Abstract
The liver plays a crucial role in maintaining homeostasis for lipid and glucose. Hepatic lipid synthesis is regulated by nutritional signals in response to fasting and refeeding. It is known that overnutrition regulates MAPK-dependent pathways that control lipid metabolism in the liver by activating MAPK phosphatase-2 (MKP-2). Uncertainty still exists regarding the regulatory mechanisms and effects of MKP-2 on hepatic response to fasting. We investigated the effect of fasting on the expression of MKP-2 and the impact on hepatic inflammatory response to feeding a high-fat diet (HFD). In this study, we show that fasting stress led to an upregulation of hepatic MKP-2 expression and a corresponding decrease in phosphorylation of p38 MAPK in mouse livers. We discovered that hepatic steatosis brought on by fasting is not effective in MKP-2-deficient livers due in part to a decrease in lipolysis and GLUT2 expression. In response to refeeding a chow or HFD, MKP-2 exhibited differential regulation of hepatic inflammatory cytokines including IL-1β. It has been demonstrated that the mitochondrial carrier uncoupling protein 2 (UCP2) plays a significant role in immune function. We discovered that MKP-2 negatively controls the expression of the UCP2 protein in the liver, modulating the expression of inflammatory cytokines. These results lend credence to the idea that upregulation of MKP-2 is a physiologically relevant response and may help the liver better utilize hepatic lipids while fasting. Collectively, these findings show that MKP-2 modulates lipolysis and hepatic inflammatory response in response to alterations in nutritional status, such as excess nutrients and fasting.
Keywords
Cytokines, Fasting, Liver inflammation, Mitogen-activated protein kinase, Protein tyrosine phosphatase, Uncoupling protein-2
Introduction
The incidence of obesity is increasing worldwide, affecting ~33% of adult Americans [1,2]. Overnutrition and obesity are closely linked to a number of metabolic diseases including type 2 diabetes (T2D), atherosclerosis, heart disease, and fatty liver disease [2,3]. The causes of obesity are complex ranging from physiological, hereditary, social, and environmental factors [4]. Interventions against obesity such as exercise, genetic manipulations, and pharmacological treatments have limited effectiveness for many patients with obesity [5]. The imbalance of energy intake and expenditure contributes significantly to the development of obesity and is being affected by signals from many peripheral tissues [6]. Therefore, the identification of ways to improve major insulin-responsive tissue function to increase the pliability of these tissues in response to stress signaling may improve the pathogenesis of obesity and its associated disorders. Although many human studies have demonstrated the beneficial effects of fasting in improving obesity and its complications [5,7], the fundamental mechanisms are unclear. Dysregulated inflammatory responses are important components of overeating and obesity that induce injury in major insulin-responsive tissues including the liver [8]. Consumption of large amounts of calories such as a high-fat diet leads to hyperglycemia and enhanced serum free fatty acids. The liver is a central organ impacted by inflammation that results from metabolic stress [9].
The liver plays a major role in glucose and lipid metabolism and is comprised of many different cells including hepatocytes, hepatic stellate cells, Kupffer cells, and cholangiocytes, and so on [10]. The liver is impacted by numerous types of stress including the western diet, exotoxins, and overuse of anti-inflammatory medication [9,10]. It has been reported that globally there are approximately two million deaths due to liver disease worldwide every year [9]. Liver injury activates the natural killer cells and Kupffer cells to release cytokines that play an important role in the adaptive immune response and stimulate the elimination of harmful pathogens [11]. However, lipid accumulation can induce liver damage and inflammation [12]. The mitogen-activated protein kinases (MAPKs), serine/threonine kinases expressed in the liver play an important role in regulating lipid accumulation and inflammation [13,14].
Studies have shown that in response to various hepatic cellular stresses, p38 MAPK promotes the progression of hepatic inflammation [13,14]. Overnutrition stimulates ERK1/2 pathway thereby stimulating the development of inflammation and fibrosis. It was reported that phosphorylated JNK1/2 plays a pro-inflammatory role in enhancing the production of inflammatory factors together with activation and infiltration of macrophages into the liver tissue [15]. The MAPK phosphatases (MKPs) counteract the MAPKs by direct dephosphorylation [16]. However, how the MKPs shift the balance towards MAPK phosphorylation to mediate the beneficial effects of fasting on obesity remains unclear.
In rodents, fasting has been reported to mitigate inflammation, fat mass and many components of metabolic disorders [17,18]. Intermittent fasting has been shown to decrease the expression levels of IL-6, IL-1β, and TNFα [17]. Other studies using the brain demonstrated that fasting and refeeding a high-fat diet reduces learning abilities by stimulating caspase-1 [19]. Whether fasting reduces and refeeding a high-fat enhances the activities of inflammatory mediators remains elusive [18]. It has been reported fasting induces differential regulation of p38 MAPK and ERK activation in different regions of the brain in response to energy metabolism [20]. On the other hand, fasting has been shown to increase the effectiveness of anti-cancer action of tyrosine kinase inhibitors by enhancing inhibition of MAPK signaling [21].
The goal of this study is to determine the effect of MKP-2 deficiency on hepatic metabolic response to fasting and refeeding-induced inflammatory conditions associated with poor eating habits. MKP-2 global knockout mice have been genetically characterized elsewhere [22].
Materials and Methods
Reagents and antibodies
All reagents were purchased from standard chemical vendors. The following antibodies phospho-p38 MAPK (#4511s), phospho-ERK1/2 (#9101s), p38 MAPK (#9228s), ERK1/2 (#4696s), beta-actin (#8457s), α-tubulin (#2125s), and UCP2 (#89326s), were obtained from Cell Signaling Technology. MKP-2 (#610850) was obtained from BD Biosciences.
