Abstract
IDH2 gene mutations, typically at residues R140 and R172, occur in 8–19% of patients with acute myeloid leukemia (AML). These mutations induce production of 2-hydroxyglutarate (2-HG), an oncometabolite that causes DNA and histone hypermethylation, and subsequent blockade of hematopoietic cell differentiation. In a phase 1b/2 trial (NCT02677922), combination therapy with azacitidine + enasidenib significantly improved overall response rate compared with azacitidine only therapy (74% vs 36%; P<0.001) in patients with newly diagnosed IDH2-mutated AML not eligible for intensive chemotherapy. We investigated the association between molecular features and clinical outcomes from that trial. In all, 101 patients were randomized to enasidenib + azacitidine (n=68) or azacitidine only (n=33); 74% of patients had IDH2-R140. Baseline 2-HG levels and IDH2 variant allele frequency (VAF) were similar between treatment arms and IDH2 variants, and were not significantly different between clinical response categories. Significant 2-HG and IDH2 VAF reductions from baseline were observed with combination therapy compared with azacitidine only. Molecular profiling revealed SRSF2 preferentially co-mutated with IDH2-R140, and DNMT3A co-mutated with IDH2-R172. IDH2 VAF was reduced to <1% in 50% of patients who achieved CR with combination therapy (18/36) and azacitidine only (2/4). VAFs of genes in the DNA methylation, receptor-tyrosine-kinase, and RAS canonical pathways were reduced in patients achieving CR. Of note, combination therapy improved event-free survival in patients with RAS-pathway mutations, which have been associated with resistance to enasidenib monotherapy.
Keywords
Acute myeloid leukemia, Newly diagnosed, IDH2-mutated, 2-hydroxyglutarate, Enasidenib, Azacitidine, Variant allele frequency, Molecular characterization
Introduction
Acute myeloid leukemia (AML) is a molecularly heterogeneous, aggressive disease that predominantly affects older patients [1]. Isocitrate dehydrogenase-2 (IDH2) gene mutations, typically at the active site arginine residues R140 and R172, occur in 8–19% of patients with AML [2,3]. Mutant-IDH2 enzymes acquire neomorphic activity that produces the oncometabolite 2-hydroxyglutarate (2-HG), which competitively inhibits α-ketoglutarate-dependent enzymes, leading to aberrant hypermethylation of DNA and histones [4,5].
Enasidenib is an oral, small-molecule inhibitor of mutant-IDH2 enzymes that suppresses 2-HG production and has been shown to reverse the leukemogenic blockade of cell differentiation [6]. Enasidenib is approved in the United States for the treatment of patients with mutant-IDH2 relapsed or refractory (R/R) AML [7]. Enasidenib monotherapy has demonstrated clinically meaningful improvements in morphologic response and event-free survival (EFS) compared with conventional salvage regimens in older patients with R/R AML [8] and clinical activity in newly diagnosed (ND) AML [9]. Azacitidine, a hypomethylating agent (HMA), has been shown to improve overall survival (OS) in patients with ND-AML [10-12].
In vitro, combining enasidenib and azacitidine led to enhanced apoptosis and leukemic-cell differentiation, leading to synergistic hypomethylating activity and enhanced differentiation [13]. In a randomized, open-label, phase 1b/2 trial (AG221-AML-005; NCT02677922), the enasidenib + azacitidine combination was associated with significant clinical benefits versus azacitidine only in patients with mutant-IDH2 ND-AML unfit for intensive chemotherapy; 74% (n=50) of patients who received enasidenib + azacitidine achieved an overall response versus 36% (n=12) of patients who received azacitidine only (odds ratio 4·9 [95% confidence interval 2.0–11.9]; P=0·0003) [14].
In this study, we investigated the molecular features associated with outcomes in the phase 1b/2 AG221-AML-005 trial [14]. The objectives of this analysis were to evaluate changes in 2-HG and IDH2 variant allele frequency (VAF) levels from baseline; to characterize the baseline genetic landscape, including tumor mutation burden; and to evaluate changes from baseline in VAF for genes in the DNA methylation, receptor-tyrosine-kinase (RTK), and RAS canonical pathways, in association with clinical response to enasidenib + azacitidine and azacitidine only.
Materials & Methods
Eligibility criteria and study design have been previously reported [14]. In the phase 2 portion of the trial, adult patients with mutant-IDH2 ND-AML were randomized 2:1 to receive enasidenib (100 mg/day) in combination with azacitidine (75 mg/m2/day × 7 days) or azacitidine only, in repeated 28-day treatment cycles. Review boards and ethics committees approved the trial protocol and amendments at all participating sites; patients provided informed consent prior to the study [14].
Measurement of 2-HG and IDH2 VAF
Total 2-HG was quantified by liquid chromatography-tandem mass spectrometry (Covance; Princeton, NJ), according to an analytically validated method. Peripheral blood plasma was isolated from patients at baseline, cycle (C)1-day (D)15, C2D1, C2D15, and on D1 of every other cycle from C3–C19.
Bone marrow mononuclear cells were isolated from patients at baseline, and on D1 of C2, C3, C5, C11, C17, and C23, and at end of treatment (EOT). IDH2 VAF was assessed in DNA by digital polymerase chain reaction (Sysmex; Baltimore, MD; lower limit of detection 0.02%–0.04%).
Extent of change in 2-HG and IDH2 VAF during treatment was evaluated in association with clinical response. Clinical response categories included complete remission (CR), less than CR (LTCR; non-CR response, including complete response with incomplete platelet recovery, complete response with incomplete blood count recovery, partial response, and morphologic leukemia-free state), per International Working Group (IWG) response criteria [15], and no response (NR; stable disease [IWG] or progressive disease).
Identification of co-occurring mutations
Bone marrow mononuclear cell DNA was isolated from patients at baseline and co-occurring gene mutations were identified by targeted next-generation sequencing using a 37-gene myeloid panel (ArcherDx; Boulder, CO) at a 1% level of detection (LOD). Manufacturer-recommended filter set was used to identify mutation variants. The baseline genetic landscape, including tumor mutational burden, was evaluated in association with clinical response (defined above). The baseline mutational burden was categorized as greater than or equal to or less than the median number of mutated genes for the entire population.
Gene pathway analysis
A gene pathway is considered mutated if there are mutations present in any of its genes. VAF aggregation was performed by taking the maximum value among all mutations in genes belonging to a given pathway. Gene VAFs were assessed at baseline and on D1 of C2, C3, C5, C11, and C17. Changes in gene VAFs during treatment were evaluated in association with clinical response (defined above).
Statistical analysis
Statistical analyses were performed using Prism version 8.0.0 (GraphPad; San Diego, CA) and the R survival (v.3.2.11), survminer (v.049) and ggplot2 (v.3.3.5) packages. Significant differences between groups were assessed using statistical methods listed in the corresponding Figure and Table footnotes. Significance analysis across longitudinal VAFs was performed by comparing each on-treatment or EOT visit with values at screening using a two-tailed Wilcoxon Rank Sum Test.
Results
Enrollment and IDH2 subtypes
In all, 101 patients were randomized to enasidenib + azacitidine (n=68) or azacitidine only (n=33). Patient characteristics have been reported [14]. Most patients (74%) had an IDH2-R140 mutation and 24% had IDH2-R172; IDH2 subtype was missing for two patients.
2-HG and IDH2 VAF
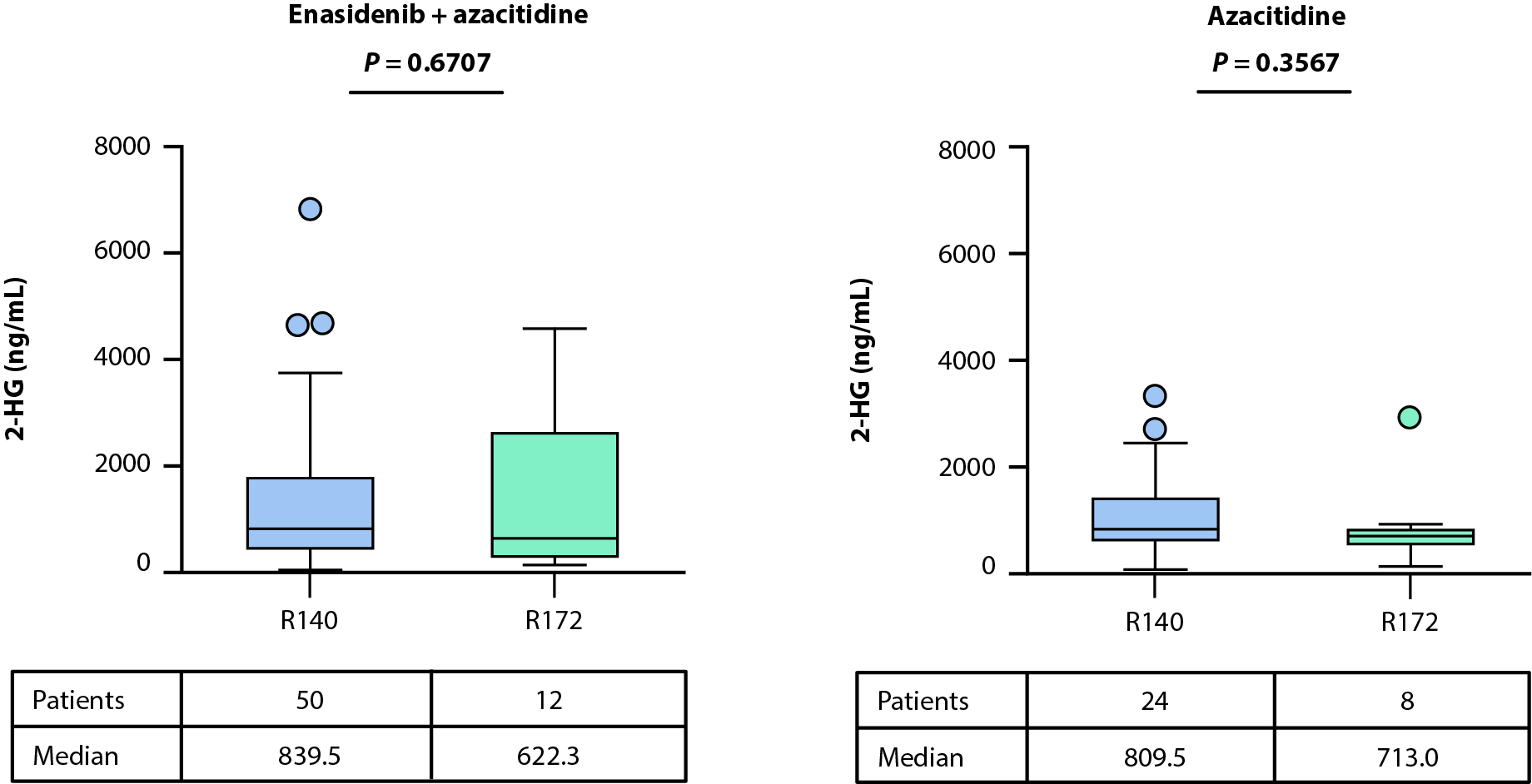
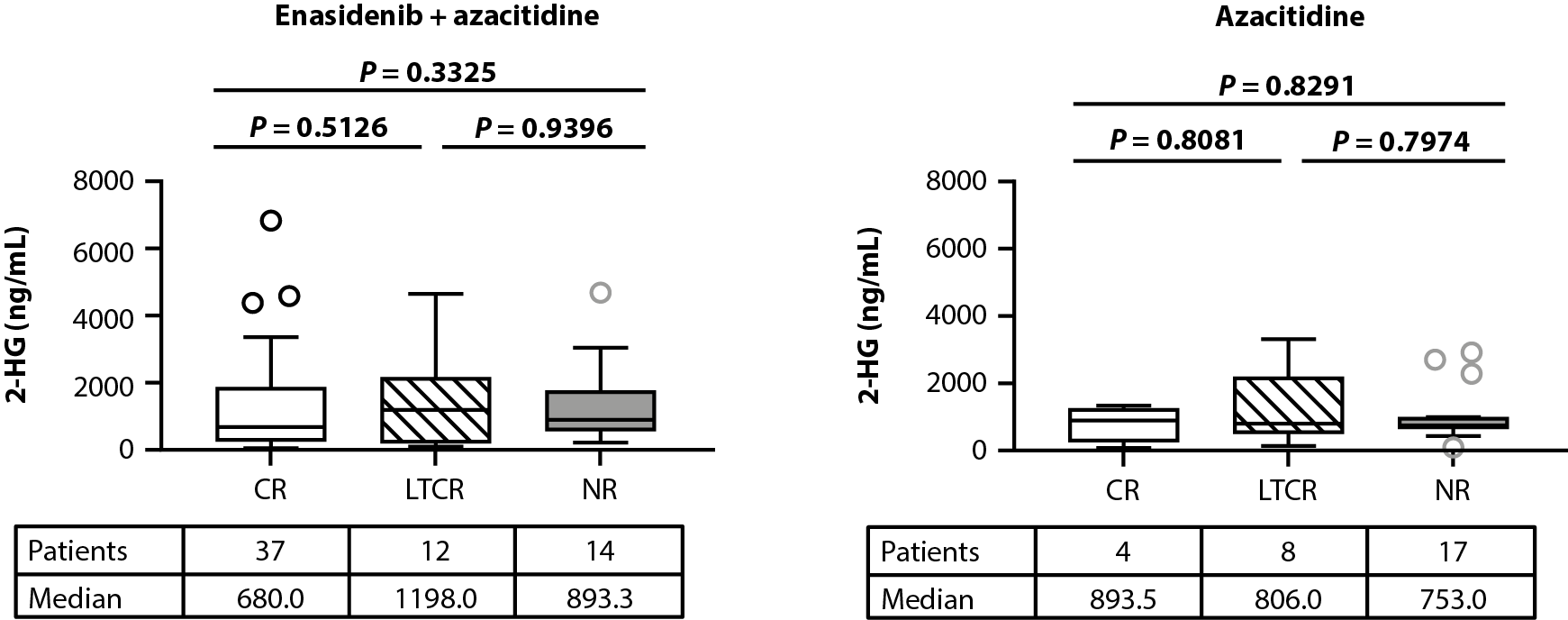
Levels of 2-HG at baseline were similar between treatment arms [14] and in patients exhibiting R140 versus R172 IDH2 variants (Figure 1A). The relationship between baseline 2-HG levels and clinical response (as defined by best response) was evaluated in biomarker-evaluable patients with available bioanalytical data treated with enasidenib + azacitidine or azacitidine only. No statistically significant differences were observed in baseline 2-HG between clinical response categories of CR, LTCR, and NR in either treatment arm (enasidenib + azacitidine: P=0.3325, azacitidine only: P=0.8291; Figures 1B, 1C).
