingly, IL-17A and IL-17E promoted resistance to docetaxel and failed to induce apoptosis, which contrasts with other studies demonstrating that IL-17E has anti-breast cancer activities [44,70-73].
Concluding Remarks
This review summarizes the literature regarding the impact of IL-17 members mainly in breast cancer development and resistance to therapies. From migration to modulation of cell phenotypes and function, IL-17 members exert a wide variety of biological effects on their target cells, which directly impact tumor growth. Taken alone, IL-17 members are discreet actors of inflammation. However, when expressed within a myriad of other mediators, IL-17 members are able to synergize either pro- or anti-inflammatory processes. In particular, IL- 17A enhances both TLR- and inflammasome-dependent activities through synergic action on TNF-α [74] and IL- 1β [75] respectively. IL-17 members may then, as well, make a link between cancer and associated comorbidities such as obesity [76], high salt intake [77] and dysbiosis [75]. Treatment with anti-IL-17 holds a promise as cancer therapy. Several treatments targeting IL-17A have been approved and are in fact currently used successfully in some chronic inflammatory diseases with no significant adverse reactions despite the reported protector role of IL- 17A in infection [78] and in cardiovascular diseases [79]. These therapeutic agents, used alone or in combination with other cytokine inhibitors, could then potentially be employed as adjuvant treatment to overcome resistance to chemotherapy, checkpoint inhibitors and radiotherapy in breast cancer.
Author Contributions Statement
References
2. Marra A, Viale G, Curigliano G. Recent advances in triple negative breast cancer: the immunotherapy era. BMC medicine. 2019 Dec 1;17(1):90.
3. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu. Rev. Immunol.. 2004 Apr 23;22:329-60.
4. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010 Mar 19;140(6):883- 99.
5. Yang C, Hayashida T, Forster N, Li C, Shen D, Maheswaran S, Chen L, Anderson KS, Ellisen LW, Sgroi D, Schmidt EV. The integrin αvβ3-5 ligand MFG-E8 is a p63/p73 target gene in triple-negative breast cancers but exhibits suppressive functions in ER+ and erbB2+ breast cancers. Cancer research. 2011 Feb 1;71(3):937-45.
6. Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumors in mice. Cancer research. 2005 May 15;65(10):4408-16.
7. Chalasani P, Marron M, Roe D, Clarke K, Iannone M, Livingston RB, Shan JS, Stopeck AT. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer medicine. 2015 Jul;4(7):1051-9.
8. Panigrahy D, Gartung A, Yang J, Yang H, Gilligan MM, Sulciner ML, Bhasin SS, Bielenberg DR, Chang J, Schmidt BA, Piwowarski J. Preoperative stimulation of resolution and inflammation blockade eradicates micrometastases. The Journal of clinical investigation. 2019 Jun 17;129(7).
9. Dvorak HF. Tumors: wounds that do not heal. New England Journal of Medicine. 1986 Dec 25;315(26):1650- 9.
10. Lyon DE, McCain NL, Walter J, Schubert C. Cytokine comparisons between women with breast cancer and women with a negative breast biopsy. Nursing research. 2008;57(1):51.
11. Kawaguchi K, Sakurai M, Yamamoto Y, Suzuki E, Tsuda M, Kataoka TR, Hirata M, Nishie M, Nojiri T, Kumazoe M, Saito K. Alteration of specific cytokine expression patterns in patients with breast cancer. Scientific reports. 2019 Feb 27;9(1):1-2.
12. Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. The Journal of Immunology. 1993 Jun 15;150(12):5445-56.
13. Fabre JA, Giustiniani J, Garbar C, Merrouche Y, Antonicelli F, Bensussan A. The interleukin-17 family of cytokines in breast cancer. International journal of molecular sciences. 2018 Dec;19(12):3880.
14. Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, Gu C, Cai G, Ouyang W, Sen G, Stark GR. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4–ERK5 axis. Journal of Experimental Medicine. 2015 Sep 21;212(10):1571-87.
15. Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, Qiu Y, Whitters MJ, Tomkinson KN, Dunussi- Joannopoulos K, Carreno BM. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. Journal of Biological Chemistry. 2007 May 4;282(18):13447-55.
16. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nature immunology. 2005 Nov;6(11):1133-41.
17. Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. The Journal of Immunology. 2008 Nov 1;181(9):5948-55.
18. Chen W, Qin Y, Wang D, Zhou L, Liu Y, Chen S, Yin L, Xiao Y, Yao XH, Yang X, Ma W. CCL20 triggered by chemotherapy hinders the therapeutic efficacy of breast cancer. PLoS biology. 2018 Jul 27;16(7):e2005869.
19. Bunte K, Beikler T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immunemediated inflammatory diseases. International journal of molecular sciences. 2019 Jan;20(14):3394.