Animal studies
The University of Alabama in Huntsville Institutional Animal Care and Use Committee approved all animal studies and experiments were conducted according to the NIH’s Guidelines on the Use of Laboratory Animals. MKP-2 global knockout mice have been genetically characterized elsewhere [22]. These mice were kindly provided by Dr. Robin Plevin, University of Strathclyde, United Kingdom. The fasting and refeeding experiments included six to seven male Mkp-2+/+ and Mkp-2-/- mice in each treatment group. The treatment groups included mice fed ad libitum with rodent lab diet (Lab supply, Nothlake, TX) or fasted for 48 h prior to sacrifice. For the refeeding experiments, Mkp-2+/+ and Mkp-2-/- mice were fasted for 48 h and refed either a chow (custom low fat purified rodent diet) or high-fat diet (#112252, Dyets, Bethlehem, PA) for 2 h ad libitum prior to sacrifice. We used eight to twelve weeks old male mice for all the fasting and refeeding experiments. All mouse works were conducted between 7:00 AM (light cycle) and 1:00 AM (dark cycle), including fasting, refeeding, and sacking mice. Consumption of large amounts of calories such as a high-fat diet leads to hyperglycemia and enhanced serum free fatty acids. Also, generally, rodents have the propensity to break their fasting by overeating high-calorie diets [23].
Therefore, refeeding a high-fat diet stimulates systemic inflammation.
RNA extraction and real-time quantitative PCR analysis
RNA was isolated from the liver tissue of Mkp-2+/+ and Mkp-2-/- mice using an RNeasy kit (Qiagen, CA, USA) according to the manufacturer’s instructions. A total of 1 μg RNA was reverse transcribed to generate cDNA using a reverse transcriptase PCR kit (Applied Biosystems, CA, USA). Real-time quantitative PCR was performed in triplicate with the Applied Biosystems 7500 Fast RT-PCR system and, SYBR Green gene expression master mix with the following primer pairs: IL-1β, 5’-GACGGACCCCAAAAGATGAAGG-3’ and 3’-GAGGTGCTGATGTACCAGTTGG-5’; GLUT2, 5’-ACGGTCTTCACGTTGGTCTC-3’ and 3’-GAAGGCCACAAAGCCAAATA-5’; TLR2, 5’-TGGCTCAAATCCTGGTTGAC-3’ and 3’-GAGAAGGGCACAGCAGACTC-5’; CXCL1, 5’-GCCTATCGCCAATGAGCTG-3’ and 3’-GAACCAAGGGAGCTTCAGG-5’; CXCL10, 5’-ATCCCGAGCCAACCTTC-3’ and 3’-CTTAGATTCCGGATTCAGACAT-5’; 18S, 5′- ACCGCAGCTAGGAATAATGGA-3′ and 3′-GCCTCAGTTCCGAAAACCA-5′.
All relative gene expression levels were analyzed using the ?Ct method and normalized to 18S. Gene expression master mix from Applied Biosystems were used. For genotyping, PCR analysis was employed using the following primers: WT forward primer 5’-CTTCAGACTGTCCCAATCAC-3’. WT reverse primer 5’-GACTCTGGATTTGGGGTCC-3’. KO forward primer 5’-TGACTAGGGGAGGAGTAGAAGGTGGC-3’. KO reverse primer 5’-ATAGTGACGCAATGGCATCTCCAGG- 3’.
Measurement of blood biochemical parameters and hepatic lipids
Male Mkp-2+/+ and Mkp-2-/- mice aged between 8 to 12 weeks old were used for the measurement of fasting plasma glucose concentrations by a glucometer (CareTouch Blood Glucose Monitoring System). Hepatic triglycerides and alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined using colorimetric assay kits (Cayman Chemical, Michigan) according to the manufacturer’s protocol.
Histological analysis of tissue samples
Liver tissues were isolated from male chow-fed and fasted male Mkp-2+/+ and Mkp-2-/- mice and then fixed in 4% paraformaldehyde in PBS and processed for paraffin sections and stained with hematoxylin and eosin.
Immunoblotting
Tissues were isolated from male Mkp-2+/+ and Mkp-2-/- mice at the end of the studies and snap frozen in liquid nitrogen and kept in – 80°C. Liver tissue was homogenized in RIPA buffer (25 mM Tris. HCl pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.1 % SDS, 1.0 % sodium deoxycholic acid), supplemented with protease and phosphatase inhibitors (5 μg/ml leupeptin, 5 μg/ml aprotinin, 1 μg/ ml pepstatin A, 1 mM PMSF, 1 mM benzamidine, 1 mM Na3VO3, and 10 mM NaF). Homogenates were lysed for 30 min on the shaker at 4°C prior to clarification at 20,800 g for 30 min at 4°C. Protein concentrations were determined by Pierce BCA Protein Assay kit (Pierce, Rockford, IL) [22]. Protein lysates were resolved by SDS-PAGE and transferred to nitrocellulose membranes, and then incubated with phospho-specific antibodies followed by enhanced chemiluminescence or fluorescent detection. All immunoblotting bands were quantified using Bio-Rad ImageLab software.
Statistical analysis
All data represent the mean ± SEM. Differences between groups were assessed using a student’s t-test or analysis of variance (ANOVA) with Bonferroni’s post-test for multiple comparisons using GraphPad Prism 9.5.1 statistical software.