A
B C
D
E
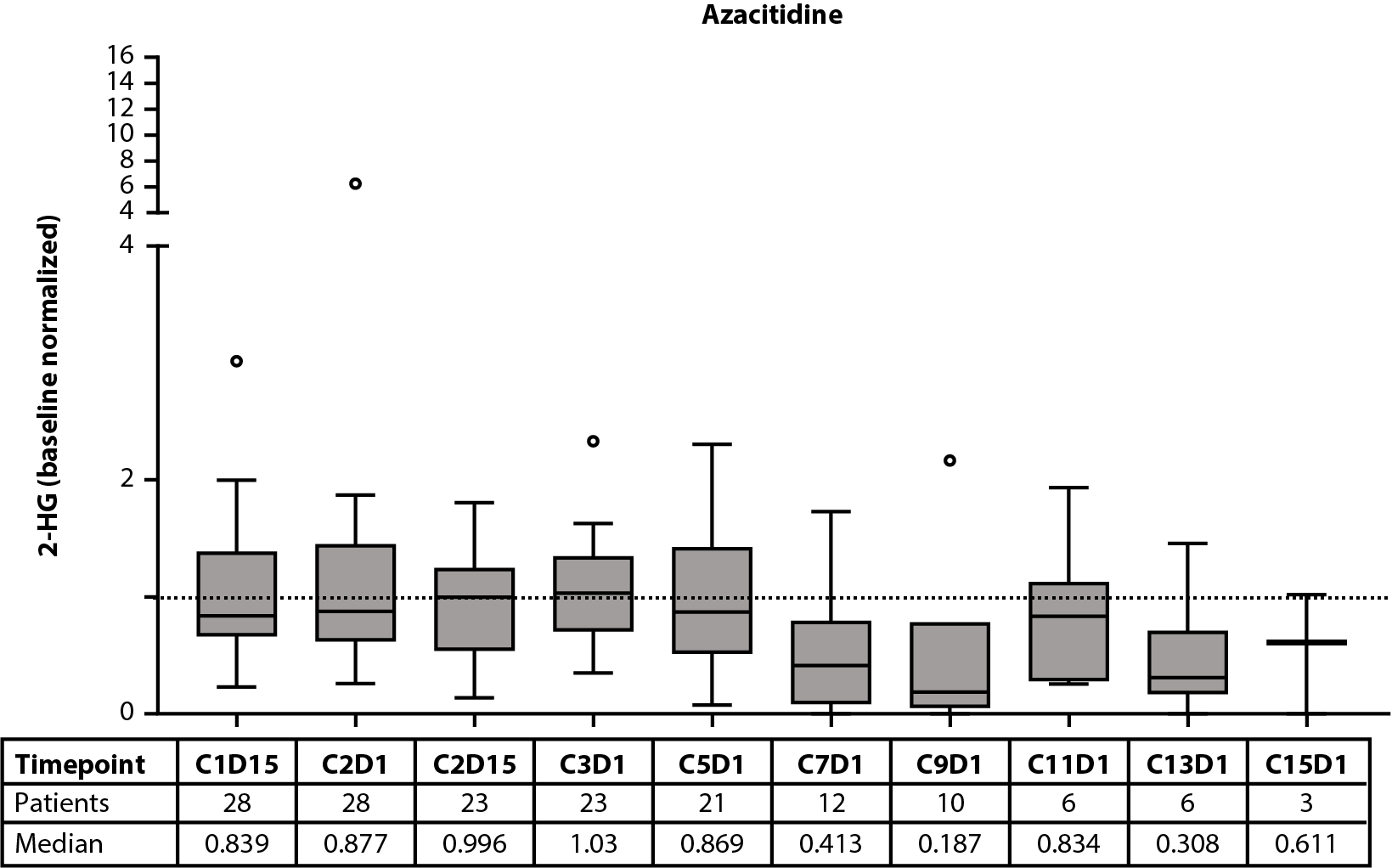
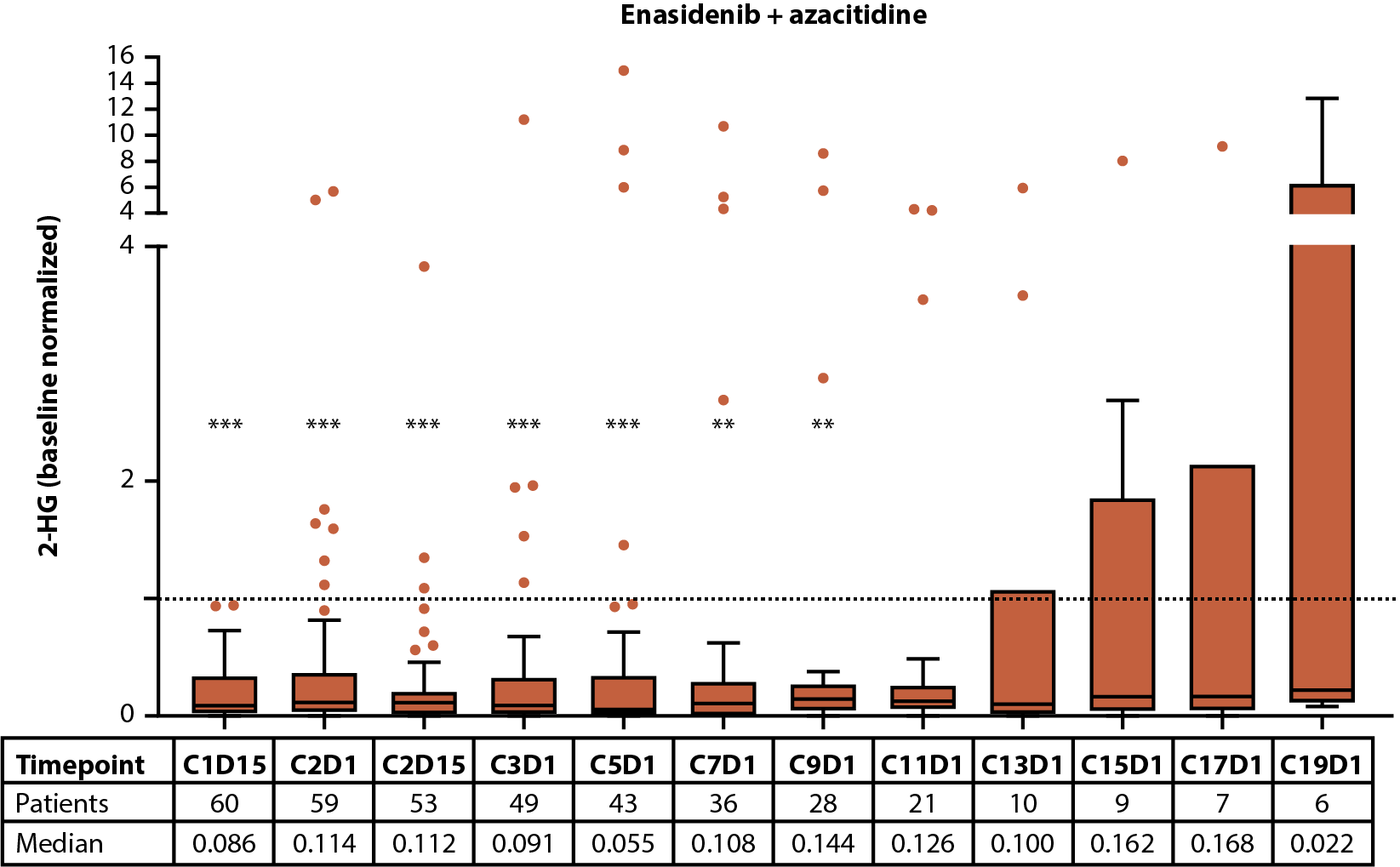
Figure 1. Baseline 2-HG concentrations in each treatment arm according to IDH2 variant (A). Relationship between baseline 2-HG concentrations and clinical response in patients treated with enasidenib + azacitidine (B) or azacitidine only (C). Baseline-normalized changes in 2-HG in patients treated with enasidenib + azacitidine (D) or azacitidine only (E). Horizontal bars indicate median; error bars indicate Tukey’s range; P values calculated using Mann-Whitney test (B, C) or Wilcoxon test (D, E). *P≤0.05; **P ≤0.01; ***P≤0.001. 2-HG: D-2-Hydroxyglutarate; CR: Complete Remission; CxDx: Cycle x, Day x; IDH2: Isocitrate Dehydrogenase-2; LTCR: Less than CR; NR: No Response.