20. Toy D, Kugler D, Wolfson M, Bos TV, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. The Journal of Immunology. 2006 Jul 1;177(1):36-9.
21. Hymowitz SG, Filvaroff EH, Yin J, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, Pan G. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. The EMBO journal. 2001 Oct 1;20(19):5332-41.
22. Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, Harder B, Okada S, Ostrander CD, Kreindler JL, Aujla SJ. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. The Journal of Immunology. 2007 Oct 15;179(8):5462-73.
23. Liu R, Lauridsen HM, Amezquita RA, Pierce RW, Jane-wit D, Fang C, Pellowe AS, Kirkiles-Smith NC, Gonzalez AL, Pober JS. IL-17 promotes neutrophilmediated immunity by activating microvascular pericytes and not endothelium. The Journal of Immunology. 2016 Sep 15;197(6):2400-8.
24. Nesmond S, Muller C, Le Naour R, Viguier M, Bernard P, Antonicelli F, Le Jan S. Characteristic pattern of IL- 17RA, IL-17RB and IL-17RC in monocytes/macrophages and mast cells from patients with bullous pemphigoid. Frontiers in immunology. 2019;10:2107.
25. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL- 17 receptor–based signaling and implications for disease. Nature immunology. 2019 Dec;20(12):1594-602.
26. Parsonage G, Filer A, Bik M, Hardie D, Lax S, Howlett K, Church LD, Raza K, Wong SH, Trebilcock E, Scheel-Toellner D. Prolonged, granulocyte–macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFalpha. Arthritis research & therapy. 2008 Apr 1;10(2):R47.
27. Silverpil E, Glader P, Hansson M, Lindén A. Impact of interleukin-17 on macrophage phagocytosis of apoptotic neutrophils and particles. Inflammation. 2011 Feb 1;34(1):1-9.
28. Zizzo G, Cohen PL. IL-17 stimulates differentiation of human anti-inflammatory macrophages and phagocytosis of apoptotic neutrophils in response to IL-10 and glucocorticoids. The Journal of Immunology. 2013 May 15;190(10):5237-46.
29. Hou X, Chen G, Bracamonte-Baran W, Choi HS, Diny NL, Sung J, Hughes D, Won T, Wood MK, Talor MV, Hackam DJ. The Cardiac Microenvironment Instructs Divergent Monocyte Fates and Functions in Myocarditis. Cell reports. 2019 Jul 2;28(1):172-89.
30. Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. The Journal of immunology. 2014 Oct 15;193(8):3925-33.
31. Tartour E, Fossiez F, Joyeux I, Galinha A, Gey A, Claret E, Sastre-Garau X, Couturier J, Mosseri V, Vives V, Banchereau J. Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer research. 1999 Aug 1;59(15):3698- 704.
32. Numasaki M, Fukushi JI, Ono M, Narula SK, Zavodny PJ, Kudo T, Robbins PD, Tahara H, Lotze MT. Interleukin-17 promotes angiogenesis and tumor growth. Blood, The Journal of the American Society of Hematology. 2003 Apr 1;101(7):2620-7.
33. Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the yin and the yang. Trends in immunology. 2017 May 1;38(5):310-22.
34. Yang L, Qi Y, Hu J, Tang L, Zhao S, Shan B. Expression of Th17 cells in breast cancer tissue and its association with clinical parameters. Cell biochemistry and biophysics. 2012 Jan 1;62(1):153-9.
35. Cochaud S, Giustiniani J, Thomas C, Laprevotte E, Garbar C, Savoye AM, Curé H, Mascaux C, Alberici G, Bonnefoy N, Eliaou JF. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Scientific reports. 2013 Dec 9;3:3456.
36. Chen WC, Lai YH, Chen HY, Guo HR, Su IJ, Chen HH. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology. 2013 Aug;63(2):225-33.
37. Benevides L, Cardoso CR, Tiezzi DG, Marana HR, Andrade JM, Silva JS. Enrichment of regulatory T cells in invasive breast tumor correlates with the upregulation of IL-17A expression and invasiveness of the tumor. European journal of immunology. 2013 Jun;43(6):1518- 28.
38. Benevides L, da Fonseca DM, Donate PB, Tiezzi DG, De Carvalho DD, de Andrade JM, Martins GA, Silva JS. IL17 promotes mammary tumor progression by changing the behavior of tumor cells and eliciting tumorigenic neutrophils recruitment. Cancer research. 2015 Sep 15;75(18):3788-99.
39. Wang L, Jiang Y, Zhang Y, Wang Y, Huang S, Wang Z, Tian B, Yang Y, Jiang W, Pang D. Association analysis of IL-17A and IL-17F polymorphisms in Chinese Han women with breast cancer. PloS one. 2012;7(3).