Results
Metabolic response to refeeding HFD in MKP-2-deficient mice after 48-h fast
Mkp-2+/+ and Mkp-2-/- mice were fasted for 48 h and referred to either a chow or high-fat diet for 2 h to initially determine the effect of fasting and refeeding on body weight, liver weight, adipose tissue weight, spleen weight, and blood glucose levels. Mkp-2-/- mice exhibited comparable body weight in the fasted and refed for 2 h with HFD to those of Mkp-2+/+ mice (Figure 1A). Mkp-2-/- mice exhibited small but significant reduction in body weight in the fed state and when refed chow diet for 2 h compared with Mkp-2+/+ mice. (Figure 1A). The liver is the main component of the inflammatory response and the target organ for many dietary liver diseases [24]. The spleen is a key organ in controlling the immune response related to inflammation [25]. Further analysis showed that when fasted for 48 h, Mkp-2-/- mice displayed a small but significant decrease in liver weight compared with Mkp-2+/+ mice (Figure 1B). Because of the liver’s function in metabolism, and considering that the Mkp-2-/- mice body’s weight decreased under fasting (Figure 1A), their liver weights must equal in order for the liver to function properly. However, the liver weights between Mkp-2+/+ and Mkp-2-/- mice were comparable under fed and 2 h chow or HFD refed conditions (Figure 1B). No difference in blood glucose levels in the fed, fasted or 2 h refed chow or HFD in Mkp-2+/+ and Mkp-2-/- mice (Figure 1C). This is consistent with our previous studies, where chow-fed Mkp-2+/+ and Mkp-2-/- mice exhibited comparable fasting blood glucose levels [26]. When fasted for 48 h, Mkp-2-/- exhibited a small but significant increase in spleen weight compared with wild type controls (Figure 1D). This may be due to an increase in splenic proliferation. However, the spleen weights were similar in Mkp-2+/+ and Mkp-2-/- mice in fed, fasted or 2 h refed chow or HFD states (Figure 1D). Although Mkp-2-/- mice are leaner under fed and chow refed conditions (Figure 1A) this has no impact on white adipose tissue weight (Figure 1E). These findings suggest that MKP-2 deficiency results in reduced body weight and 48 h of fasting did not exacerbate the phenotype.
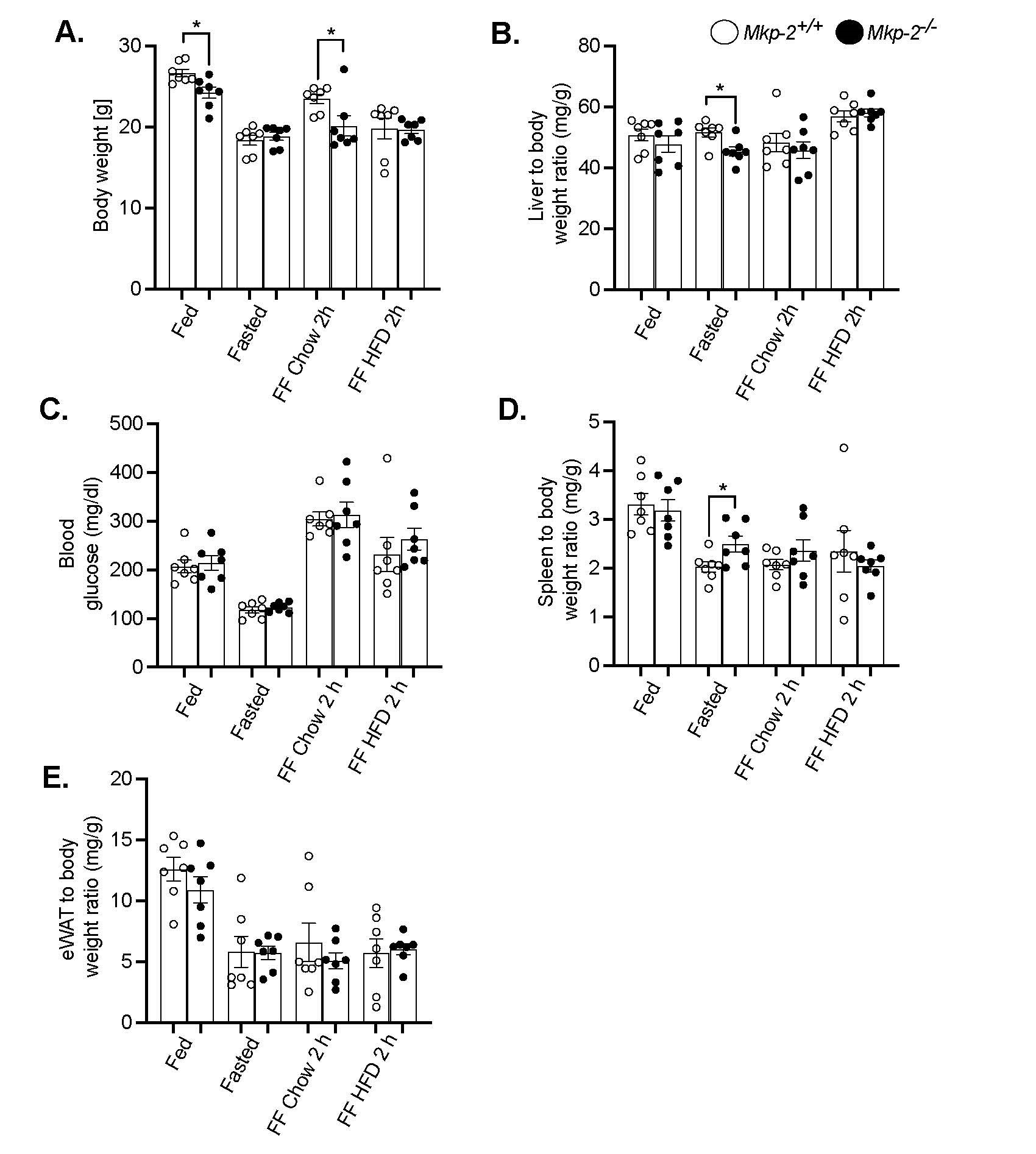
Figure 1. Metabolic response to refeeding a HFD in MKP-2-deficient mice after 48-h fast. (A) Body weight of fasted, chow- or high-fat diet fed male Mkp-2+/+ and Mkp-2-/- mice (B) Liver to body weight ratio (C) Fasting blood glucose (D) Spleen to body weight ratio (E) WAT body weight ratio. Mkp-2+/+ and Mkp-2-/- mice (n= 7 per genotype). Data are represented as mean ± SEM; *; p <0.05, as determined by analysis of variance (ANOVA) with Bonferroni’s post-test for multiple comparisons. FF; Refed. Open bars, Mkp-2+/+ mice; closed bars, Mkp-2-/- mice.