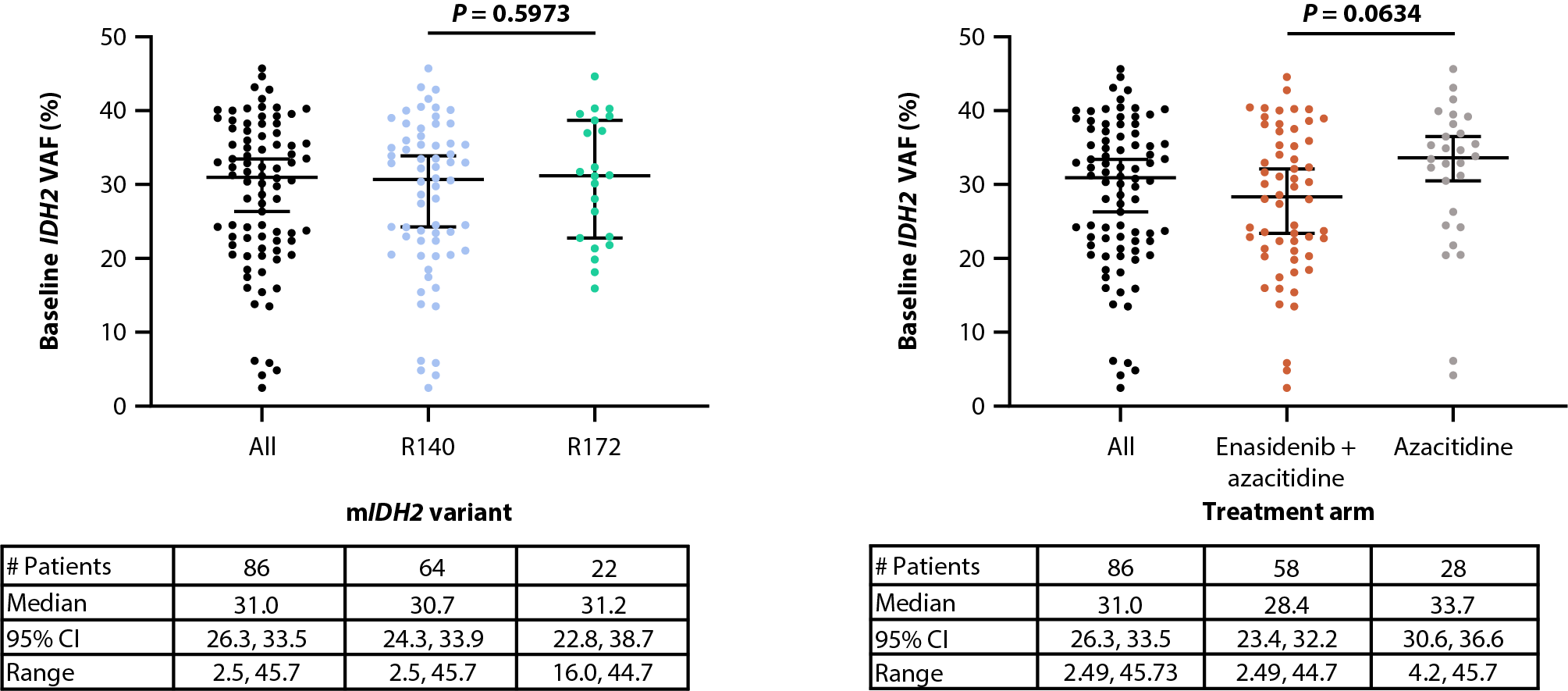
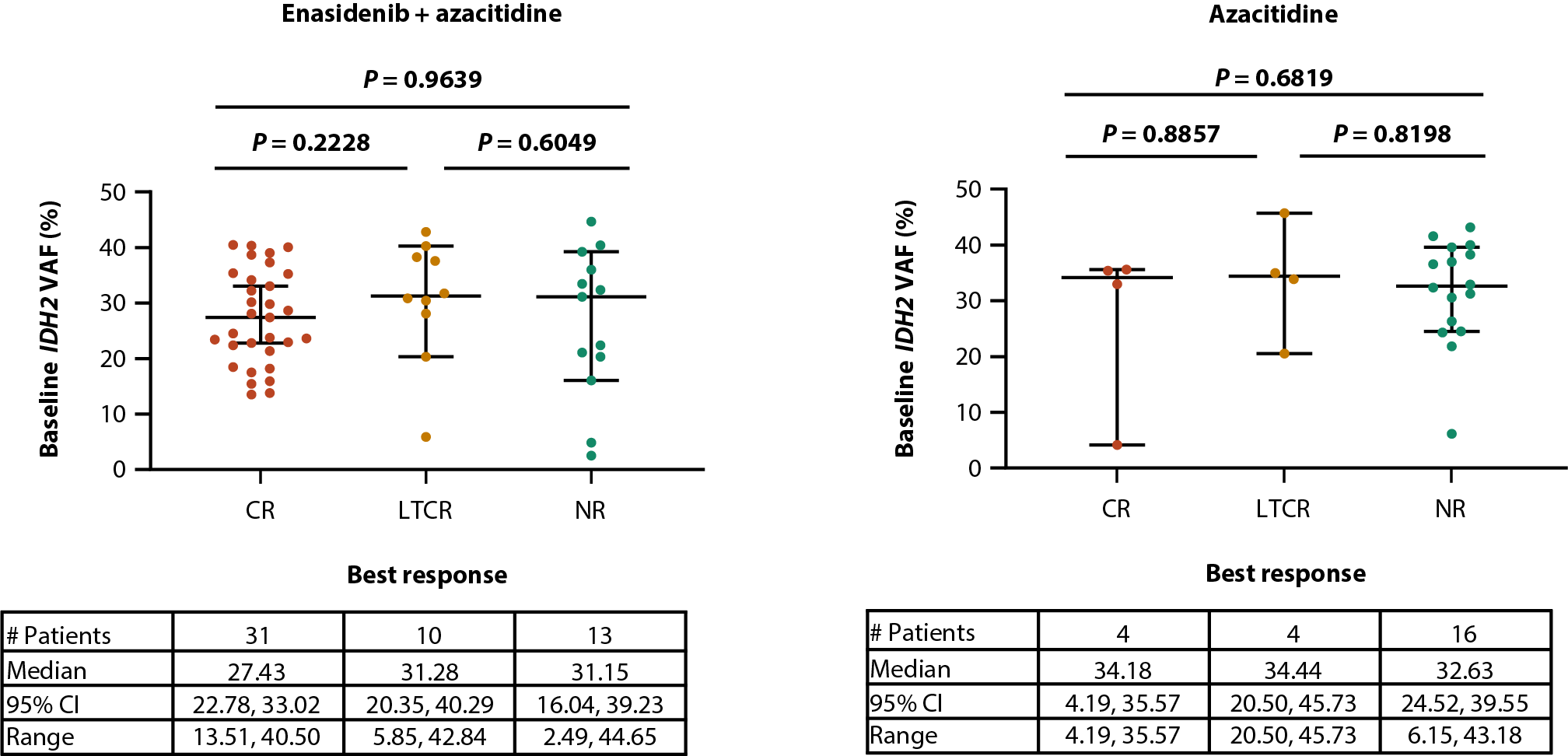
Similarly, baseline IDH2 VAFs were comparable between IDH2 variants (median IDH2 VAF of 30.7% and 31.2% for R140 and R172, respectively [P=0.5973]) and treatment arms [14] (28.4% and 33.7% for enasidenib + azacitidine and azacitidine only, respectively [P=0.0634]; Figures 2A, 2B). Furthermore, there was no relationship between baseline IDH2 VAF and clinical response in the combination arm (median IDH2 VAF of 27.4%, 31.3% and 31.2% for CR, LTCR, and NR, respectively [P=0.9639]; Figure 2C) or the azacitidine-only arm (34.2%, 34.4%, and 32.6% for CR, LTCR, and NR, respectively [P=0.6819]; Figure 2D).
A B
C D
E
F
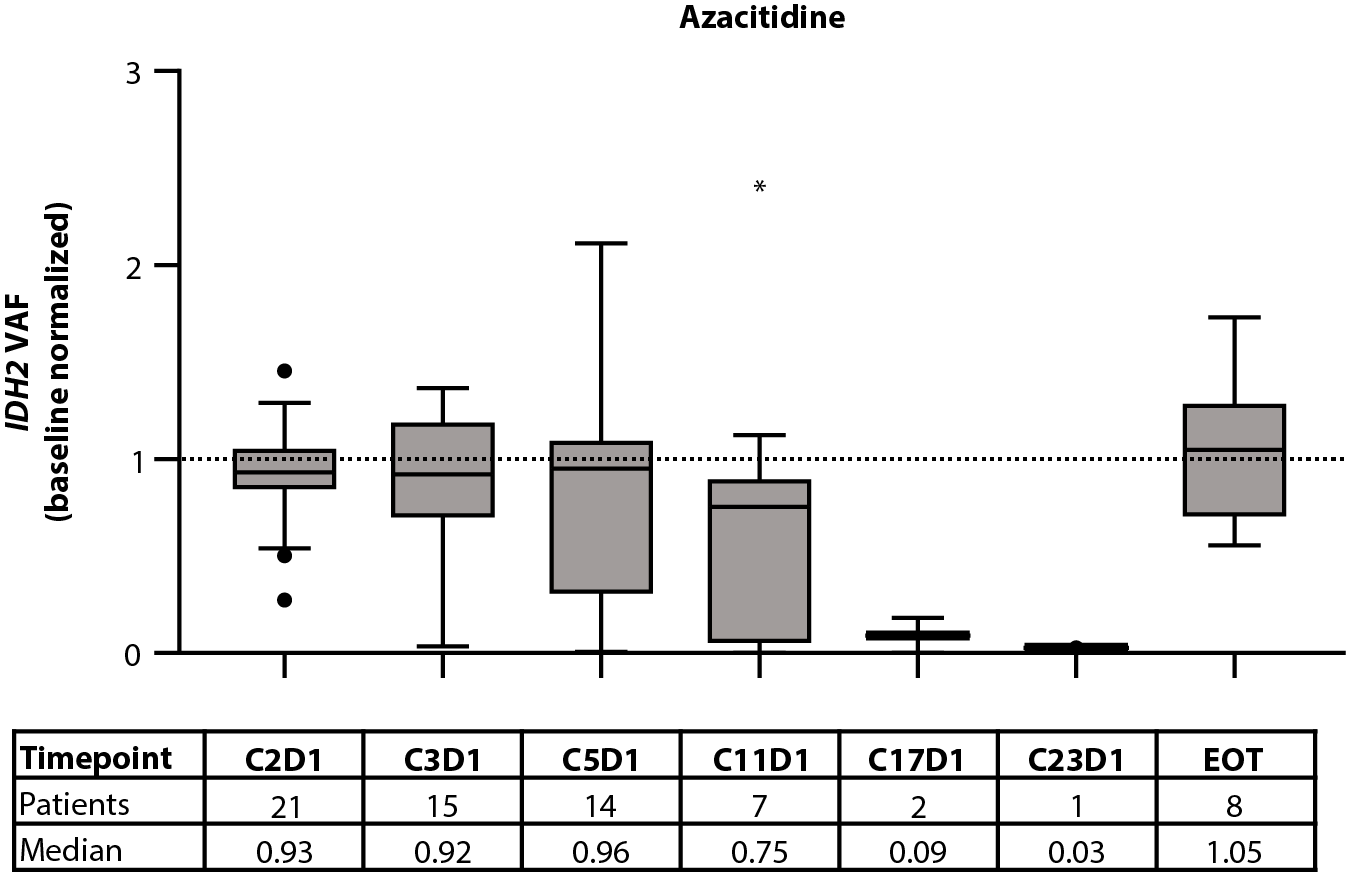
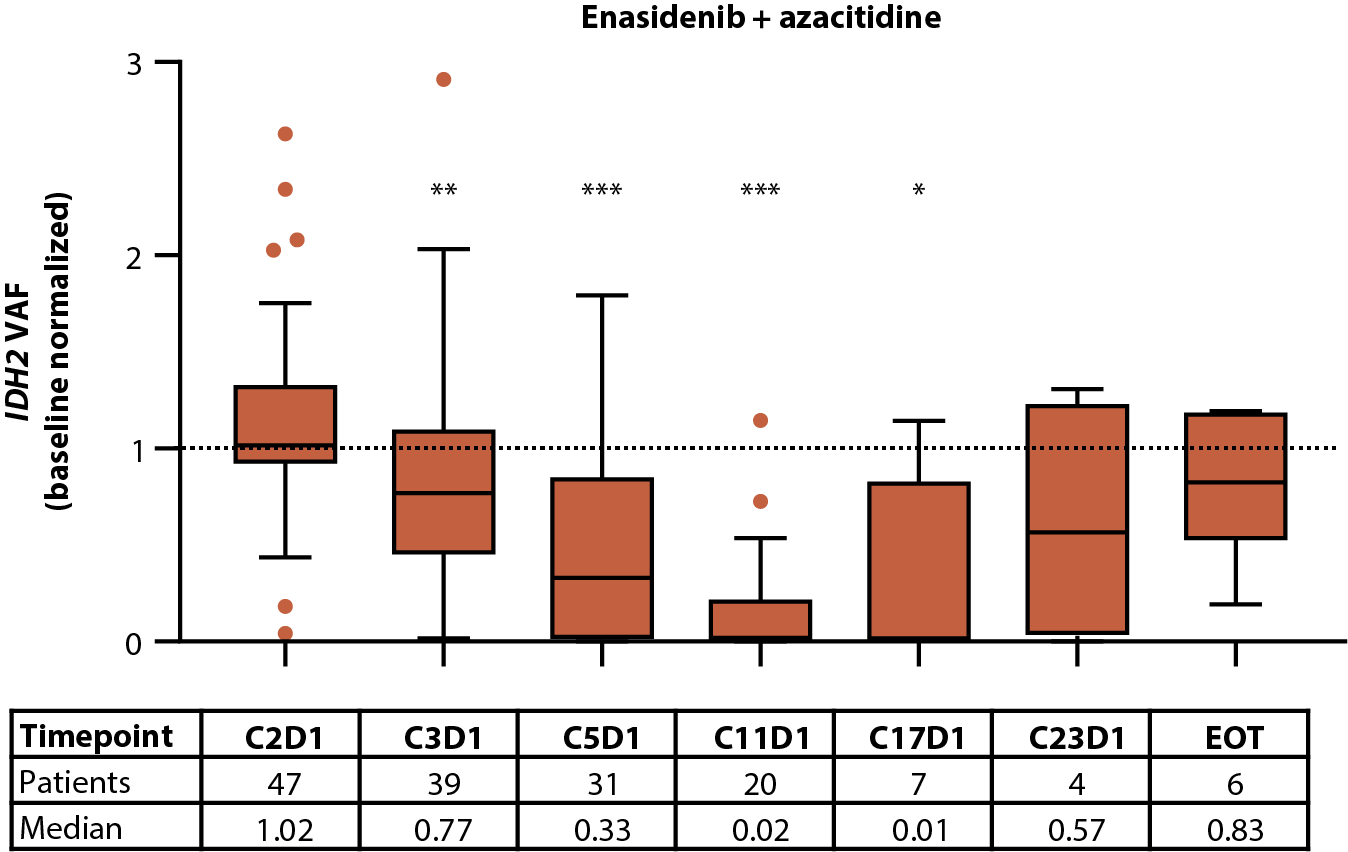
Figure 2. Baseline IDH2 VAF by IDH2 variant (A) and treatment arm (B). Relationship between baseline IDH2 VAFs and clinical response in patients treated with enasidenib + azacitidine (C) or azacitidine only (D). Baseline-normalized changes in IDH2 VAF in patients treated with enasidenib + azacitidine (E) or azacitidine only (F). Horizontal bars indicate median; error bars indicate Tukey’s range; P values calculated using Mann-Whitney test (B, C) or Wilcoxon test (D, E). *P≤0.05; **P≤0.01; ***P≤0.001. CI: Confidence Interval; CR: Complete Remission; CxDx: Cycle x, Day x; EOT: End of Treatment; IDH2: Isocitrate Dehydrogenase-2; LTCR: Less than CR; NR: No Response; VAF: Variant Allele Frequency.
Associations between 2-HG and IDH2 VAF were evaluated using Spearman nonparametric correlation. A modest positive correlation was noted between baseline 2-HG and IDH2 VAF in patients with ND-AML with paired baseline 2-HG and IDH2 VAF data (Spearman r=0.4199, P<0.0001; Figure 3A).
The effect of treatment with enasidenib + azacitidine, or azacitidine only, on 2-HG and IDH2 VAF was assessed longitudinally in a subset of patients with ND-AML (2-HG: n=60 and n=28, respectively; IDH2 VAF: n=47 and n=21, respectively). In the combination arm, a significant (P≤0.001) reduction in median 2-HG from baseline was observed by C1D15, and 2-HG levels remained decreased throughout treatment; decreases in 2-HG were statistically significant (P≤0.01) through C9D1 