40. Horlock C, Stott B, Dyson PJ, Morishita M, Coombes RC, Savage P, Stebbing J. The effects of trastuzumab on the CD4+ CD25+ FoxP3+ and CD4+ IL17A+ T-cell axis in patients with breast cancer. British journal of cancer. 2009 Apr;100(7):1061-7.
41. Nam JS, Terabe M, Kang MJ, Chae H, Voong N, Yang YA, Laurence A, Michalowska A, Mamura M, Lonning S, Berzofsky JA. Transforming growth factor β subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer research. 2008 May 15;68(10):3915-23.
42. Benevides L, da Fonseca DM, Donate PB, Tiezzi DG, De Carvalho DD, de Andrade JM, Martins GA, Silva JS. IL17 promotes mammary tumor progression by changing the behavior of tumor cells and eliciting tumorigenic neutrophils recruitment. Cancer research. 2015 Sep 15;75(18):3788-99.
43. Novitskiy SV, Pickup MW, Gorska AE, Owens P, Chytil A, Aakre M, Wu H, Shyr Y, Moses HL. TGF-β receptor II loss promotes mammary carcinoma progression by Th17-dependent mechanisms. Cancer discovery. 2011 Oct 1;1(5):430-41.
44. Welte T, Zhang XH. Interleukin-17 could promote breast cancer progression at several stages of the disease. Mediators of inflammation. 2015;2015.
45. Kim G, Khanal P, Lim SC, Yun HJ, Ahn SG, Ki SH, Choi HS. Interleukin-17 induces AP-1 activity and cellular transformation via upregulation of tumor progression locus 2 activity. Carcinogenesis. 2013 Feb 1;34(2):341-50.
46. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ. Human IL-17: a novel cytokine derived from T cells. The Journal of Immunology. 1995 Dec 15;155(12):5483-6.
47. Du JW, Xu KY, Fang LY, Qi XL. Interleukin-17, produced by lymphocytes, promotes tumor growth and angiogenesis in a mouse model of breast cancer. Molecular medicine reports. 2012 Nov 1;6(5):1099-102.
48. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, Ouyang W. An interleukin-17–mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nature medicine. 2013 Sep;19(9):1114.
49. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels LJ, Jonkers J, de Visser KE. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015 Jun;522(7556):345-8.
50. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nature immunology. 2018 Feb;19(2):108-19.
51. Ma M, Huang W, Kong D. IL-17 inhibits the accumulation of myeloid-derived suppressor cells in breast cancer via activating STAT3. International immunopharmacology. 2018 Jun 1;59:148-56.
52. Wanke F, Tang Y, Gronke K, Klebow S, Moos S, Hauptmann J, Shanmugavadivu A, Regen T, Mufazalov IA, Gabriel LA, Reißig S. Expression of IL-17F is associated with non-pathogenic Th17 cells. Journal of Molecular Medicine. 2018 Aug 1;96(8):819-29.
53. Kouri VP, Olkkonen J, Ainola M, Li TF, Björkman L, Konttinen YT, Mandelin J. Neutrophils produce interleukin-17B in rheumatoid synovial tissue. Rheumatology. 2014 Jan 1;53(1):39-47.
54. Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, Dowd P, Gurney AL, Wood WI. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proceedings of the National Academy of Sciences. 2000 Jan 18;97(2):773-8.
55. Ferretti E, Ponzoni M, Doglioni C, Pistoia V. IL-17 superfamily cytokines modulate normal germinal center B cell migration. Journal of leukocyte biology. 2016 Nov;100(5):913-8.
56. Shi Y, Ullrich SJ, Zhang J, Connolly K, Grzegorzewski KJ, Barber MC, Wang W, Wathen K, Hodge V, Fisher CL, Olsen H. A novel cytokine receptor-ligand pair identification, molecular characterization, and in vivo immunomodulatory activity. Journal of Biological Chemistry. 2000 Jun 23;275(25):19167-76.
57. Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harbor perspectives in biology. 2018 Apr 1;10(4):a028522.
58. Bie Q, Jin C, Zhang B, Dong H. IL-17B: A new area of study in the IL-17 family. Molecular immunology. 2017 Oct 1;90:50-6.
59. Laprevotte E, Cochaud S, Du Manoir S, Lapierre M, Dejou C, Philippe M, Giustiniani J, Frewer KA, Sanders AJ, Jiang WG, Michaud HA. The IL-17B-IL-17 receptor B pathway promotes resistance to paclitaxel in breast tumors through activation of the ERK1/2 pathway. Oncotarget. 2017 Dec 26;8(69):113360.