Fasting increases Mkp-2 expression in the livers of mice
By evaluating the levels of Mkp-2 mRNA expression in the livers of mice using fasting- refeeding paradigm, changes in expression were measured to determine whether Mkp-2 is metabolically regulated. Figure 2A shows an experiment in which Mkp-2+/+ were fasted for 48 h and refed for 2 h with either a chow or HFD diet. Liver tissues were assayed by quantitative real-time PCR for Mkp-2 mRNA expression. Mkp-2 mRNA expression levels were low under fed conditions (Figure 2A). However, Mkp-2 mRNA levels were significantly increased ~3 fold in liver after a 48-h fast (Figure 2A). Refeeding for 2 h with chow diet decreased Mkp-2 mRNA to below prefasting levels (Figure 2A). Interestingly, refeeding for 2 h with HFD after the 48-h fast increased hepatic Mkp-2 mRNA levels above the fed state levels, however, this was not statistically significant (Figure 2A). To validate the MKP-2 mRNA data we examined the protein expression of MKP-2 using immunoblotting under the same conditions. Consistent with the MKP-2 mRNA expression, MKP-2 protein expression significantly increased ~2 fold in the liver after a 48-h fast (Figures 2B and 2C). These data confirmed the MKP-2 mRNA expression. Additionally, the phosphorylation of p38 MAPK decreases concurrently with the rise in Mkp-2 levels during fasting (Figures 2D and 2F). These results demonstrate that Mkp-2 is induced during fasting in the livers of wild type mice and is associated with down-regulation of the activity of p38 MAPK.
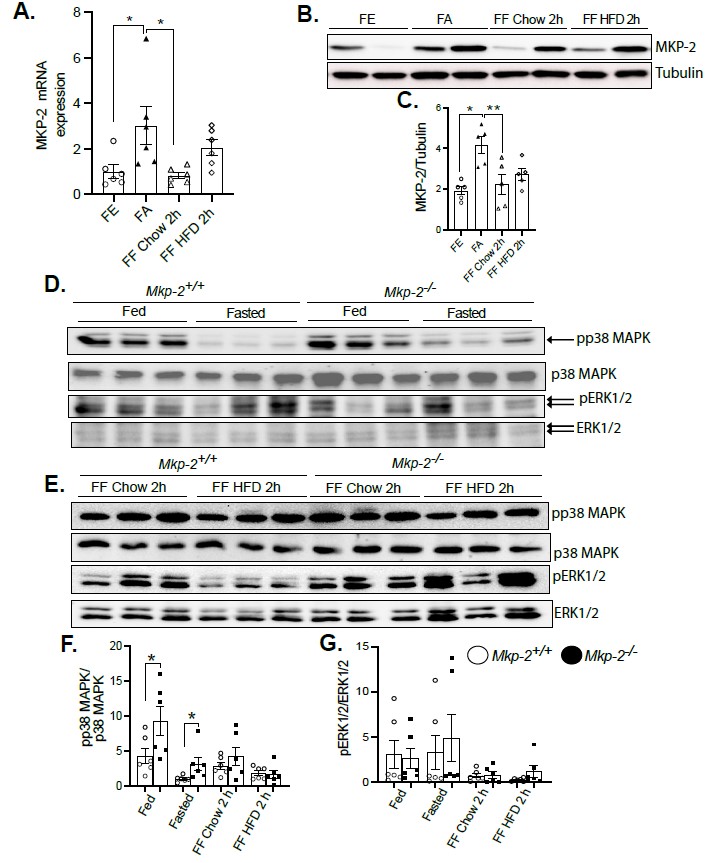
Figure 2. Fasting increases DUSP4 (MKP-2) expression in the livers of mice. mRNA and protein expression of hepatic MKP-2 from fasted and chow- and high-fat diet refed wild type livers. Liver lysates from fasted and chow- and high fat diet fed Mkp-2+/+ and Mkp-2-/- mice were analyzed by immunoblotting (B, C, D, E, F and G) MKP-2, pp38 MAPK, pERK1/2. Representative immunoblots were exposed to enhanced chemiluminescence (ECL) and quantitated by densitometry for the levels of MKP-2/β-actin (B and C) phospho-p38 MAPK/p38 MAPK, phospho-ERK1/2/ERK1/2. Results represent n = 5-6 per genotype and data shown are the mean ± SEM; *; p <0.05 as determined by analysis of variance (ANOVA) with Bonferroni’s post-test for multiple comparisons. FE: Fed; FA: Fasted; FF: Refed. Open bars, Mkp-2+/+ mice; closed bars, Mkp-2-/- mice.