when compared with baseline (Figure 1D). In the azacitidine-only arm, 2-HG was not significantly reduced from baseline at any time (Figure 1E). Significant (P≤0.05) reductions from baseline were observed for IDH2 VAF by C3D1 through C17D1 in the combination arm (Figure 2E). Increasing IDH2 VAF was seen at C17D1 and beyond, with median EOT VAFs comparable with those at baseline. In contrast, IDH2 VAF was significantly reduced only at C11D1 with azacitidine only P≤0.05; Figure 2F).
There was no definitive correlation between minimum on-study 2-HG and IDH2 VAF in the combination arm (r=0.2464, P=0.0671; Figure 3B), perhaps because most patients had robust reductions in 2-HG in response to treatment, whereas IDH2 VAF reductions were variable and required longer treatment duration.
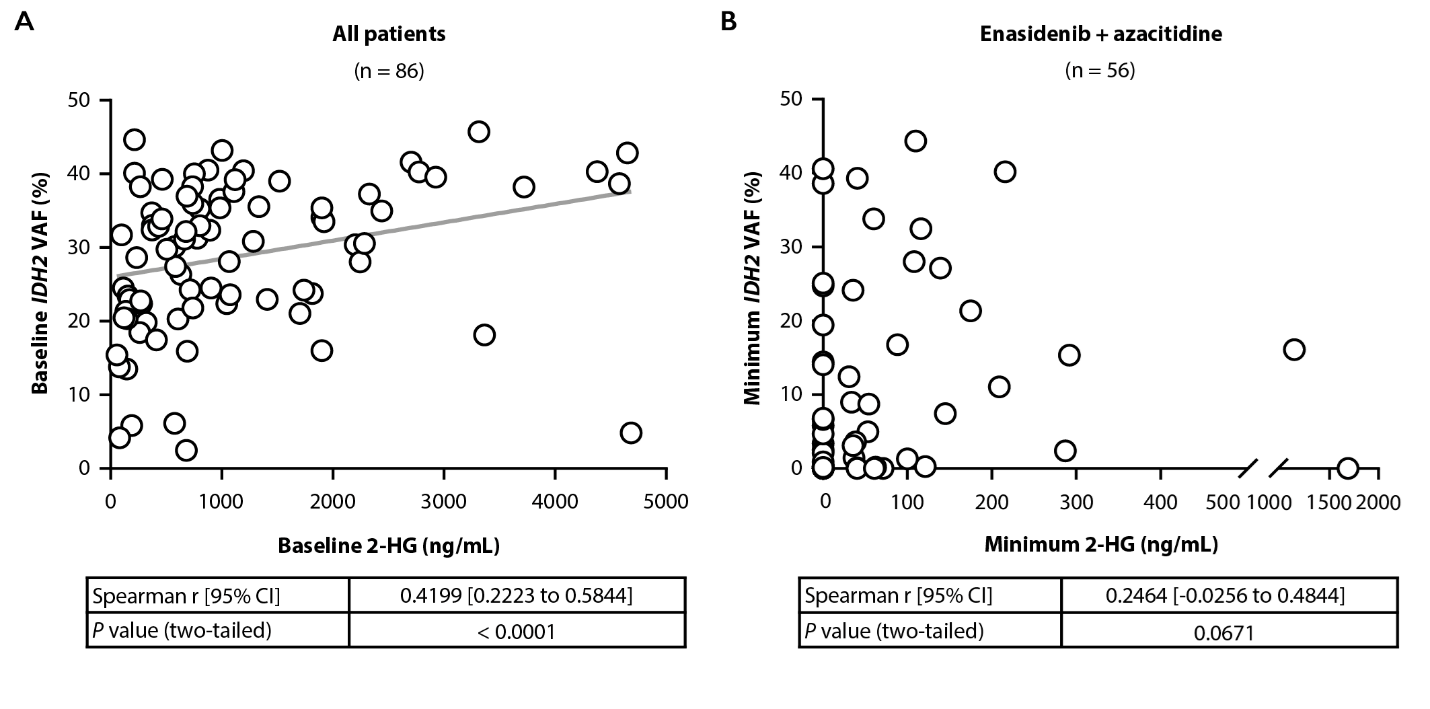
Figure 3. Correlation between baseline 2-HG and IDH2 VAF in all patients (A). Correlation between minimum on-treatment 2-HG and IDH2 VAF achieved in the combination arm (B). r value calculated using Spearman correlation; P values calculated using two-tailed test. 2-HG, D-2-Hydroxyglutarate; CI: Confidence Interval; IDH2: Isocitrate Dehydrogenase-2; VAF: Variant Allele Frequency.
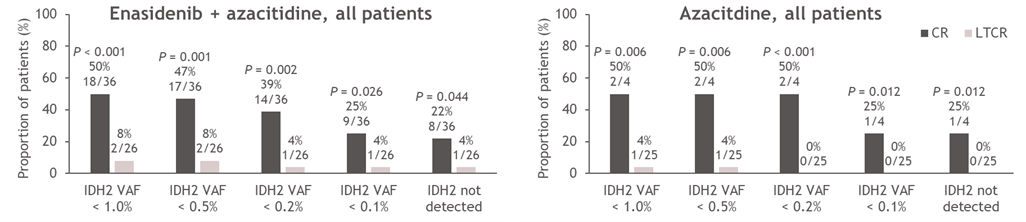
The relationship between molecular clearance, defined as IDH2 VAF clearance below the assay limit of detection of 0.02–0.04%, and clinical response was further investigated (Figure 4). Significantly more patients who achieved CR versus LTCR had complete IDH2 VAF clearance (enasidenib + azacitidine: 22% and 4% for CR and LTCR, respectively [P=0.044], azacitidine only: 25% and 0% for CR and LTCR, respectively [P=0.012]; Figure 4A). Notably, all patients who achieved molecular clearance harbored the IDH2-R140 mutation (Figure 4B). Fifty percent (18/36) of patients who achieved CR in the combination arm also achieved reductions in IDH2 VAF to <1% (P<0.001 vs LTCR; Figure 4A), and IDH2 VAF reductions to <1% were proportionally more common in the IDH2-R140 subgroup (59%) than in the IDH2-R172 subgroup (22%; Figure 4B). In the azacitidine-only arm, two out of four patients who achieved CR had IDH2 VAF <1% on-study (P=0.006 vs LTCR; both had IDH2-R140 mutations; Figure 4A, 4B).
A.
B.