60. Huang CK, Yang CY, Jeng YM, Chen CL, Wu HH, Chang YC, Ma C, Kuo WH, Chang KJ, Shew JY, Lee WH. Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-κB-mediated antiapoptotic pathway. Oncogene. 2014 Jun;33(23):2968-77.
61. Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, Modrusan Z. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nature immunology. 2011 Dec;12(12):1159.
62. Hwang SY, Kim HY. Expression of IL-17 homologs and their receptors in the synovial cells of rheumatoid arthritis patients. Molecules & Cells (Springer Science & Business Media BV). 2005 Apr 1;19(2).
63. Jungnickel C, Schmidt LH, Bittigkoffer L, Wolf L, Wolf A, Ritzmann F, Kamyschnikow A, Herr C, Menger MD, Spieker T, Wiewrodt R. IL-17C mediates the recruitment of tumor-associated neutrophils and lung tumor growth. Oncogene. 2017 Jul;36(29):4182-90.
64. Starnes T, Broxmeyer HE, Robertson MJ, Hromas R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. The Journal of Immunology. 2002 Jul 15;169(2):642-6.
65. O’Sullivan T, Saddawi-Konefka R, Gross E, Tran M, Mayfield SP, Ikeda H, Bui JD. Interleukin-17D mediates tumor rejection through recruitment of natural killer cells. Cell reports. 2014 May 22;7(4):989-98.
66. Chang SH, Dong C. Signaling of interleukin-17 family cytokines in immunity and inflammation. Cellular signalling. 2011 Jul 1;23(7):1069-75.
67. Wu L, Zepp JA, Qian W, Martin BN, Ouyang W, Yin W, Bunting KD, Aronica M, Erzurum S, Li X. A novel IL-25 signaling pathway through STAT5. The Journal of Immunology. 2015 May 1;194(9):4528-34.
68. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, Kastelein RA. IL-25 regulates Th17 function in autoimmune inflammation. The Journal of experimental medicine. 2007 Jan 22;204(1):161-70.
69. Merrouche Y, Fabre J, Cure H, Garbar C, Fuselier C, Bastid J, Antonicelli F, Al-Daccak R, Bensussan A, Giustiniani J. IL-17E synergizes with EGF and confers in vitro resistance to EGFR-targeted therapies in TNBC cells. Oncotarget. 2016 Aug 16;7(33):53350.
70. Furuta S, Jeng YM, Zhou L, Huang L, Kuhn I, Bissell MJ, Lee WH. IL-25 causes apoptosis of IL- 25R–expressing breast cancer cells without toxicity to nonmalignant cells. Science translational medicine. 2011 Apr 13;3(78):78ra31.
71. Benatar T, Cao MY, Lee Y, Lightfoot J, Feng N, Gu X, Lee V, Jin H, Wang M, Wright JA, Young AH. IL- 17E, a proinflammatory cytokine, has antitumor efficacy against several tumor types in vivo. Cancer Immunology, Immunotherapy. 2010 Jun 1;59(6):805-17.
72. Younesi V, Nejatollahi F. Induction of anti-proliferative and apoptotic effects by anti-IL-25 receptor single chain antibodies in breast cancer cells. International immunopharmacology. 2014 Dec 1;23(2):624-32.
73. Benatar T, Cao MY, Lee Y, Li H, Feng N, Gu X, Lee V, Jin H, Wang M, Der S, Lightfoot J. Virulizin® induces production of IL-17E to enhance antitumor activity by recruitment of eosinophils into tumors. Cancer Immunology, Immunotherapy. 2008 Dec 1;57(12):1757- 69.
74. Hot A, Lenief V, Miossec P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Annals of the rheumatic diseases. 2012 May 1;71(5):768- 76.
75. Calcinotto A, Brevi A, Chesi M, Ferrarese R, Perez LG, Grioni M, Kumar S, Garbitt VM, Sharik ME, Henderson KJ, Tonon G. Microbiota-driven interleukin- 17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nature communications. 2018 Dec 3;9(1):1-3.
76. Gislette T, Chen J. The possible role of IL-17 in obesityassociated cancer. The Scientific World JOURNAL. 2010;10:2265-71.
77. Amara S, Tiriveedhi V. Inflammatory role of high salt level in tumor microenvironment. International journal of oncology. 2017 May 1;50(5):1477-81.
78. Das S, Khader S. Yin and yang of interleukin-17 in host immunity to infection. F1000Research. 2017;6.
79. Simon T, Taleb S, Danchin N, Laurans L, Rousseau B, Cattan S, Montely JM, Dubourg O, Tedgui A, Kotti S, Mallat Z. Circulating levels of interleukin-17 and cardiovascular outcomes in patients with acute myocardial infarction. European heart journal. 2013 Feb 21;34(8):570-7.