Since fasting increased the expression of Mkp-2 in mouse livers, it was important to understand how fasting’s metabolic response affected the activation of MAPKs. Immunoblot analysis confirmed the deletion of MKP-2 in a major metabolic organ, liver. As expected, Mkp-2-/- mice exhibited >80% reduction in MKP-2 protein in the liver (Figure S1). We fasted Mkp-2-/- and Mkp-2+/+ mice for 48 h and refed for 2 h with either a chow or HFD diet. When MKP-2 levels rose in Mkp-2+/+ mice while they were fasting, p38 MAPK and but not ERK phosphorylation also reduced at the same time (Figures 2D-2G). In the fed state, we found enhanced hepatic phosphorylation of p38 MAPK but not ERK in Mkp-2-/- compared with Mkp-2+/+ mice (Figures 2D-2G). Refeeding a chow diet (2 h) after the 48 h fast returned hepatic phosphorylation of p38 MAPK in Mkp-2-/- mice to levels comparable to the non-fasted levels in Mkp-2+/+ (Figures 2D-2G), however, ERK phosphorylation returned to levels above the non-fasted levels (Figures 2D-2G). Furthermore, there was no difference in p38 MAPK and ERK phosphorylation between the two genotypes after refeeding with chow diet (2 h) (Figures 2D-2G). After 48 h fast, refeeding a HFD for 2 h reduced hepatic phosphorylation of p38 MAPK below non-fasted levels in both Mkp-2-/- and Mkp-2+/+ mice, however, ERK phosphorylation returned to above non-fasted levels (Figures 2D-2G). These findings show that fasting induces downregulation of hepatic p38 MAPK phosphorylation in Mkp-2+/+ mice, which is consistent with elevated hepatic MKP-2 expression. It appears that the effect of MKP-2 on p38 MAPK phosphorylation is more pronounced under basal conditions compared with fasting, suggesting under fasting that there may be MKP-2-independent pathways that regulate p38 MAPK. This is the first study to demonstrate that stress brought on by fasting increased MKP-2 expression and MAPK regulation in mouse livers.
Resistance to fasting-induced hepatic steatosis in MKP-2-deficient mice
Studies have shown that fatty acids transported to the liver serve as a substitute source of energy during fasting [27]. We next examine the effects of MKP-2 deficiency on fasting-induced hepatic steatosis. We fasted Mkp-2+/+ and Mkp-2-/- mice for 48 h. In the fed state, the livers of Mkp-2-/- and Mkp-2+/+ mice were comparable (Figure 3A, left panel). Interestingly, after 48 h fast, the livers of Mkp-2-/- mice displayed protection from the development of fasting-induced hepatic steatosis compared with Mkp-2+/+ control mice as analyzed by hematoxylin and eosin staining (Figure 3A, right panel). This is consistent with reduced liver weight in Mkp-2-/- mice (Figure 1B). Consistent with protection from hepatic steatosis, hepatic triglycerides (TGs) were significantly reduced in Mkp-2-/- compared with Mkp-2+/+ mice (Figure 3B). To determine the effects of fasting on liver injury in MKP-2-deficient mice, we examined the serum levels of AST and ALT. In the fed state, Mkp-2-/- exhibited reduced AST and ALT compared with Mkp-2+/+ mice (Figures 3C and 3D). Interestingly, fasted Mkp-2-/- mice displayed a dramatic reduction in serum levels of AST (~50%) and ALT (~70%) compared with Mkp-2+/+ mice (Fig. 3C and D). This is consistent with protection from hepatic steatosis observed in Mkp-2-/- mice. GLUT2 