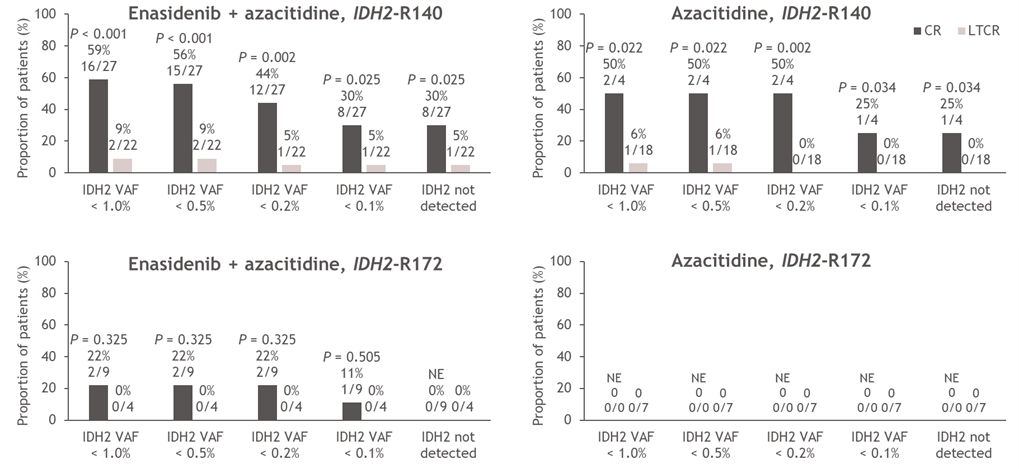
Figure 4. Minimum on-treatment IDH2 VAF achieved according to treatment arm and response status (CR vs LTCR) (A) and treatment arm, response status (CR vs LTCR) and IDH2 variant (B). P values calculated using Chi-squared test. AZA: Azacitidine; CR: Complete Remission; ENA: Enasidenib; IDH2: Isocitrate Dehydrogenase-2; LTCR: Less than CR; VAF: Variant Allele Frequency.
Baseline mutational profiling
Of the 86 patients with available baseline genomic data, all exhibited at least one mutation in a gene other than IDH2. The most common co-occurring mutations were ASXL1 (53%), SRSF2 (50%), and DNMT3A (49%) (Supplemental Table 1) [14]. SRSF2 was significantly preferentially co-mutated with IDH2-R140 (vs IDH2-R172; P<0.0001), while DNMT3A (P=0.0002) and ETV6 (P=0.0098) were significantly co-mutated with IDH2-R172 (Figure 5A). Baseline mutational burden was similar between IDH2 variants (P=0.0555; Figure 5B). The median number of baseline mutations was six for the entire patient population. When categorized as ≥6 mutations or <6 mutations at baseline, mutation burden was not associated with clinical response on-study (Figure 5C).
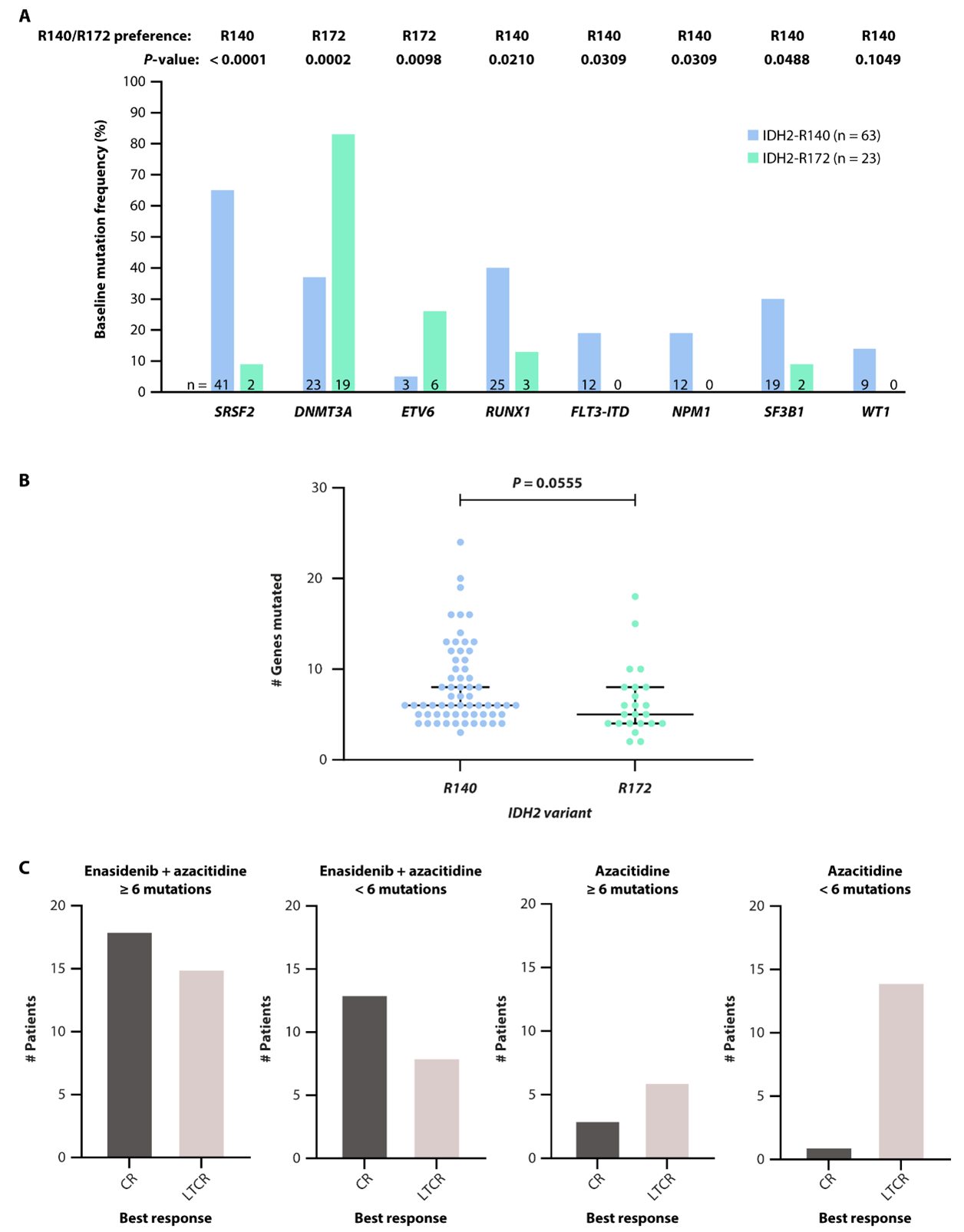
Figure 5. Baseline co-mutations preferentially co-mutated with IDH2-R140 or IDH2-R172 (A). Number of baseline gene mutations according to IDH2 variant (B). Response to enasidenib + azacitidine or azacitidine only according to baseline mutation burden (<6 vs ≥6 mutations) (C). P values calculated using Fisher’s exact test (A) or Mann-Whitney test (B, C). AZA: Azacitidine; CR: Complete Remission; IDH2: Isocitrate Dehydrogenase-2; LTCR: Less than CR.
IDH2 clonality
Clonal hierarchy and clonal evolution can potentially influence treatment outcomes [16-19]. Thus, we evaluated the position of mutant-IDH2 in the clonal hierarchy by exploring baseline VAFs of co-occurring gene mutations relative to IDH2, and explored changes in the clonal landscape during treatment with enasidenib + azacitidine. IDH2 was defined as subclonal if any co-mutation VAF was greater than IDH2 VAF; otherwise, it was considered clonal. While IDH2 was primarily clonal to most other genes (but subclonal to DNMT3A) at baseline, IDH2 VAF reductions during combination treatment altered tumor clonality, with other driver genes (e.g., ASXL1, CEBPA, TET2) becoming clonal to IDH2 on-study (Figure 6).
A
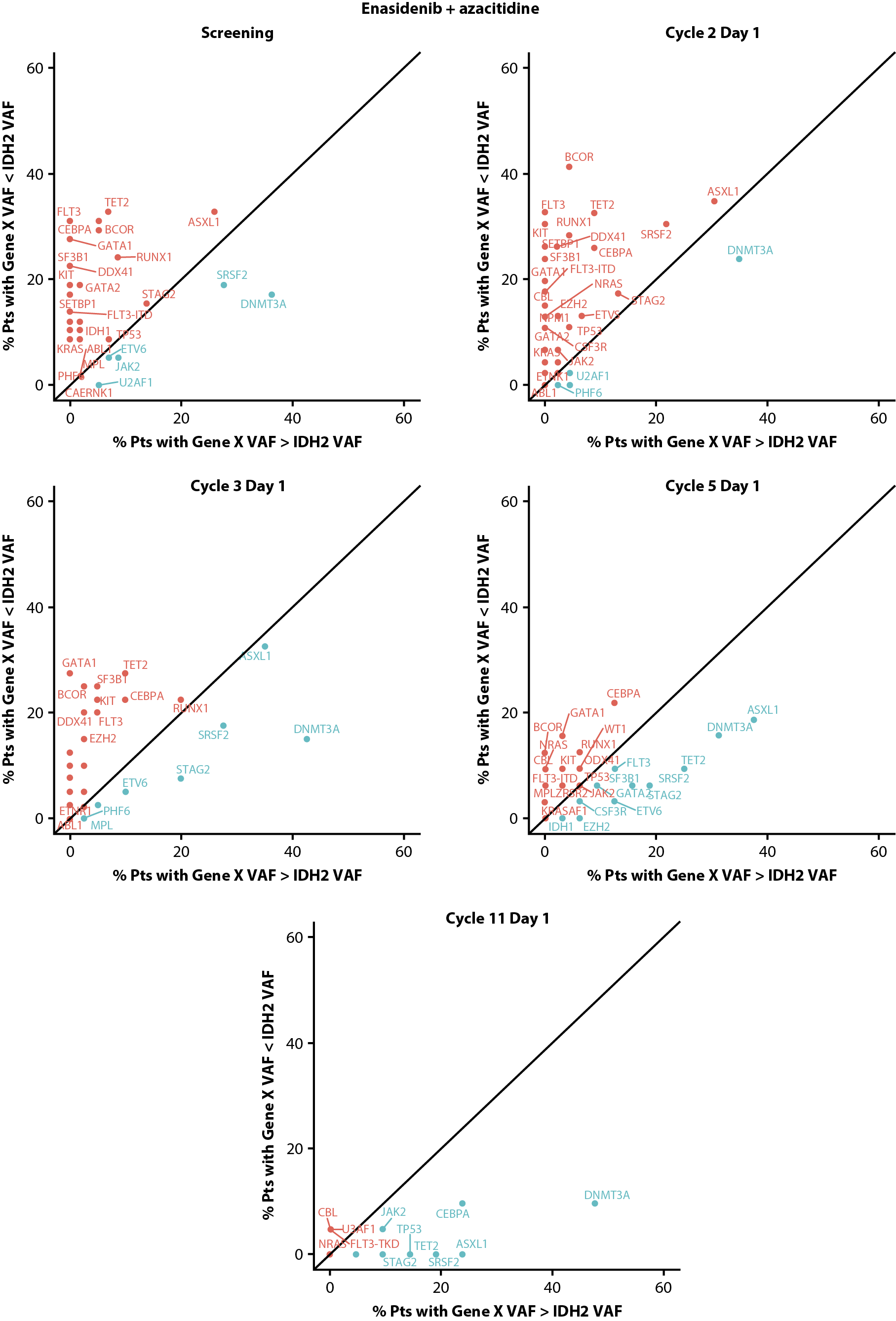
B
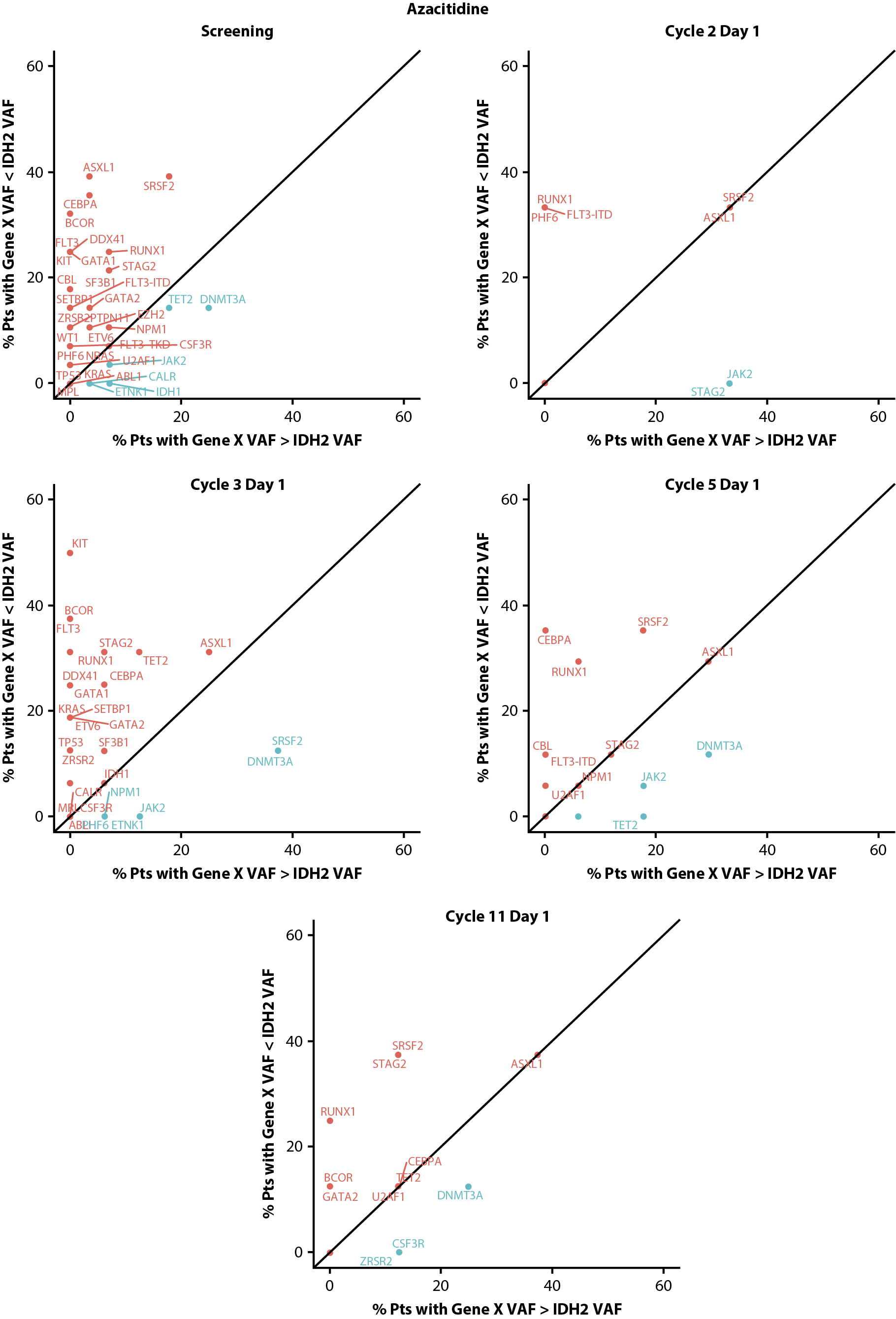
Figure 6. Clonal hierarchy of mutated IDH2 at screening and during treatment with enasidenib + azacitidine (A) or azacitidine only (B). Blue text represents gene mutations that are predominantly clonal compared with IDH2 and red text represents gene mutations that are mostly subclonal to IDH2. IDH2: Isocitrate Dehydrogenase-2; pts: patients; VAF: Variant Allele Frequency.
Gene pathway analyses
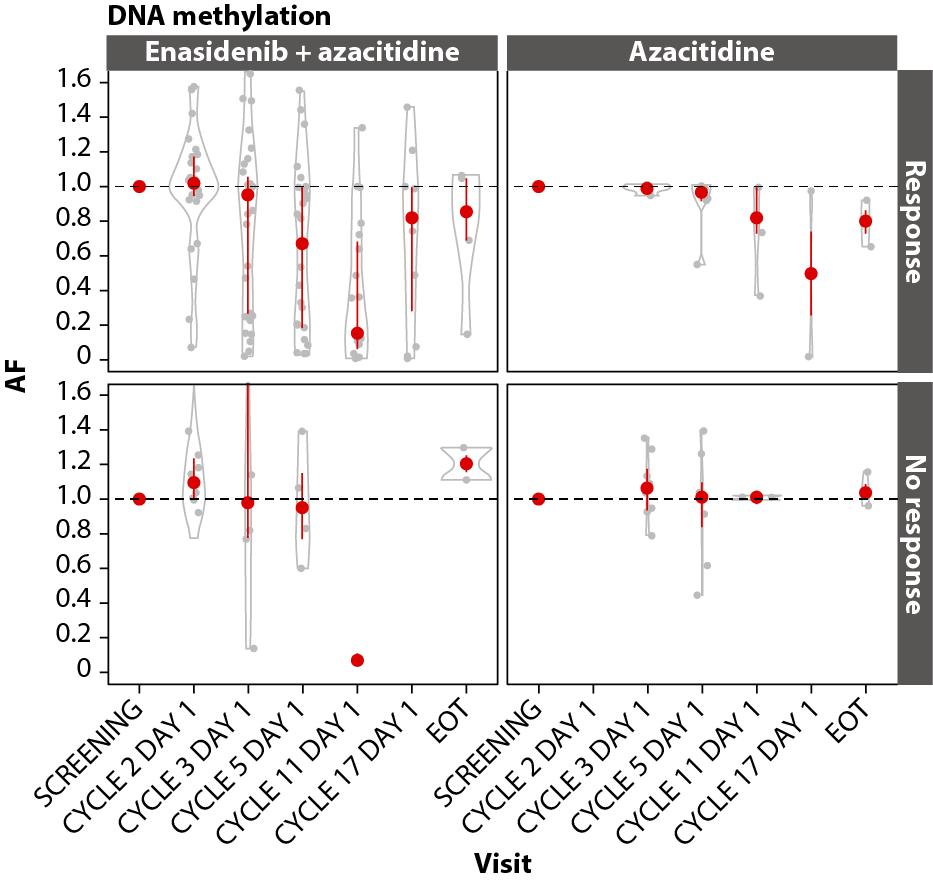
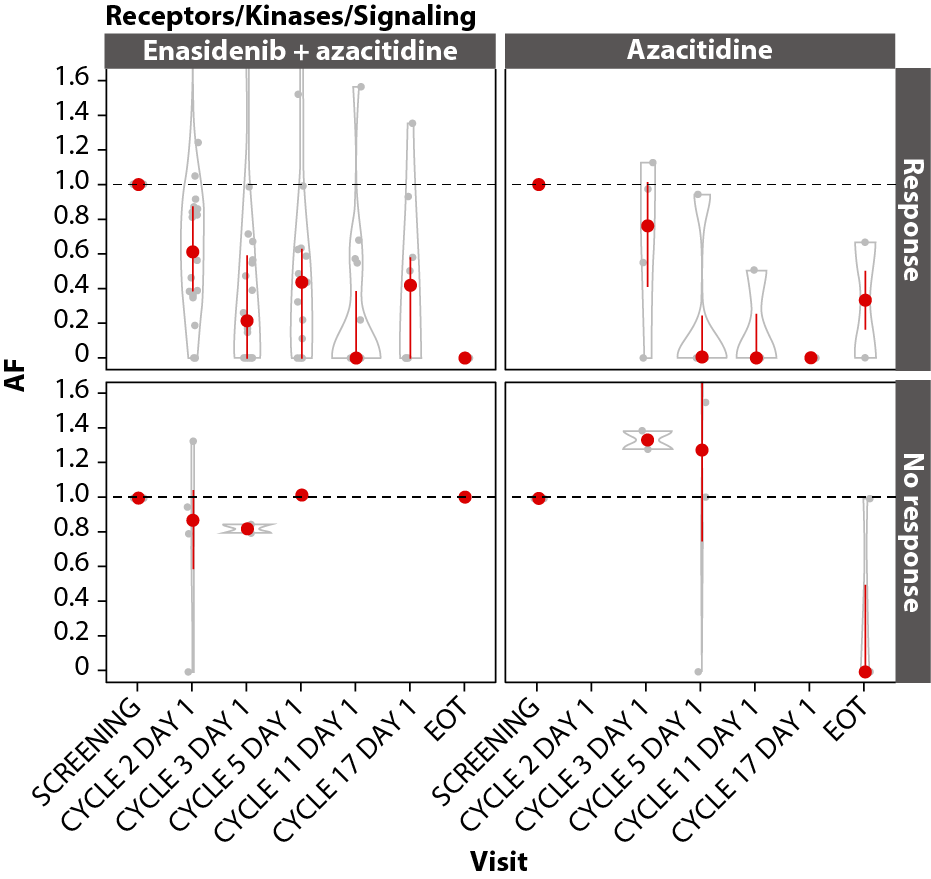
In both arms, achievement of CR was associated with VAF reductions in the DNA methylation (IDH1, IDH2, DNMT3A, TET2; Figure 7A), RTK (KIT, CSF3R, FLT3, JAK2; Figure 7B), and RAS (CBL, PTPN11, NRAS, KRAS; Figure 7C) gene pathways, with significant (P≤0.001) maximal reductions from baseline in the combination arm by C11D1 of ~85% for DNA methylation (n=19), 100% for RTK (n=15), and 100% for RAS (n=11). Maximal VAF reductions in the azacitidine-only arm are not reported due to small numbers.