is the principal glucose transporter in humans and mice [28]. To determine the effect of MKP-2 deficiency on glucose uptake in the liver we measured the gene expression of GLUT2. In the fed state, Mkp-2-/- and Mkp-2+/+ mice showed comparable GLUT2 mRNA expression levels in the liver (Figure 4A). However, in the fasted state Mkp-2-/- exhibited significantly reduced hepatic GLUT2 mRNA expression levels compared with Mkp-2+/+ controls (Figure 4A). This suggests that the uptake of newly synthesized glucose in Mkp-2-/- mice was reduced thereby limiting substrates for hepatic lipid synthesis and hence protection from fat accumulation in the liver.
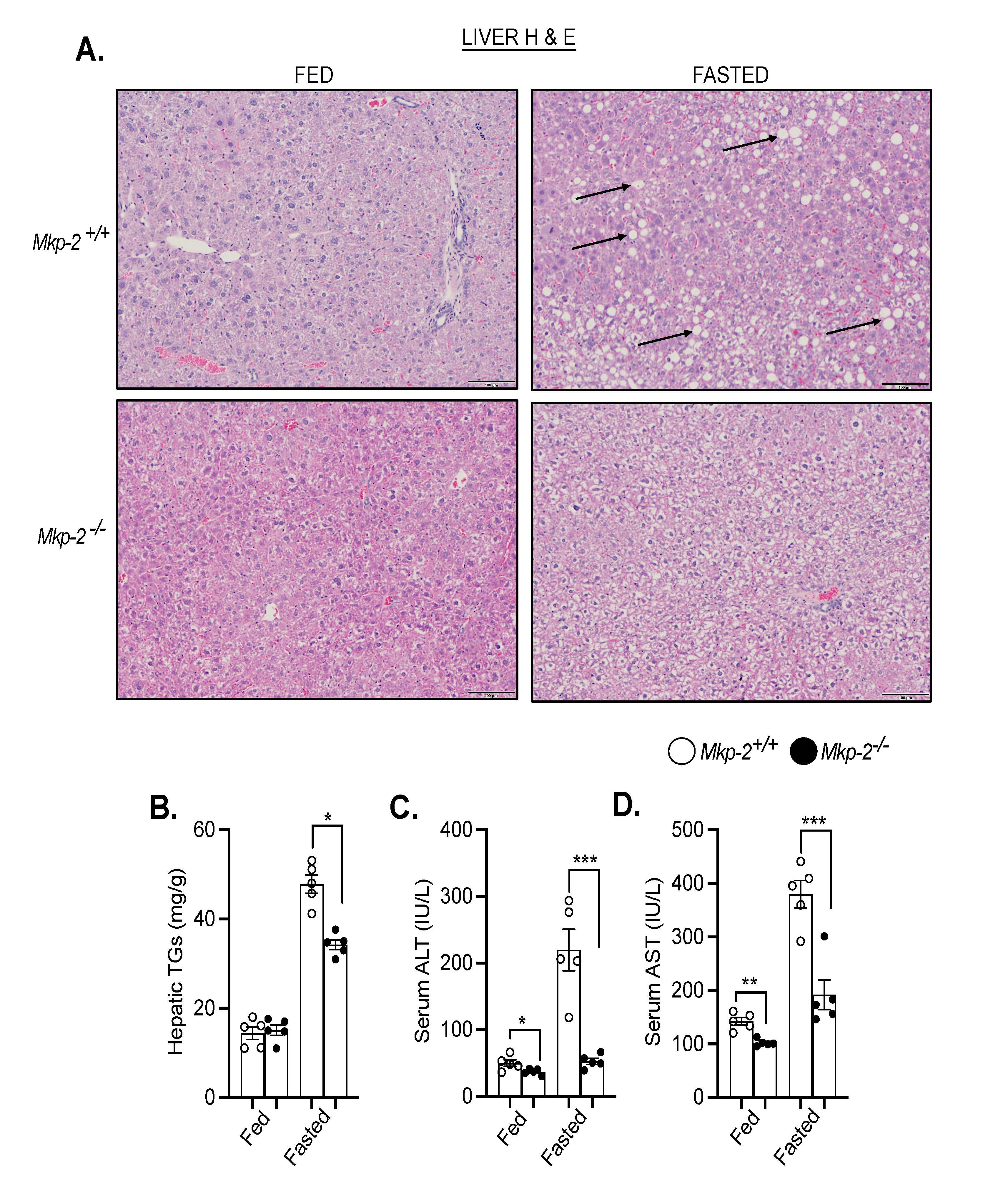
Figure 3. Protection from fasting-induced hepatic steatosis in MKP-2-deficient mice. Representative hematoxylin and eosin staining of liver sections of chow-fed (left panel) and fasted (right panel) Mkp-2+/+ and Mkp-2-/- mice (A) (n = 5 per genotype). Arrows show lipid droplets. (100 x Magnification). (B) Hepatic triglycerides (TG) (C) Serum alanine amino- transferase (ALT), (D) Serum aspartate aminotransferase (AST) from chow-fed and fasted Mkp-2+/+ and Mkp-2-/- mice (n = 5 per genotype). Data shown are the mean ± SEM; *; p <0.05,**; p <0.01, ***; p <0.0001, as determined by analysis of variance (ANOVA). Open bars, Mkp-2+/+ mice; closed bars, Mkp-2-/- mice.
One of the earliest responses of lipid metabolism to fasting is peripheral lipolysis, which explains the elevated serum levels of fatty acids [29]. A key regulator of lipolysis, the LIPE gene encodes hormone-sensitive lipase (HSL) [30]. The expression of LIPE mRNA in the EWAT of fed and fasted Mkp-2-/- and Mkp-2+/+ mice was measured using quantitative real- time PCR analysis in order to evaluate the lipolytic activity in adipose tissue. The weight of EWAT in fed and fasted Mkp-2-/- and Mkp-2+/+ mice were comparable (Figure 1E). The adipose LIPE mRNA levels in the fed state in Mkp-2+/+ and Mkp-2-/- mice were comparable (Figure 4B). However, after a 48 h fast, LIPE mRNA levels significantly increase in Mkp-2+/+ mice, however, in Mkp-2-/- mice, LIPE expression was significantly reduced compared with Mkp- 2+/+ controls (Figure 4B) suggesting reduced adipose lipolysis. Consequently, Mkp-2-/- mice exhibit protection from fasting-induced lipolysis (Figure 3). Next, we examined adipose triglyceride lipase (ATGL) known to be a critical regulator of lipolysis. The adipose ATGL mRNA levels in the fed state in Mkp-2+/+ and Mkp-2-/- mice were comparable (Figure 4C).
Consistent with the LIPE data, after 48 h fast, the expression of adipose ATGL was significantly reduced in Mkp-2-/- mice compared with Mkp-2+/+ counterparts (Figure 4C). We further examined the PLIN1 adipose mRNA levels. The adipose PLIN1 mRNA levels in the fed state in Mkp-2+/+ and Mkp-2-/- mice were comparable (Figure 4D). We found that PLIN1 was significantly reduced in Mkp-2-/- mice compared with Mkp-2+/+ mice after 48 h fast (Figure 4D). These findings demonstrate that MKP-2 deficient are protected from fasting-induced hepatic steatosis and that decrease in expression of key adipose lipolytic enzymes and hepatic GLUT2 contributes to the reduced fat accumulation in the liver and MKP-2 in part is an important regulator of these lipolytic genes.