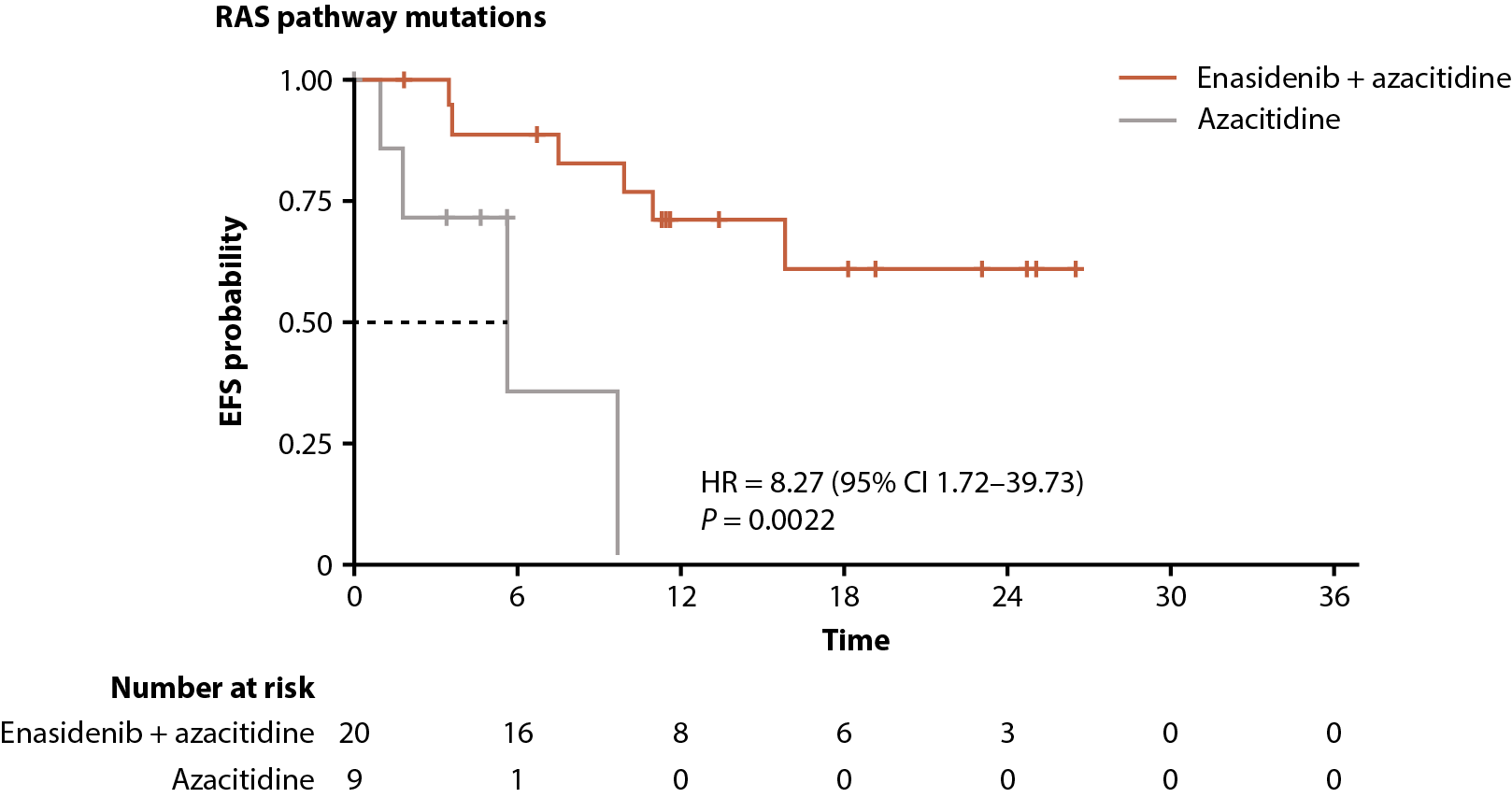
Among all patients with RAS-pathway mutations, enasidenib + azacitidine significantly prolonged EFS compared with azacitidine only (median not reached vs 5.6 months, respectively; P=0.0022); median OS was not reached versus 9.3 months (P=0.52; Figure 7D, 7E).
A
B
C
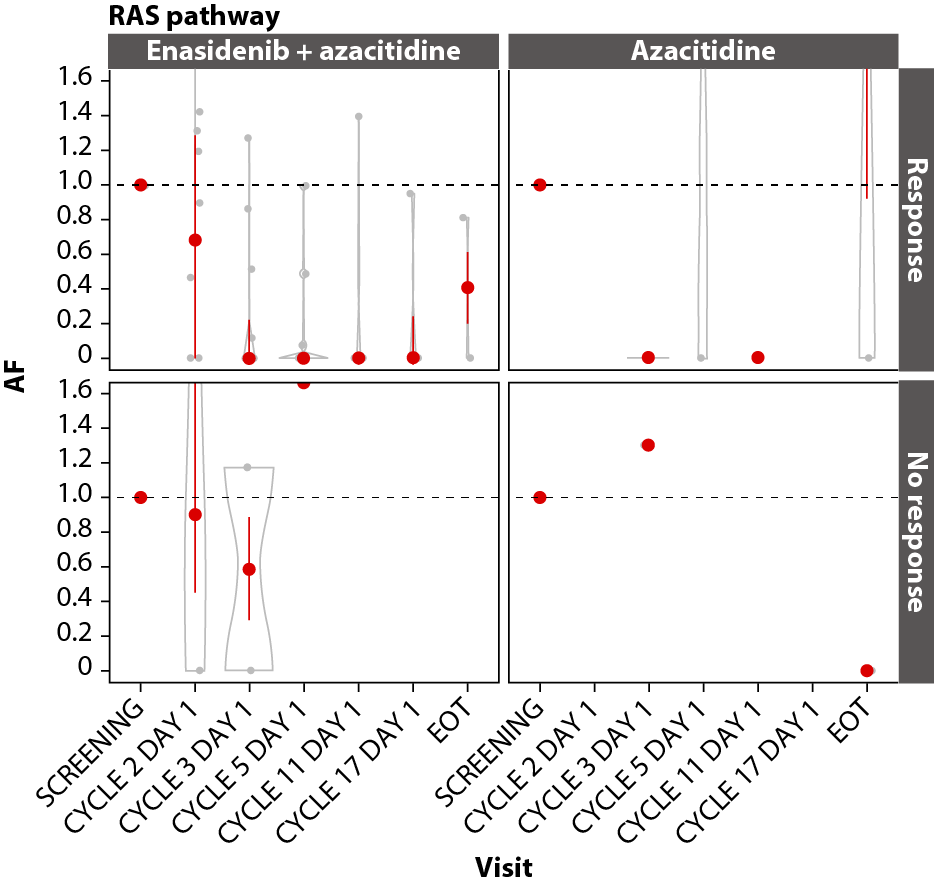
D
E
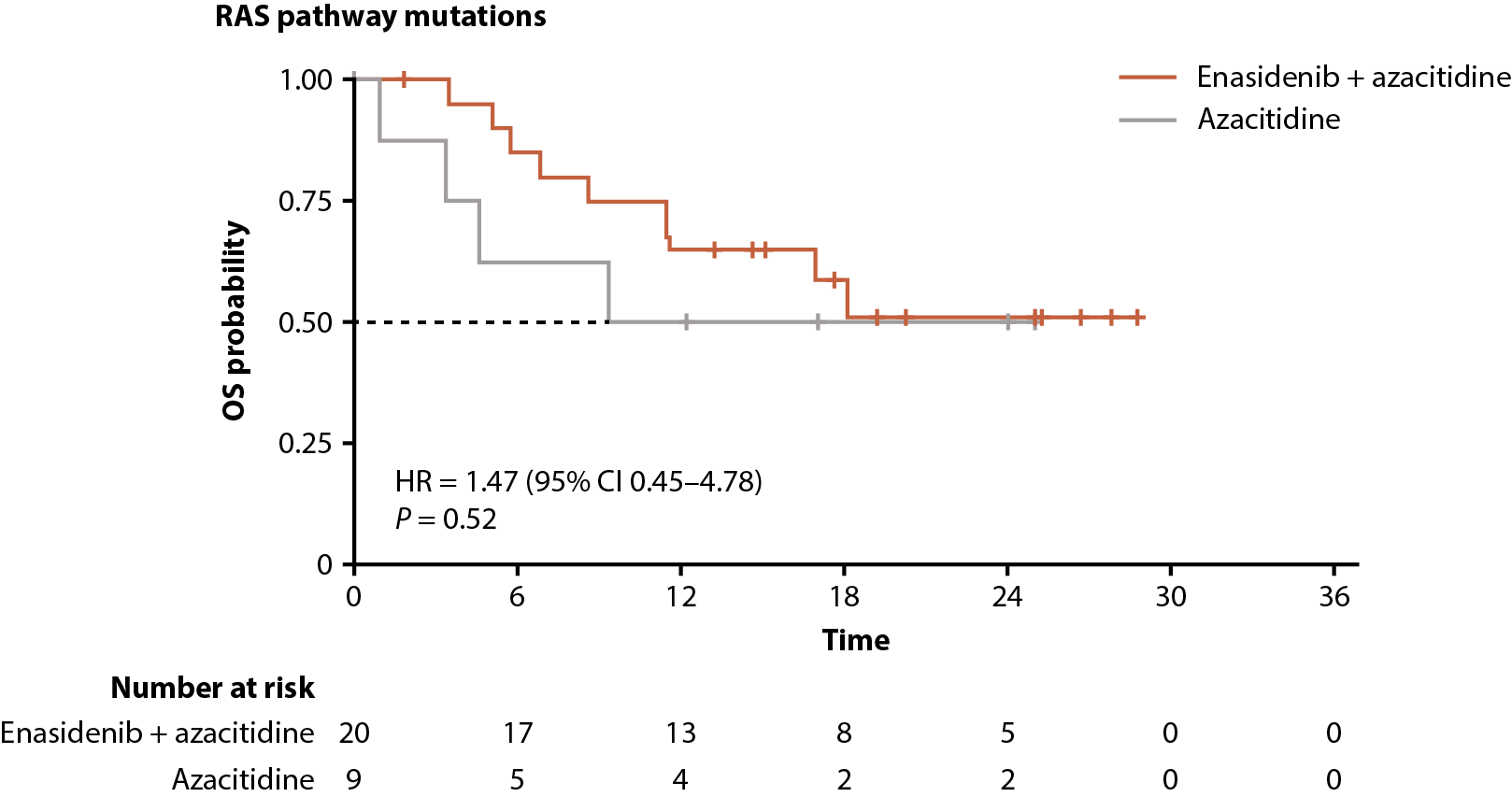
Figure 7. Gene pathway analyses. Baseline-normalized changes in VAFs for genes in the DNA methylation (A), RTK (B), and RAS (C) gene pathways over time by treatment arm and response status (CR vs LTCR or no response). Event-free survival (D) and overall survival (E) by treatment arm in patients with RAS pathway mutations. P values calculated using Wilcoxon test. AF: Allele Frequency; CI: Confidence Interval; EFS: Event-Free Survival; EOT: End of Treatment; HR: Hazard Ratio; IDH2: Isocitrate Dehydrogenase-2; LTCR: Less than CR; OS: Overall Survival.
Discussion
The current investigation characterizes molecular features associated with clinical response to combination therapy with enasidenib + azacitidine in patients with ND-AML. Our findings demonstrate a robust clinical validation of the synergistic effects of enasidenib + azacitidine seen in vitro [13]. Baseline levels of 2-HG or IDH2 VAFs were similar irrespective of IDH2 variant (R140 or R172) and were not predictive of best clinical response to enasidenib + azacitidine or azacitidine only. Longitudinal analysis revealed rapid reductions in 2-HG and IDH2 VAF during treatment with enasidenib + azacitidine; reductions were more rapid and robust with combination treatment versus azacitidine only. While higher levels of 2-HG were associated with higher IDH2 VAFs at baseline (possible indicator of disease burden), no correlation was observed between minimum 2-HG and IDH2 VAF in patients treated with enasidenib + azacitidine; levels of 2-HG were robustly reduced in most patients and IDH2 VAFs were more variable in response to treatment.
IDH2 VAF clearance was significantly more likely in patients with CR versus LTCR in both treatment arms, and more common in those with IDH2-R140 mutations than IDH2-R172. In both combination and azacitidine-only treatment arms, 50% of those who achieved CR versus 4–8% who did not achieve CR had minimum on-treatment IDH2 VAFs <1% (P≤0.01). The observed association between IDH2 VAF clearance and CR suggests that clinical response is likely driven by clearance of IDH-mutated blasts regardless of the type of targeted therapy. However, it is important to note that the cohort size was limited.
As part of this analysis, we characterized the baseline genetic landscape of patients with IDH2-mutated ND-AML, including tumor mutation burden, and evaluated the association with clinical response. Consistent with other reports [20,21], IDH2-R140 and IDH2-R172 showed distinct co-mutational profiles at baseline, with SRSF2 preferentially co-mutated with IDH2-R140 and DNMT3A preferential for IDH2-R172. Mutational burden at baseline was similar in those with IDH2-R140 and IDH2-R172. CRs were attained in patients treated with enasidenib + azacitidine irrespective of co-occurring gene mutations, suggesting no significant association between co-mutations and response to therapy.