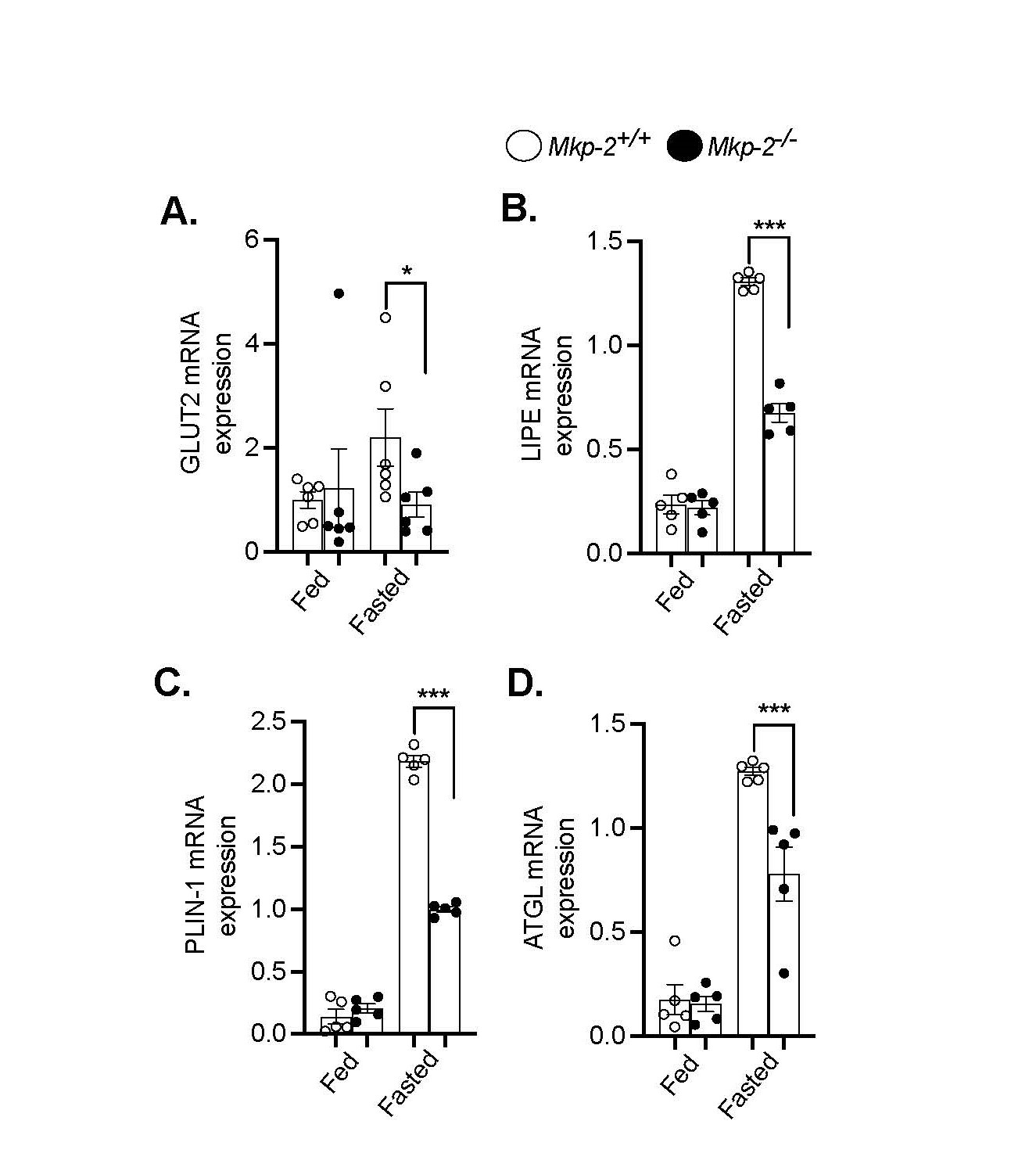
Figure 4. Reduced fasting-induced lipolysis in MKP-2-deficient mice. Hepatic mRNA expression of GLUT2 (A), Adipose mRNA expression of LIPE (B), PLIN1 (C) and ATGL (D) from chow-fed and fasted Mkp-2+/+ and Mkp-2-/- mice (n = 5 per geno- type). Data shown are the mean ± SEM; *; p <0.05, **; p <0.01, ***; p <0.0001, as determined by analysis of variance (ANOVA). Open bars, Mkp-2+/+ mice; closed bars, Mkp-2-/- mice.
Altered hepatic inflammatory gene expression in response to refeeding a HFD in MKP-2-deficient mice
Next, we sought to determine the effects of refeeding a chow or HFD on pro- and anti- inflammatory gene expression in the livers of MKP-2-deficient mice. Interleukin 1 beta (IL-1β) is involved in many inflammatory diseases including obesity and liver diseases [31]. One of the major families of pattern recognition receptors, toll-like receptors (TLRs) trigger signaling cascades that control inflammatory responses [32]. Chemokines play a part in coordinating the influx of immune cells to the site of damage or inflammation [33]. We fasted Mkp-2+/+ and Mkp-2-/- mice for 48 h. The levels of IL-1β was decreased in the fasted state compared with fed conditions in the livers of Mkp-2+/+ mice (Figure 5A). However, in the fasted state, hepatic IL-1β mRNA levels were significantly increased in Mkp-2-/- mice compared with Mkp-2+/+ mice (Figure 5A). Upon 2 h refed with chow or HFD, the livers of Mkp-2-/- mice displayed significantly elevated levels of IL-1β compared with Mkp-2+/+ mice (Figure 5A). The hepatic TLR2 mRNA levels in the fed, fasted or 2 h refed chow in Mkp-2+/+ and Mkp-2-/- mice were comparable (Figure 5B). However, following 2 h refed with HFD the levels of hepatic TLR2 mRNA levels were enhanced compared with prefasting levels and 2 h chow refed groups in the Mkp-2+/+ mice (Figure 5B). Although the hepatic TLR2 mRNA levels were reduced in Mkp-2-/- mice after refeeding with HFD for 2 h but this was not significant compared with Mkp-2+/+ mice (Figure 4B). Next, we examined the CXC chemokine family of proinflammatory mediators. Our analysis revealed that in the fed, fasted or 2 h refed with chow the mRNA levels of CXCL1 and CXCL10 were comparable in the livers of Mkp-2+/+ mice (Figures 5C and 5D). However, refeeding with a HFD decreased CXCL1 expression in Mkp-2+/+ mice (Figure 5C). The livers of Mkp-2-/- mice tended to reduce CXCL10 expression in the fed, fasted and 2 h refed with HFD compared with Mkp-2+/+ mice (Figure 5D). These findings imply that in response to refeeding a chow or HFD, MKP-2 exhibits differential regulation of hepatic inflammatory cytokines.