In patients with RAS-pathway mutations, combination treatment led to significant VAF reductions for those who attained CR and significantly prolonged EFS compared with azacitidine only. Mutations in RAS pathway genes (PTPN11, NRAS, KRAS), which occur in ~10–25% of patients with AML, are historically associated with poor outcomes and resistance to targeted therapies [6,22-25], including primary resistance to enasidenib in R/R AML [6]. In ND-AML, a study reported no association between the RAS-pathway mutations and decreased likelihood of response to enasidenib; however, the patient number was low [9]. In this trial, the combination of enasidenib + azacitidine promoted morphologic response across all gene pathways, including RAS [14], and no individual gene in the RAS pathway was significantly associated with achievement of CR. As RAS-pathway mutations are known to induce resistance to enasidenib [6,25], but not azacitidine, the addition of azacitidine may reduce the risk of resistance to enasidenib in this setting. This hypothesis is further supported by the data from the AGILE study which showed that a subgroup of patients with IDH1-mutated AML plus RAS-pathway mutations were more likely to achieve CR to combination therapy with ivosidenib (a mutant IDH1 inhibitor) and azacitidine versus placebo and azacitidine [26].
Azacitidine has previously demonstrated efficacy in older patients with AML ineligible for intensive chemotherapy, and in combination with enasidenib in newly diagnosed patients and those with prior exposure to HMAs or enasidenib [10-12,14,27]. However, to date, no long-term data have been published evaluating enasidenib + azacitidine with respect to extended durations of response, disease recurrence, and clonal selection in patients with IDH2-mutated AML (median follow-up of previous studies: 13.1–18.5 months) [14,27]. Selection pressure leading to acquired drug resistance has been reported for several targeted AML agents, including enasidenib and ivosidenib [28]. In a subanalysis of the phase 1/2 AG221-C-001 trial (NCT01915498) assessing enasidenib monotherapy in patients with IDH2-mutated AML, it was shown that clonal selection or evolution of terminal or ancestral clones, as opposed to second-site mutations in the same IDH2 allele, resulted in resistance to enasidenib [29]. In addition, several studies have demonstrated that resistance to azacitidine can also arise, yet the exact molecular mechanism is currently unknown [30]. As such, doublet and triplet combination strategies that can target multiple mechanisms or clones driving AML disease progression are of particular interest [28-31]. Recently, a triplet combination of decitabine (an oral HMA) + venetoclax + enasidenib prolonged the duration of remission in a small number of patients who experienced disease progression with decitabine + venetoclax doublet therapy in a prospective phase 2 study (NCT03404193) [32], and is currently being evaluated in patients with R/R IDH2-mutated AML (NCT04774393). The development of alternative combination treatment options may help address complications relating to drug resistance in IDH2-mutated AML. Finally, further evaluation is needed to establish the long-term efficacy and safety of, and potential acquired resistance to, enasidenib + azacitidine in this patient population.
Conclusion
In conclusion, outcomes in this IDH2-mutated ND-AML cohort clinically validate the synergistic effects of enasidenib + azacitidine previously observed in vitro. One-half of patients who achieved CR with enasidenib + azacitidine and azacitidine only had IDH2 VAF reductions to <1%, suggesting that IDH2 VAF clearance may be associated with CR irrespective of therapy. Unlike prior reports of enasidenib or azacitidine monotherapy, combining these agents improved survival in patients with RAS mutations. Given the modest sample size in the current analyses, additional studies are needed to further dissect the molecular mechanisms driving the clinical benefit seen with this combination in patients with RAS-pathway mutations.
Conflicts of Interest
AR reports current employment, equity ownership, and patents with Bristol Myers Squibb. WLS is a contractor with Bristol Myers Squibb. TP, AG, and MH report current employment and equity ownership with Bristol Myers Squibb. CDD reports receiving research funding from AbbVie, Astex, Bristol Myers Squibb, Cleave, Forma, Foghorn, ImmuneOnc, LOXO, and Servier; receiving honoraria from Astellas, Bluebird Bio, Bristol Myers Squibb, Foghorn, Gilead, ImmuneOnc, Jazz Pharmaceuticals, Kura, Novartis, Servier, and Takeda; receiving support as a LLS Scholar in Clinical Research; serving in an advisory position for GenMab, GlaxoSmithKline, Kura, and Notable Labs; serving in a consultancy position for AbbVie and Servier; and equity ownership with Notable Labs. HD reports receiving research funding from AbbVie, Agios, Amgen, Astellas, AstraZeneca, Berlin-Chemie, Bristol Myers Squibb, Celgene, Jazz Pharmaceuticals, Kronos Bio, and Novartis; and serving on advisory boards for AbbVie, Agios, Amgen, Astellas, AstraZeneca, Berlin-Chemie, Bristol Myers Squibb, Celgene, Daiichi Sankyo, Gilead, Janssen, Jazz Pharmaceuticals, Novartis, Servier, Stemline, and Syndax. EMS reports serving on advisory boards for AbbVie, Agios, Aptose, Astellas, Blueprint, Bristol Myers Squibb, Calithera, CTI Biopharma, Daiichi Sankyo, Foghorn, Genentech, Genesis, Gilead, Janssen, Jazz Pharmaceuticals, Menarini, Neoleukin, Novartis, OnCusp, Ono Pharma, PinotBio, Servier, Syndax, and Syros; receiving honoraria from Kura; serving on safety monitoring committees for Cellectis and Epizyme; receiving research funding from Bristol Myers Squibb and Eisai; and equity ownership with Auron. ATF reports receiving clinical trial support from AbbVie, Agios/Servier, and Celgene/Bristol Myers Squibb; advisory board participation with AbbVie, Agios/Servier, Amgen, Astellas, Blueprint, Celgene/Bristol Myers Squibb, Daiichi Sankyo, EnClear, Foghorn, Genentech, Immunogen, Kite, Kura Oncology, Mablytics, Menarini, Morphosys, Novartis, Orum, Pfizer, PureTech, Remix, Rigel, Seattle Genetics, Takeda, and Trillium; and consulting for Daiichi Sankyo, Forma, Ipsen, Menarini, Remix, and Rigel. PV and LQ report receiving research funding from Bristol Myers Squibb.
Funding Statement
This study was funded by Celgene, a Bristol-Myers Squibb Company.
Acknowledgments
Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Brian Kaiser and Eva Polk of Excerpta Medica, and Clair Clowes and Keri Davies of Ashfield MedComms, an Inizio Company, funded by Bristol Myers Squibb.
Authorship Contributions
AR, WLS, and MH designed the translational analyses reported in this publication. Data were collected by AR, WLS, CDD, HD, EMS, ATF, PV, LQ, and MH. Data analysis was performed by AR, WLS, TP, AG, LQ, and MH. AR, WLS, TP, AG, and MH wrote the first draft of the manuscript. All authors contributed to data interpretation, revised the manuscript, and reviewed and approved the final version.
References
2. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373:1136-52.
3. Chou WC, Lei WC, Ko BS, Hou HA, Chen CY, Tang JL, et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. 2011;25:246-53.
4. Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong W-J, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21:178-84.
5. Heuser M, Araujo Cruz MM, Goparaju R, Chaturvedi A. Enigmas of IDH mutations in hematology/oncology. Exp Hematol. 2015;43:685-97.
6. Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130:732-41.
7. IDHIFA [package insert]. Summit, NJ: Celgene Corporation; 2017.
8. De Botton S, Montesinos P, Schuh AC, Papayannidis C, Vyas P, Wei AH, et al. Enasidenib vs conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: a randomized phase 3 trial. Blood. 2023;141:156-67.
9. Pollyea DA, Tallman MS, De Botton S, Kantarjian HM, Collins R, Stein AS, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019;33:2575-84.
10. Dombret H, Seymour JF, Butrym A, Wierzbowska A, Selleslag D, Jang JH, et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood. 2015;126:291-9.
11. Van Der Helm LH, Veeger NJ, Kooy M, Beeker A, De Weerdt O, De Groot M, et al. Azacitidine results in comparable outcome in newly diagnosed AML patients with more or less than 30% bone marrow blasts. Leuk Res. 2013;37:877-82.
12. Bories P, Bertoli S, Bérard E, Laurent J, Duchayne E, Sarry A, et al. Intensive chemotherapy, azacitidine, or supportive care in older acute myeloid leukemia patients: an analysis from a regional healthcare network. Am J Hematol. 2014;89:E244-52.
13. Macbeth KJ, Chopra VS, Tang L, Zheng B, Avanzino B, See WL, et al. Combination of azacitidine and enasidenib enhances leukemic cell differentiation and cooperatively hypomethylates DNA. Experimental Hematology. 2021;98:47-52.e6.
14. DiNardo CD, Schuh AC, Stein EM, Montesinos P, Wei AH, De Botton S, et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukaemia (AG221-AML-005): a single-arm, phase 1b and randomised, phase 2 trial. Lancet Oncol. 2021;22:1597-608.
15. Cheson BD, Bennett JM, Kopecky KJ, Büchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21:4642-9.
16. Herudkova Z, Culen M, Folta A, Jeziskova I, Cerna J, Loja T, et al. Clonal hierarchy of main molecular lesions in acute myeloid leukaemia. Br J Haematol. 2020;190:562-72.
17. Morita K, Wang F, Jahn K, Hu T, Tanaka T, Sasaki Y, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun. 2020;11:5327.
18. Saygin C, Hu E, Zhang P, Sher S, Lozanski A, Doong T-J, et al. Genomic analysis of cellular hierarchy in acute myeloid leukemia using ultrasensitive LC-FACSeq. Leukemia. 2021;35:3406-20.
19. Duchmann M, Laplane L, Itzykson R. Clonal Architecture and Evolutionary Dynamics in Acute Myeloid Leukemias. Cancers (Basel). 2021;13
20. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374:2209-21.
21. Bill M, Jentzsch M, Bischof L, Kohlschmidt J, Grimm J, Schmalbrock LK, et al. Impact of IDH1 and IDH2 mutation detection at diagnosis and in remission in patients with AML receiving allogeneic transplantation. Blood Adv. 2023;7:436-44.
22. Mcmahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJD, Eastburn DJ, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9:1050-63.
23. Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’alessandro A, Winters A, et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer. 2020;1:1176-87.
24. DiNardo CD, Cortes JE. Mutations in AML: prognostic and therapeutic implications. Hematology. 2016;2016:348-55.
25. Short NJ, Konopleva M, Kadia TM, Borthakur G, Ravandi F, DiNardo CD, et al. Advances in the Treatment of Acute Myeloid Leukemia: New Drugs and New Challenges. Cancer Discov. 2020;10:506-25.
26. Montesinos P, Recher C, Vives S, Zarzycka E, Wang J, Bertani G, et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med. 2022;386:1519-31.
27. Venugopal S, Takahashi K, Daver N, Maiti A, Borthakur G, Loghavi S, et al. Efficacy and safety of enasidenib and azacitidine combination in patients with IDH2 mutated acute myeloid leukemia and not eligible for intensive chemotherapy. Blood Cancer J. 2022;12:10.
28. Desikan SP, Daver N, DiNardo C, Kadia T, Konopleva M, Ravandi F. Resistance to targeted therapies: delving into FLT3 and IDH. Blood Cancer J. 2022;12:91.
29. Quek L, David MD, Kennedy A, Metzner M, Amatangelo M, Shih A, et al. Clonal heterogeneity of acute myeloid leukemia treated with the IDH2 inhibitor enasidenib. Nat Med. 2018;24:1167-77.
30. Stomper J, Rotondo JC, Greve G, Lubbert M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: mechanisms of resistance and novel HMA-based therapies. Leukemia. 2021;35:1873-89.
31. Oyogoa E, Traer E, Tyner J, Lachowiez C. Building on foundations: Venetoclax-based combinations in the treatment of acute myeloid leukemia. Cancers (Basel). 2023;15
32. Venugopal S, Maiti A, DiNardo CD, Loghavi S, Daver NG, Kadia TM, et al. Decitabine and venetoclax for IDH1/2-mutated acute myeloid leukemia. Am J Hematol. 2021;96:E154-E7.