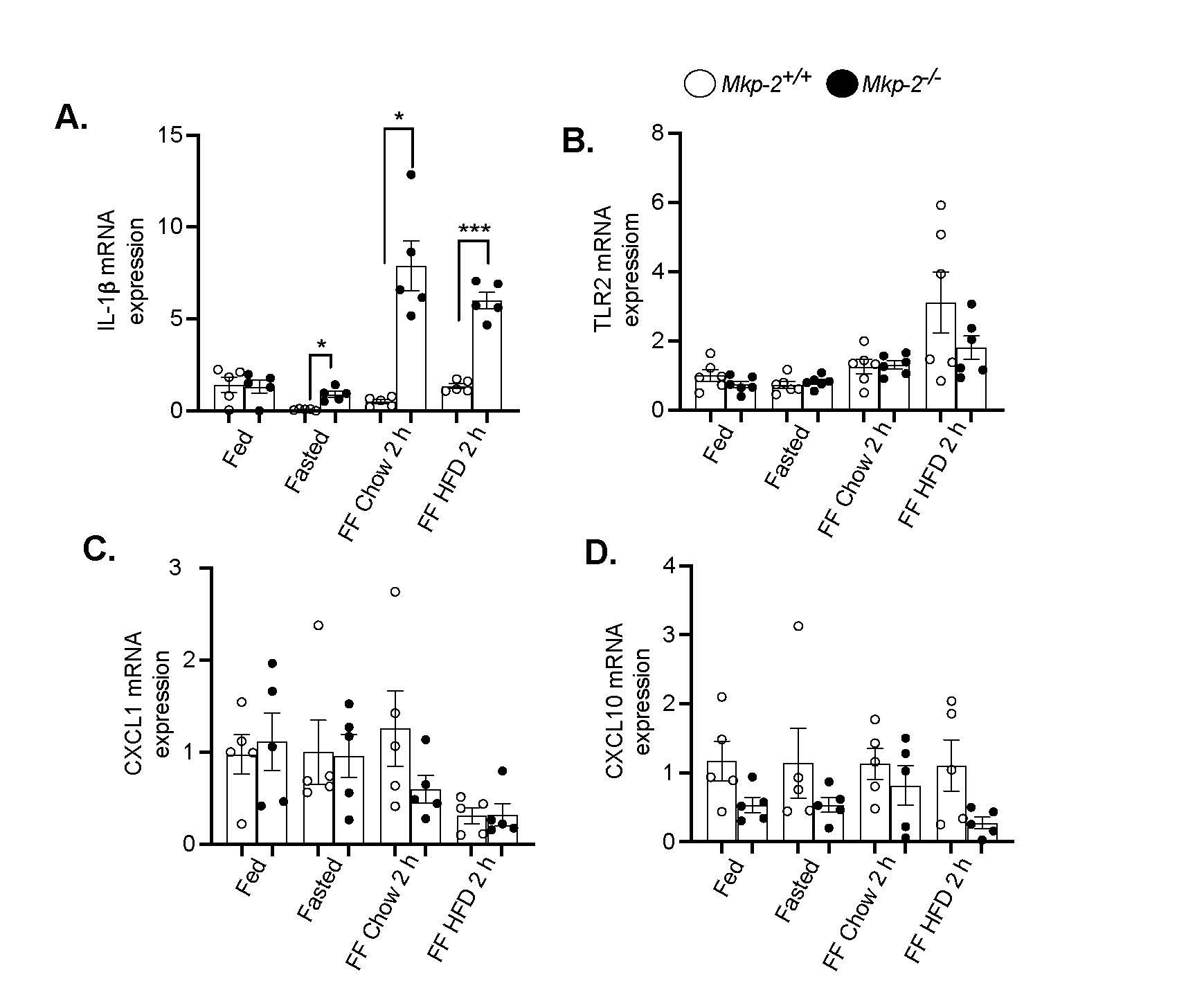
Figure 5. Altered expression of hepatic inflammatory genes in response to refeeding a HFD in MKP-2-deficient mice after 48-h fast. mRNA expression of hepatic inflammatory genes from fasted and chow- and high-fat diet refed Mkp-2+/+ and Mkp-2-/- mice, IL-1β (A), TLR2 (B), CXCL1 (C) CXCL10 (D) mRNA. Results represent n = 5-6 per genotype and data shown are the mean ± SEM; *; p <0.05, ***; p <0.0001 as determined by analysis of variance (ANOVA) with Bonferroni’s post-test for multiple comparisons. FF; Refed. Open bars, Mkp-2+/+ mice; closed bars, Mkp-2-/- mice.
MKP-2 deficiency reduced uncoupling protein-2 to mediate the altered expression of inflammatory cytokines
In order to understand the mechanism of altered expression of inflammatory cytokines in Mkp-2-/- mice, we determined the hepatic protein expression of uncoupling protein-2 (UCP- 2), a mitochondrial carrier protein and key player in stimulation of immune cells [34]. We fasted Mkp-2+/+ and Mkp-2-/- mice for 48 h and refed a chow or HFD for 2 h. In the fed, fasted, and 2 h chow or HFD refed states, we found comparable levels of UCP-2 protein expression levels in the livers of Mkp-2+/+ mice (Figures 6A-6C). Interestingly, Mkp-2-/- mice exhibited significantly reduced hepatic UCP-2 protein expression levels in the fed, fasted, and 2 h HFD refed compared with Mkp-2+/+ mice (Figures 6A-6C). Many cell types secrete cytokines and various studies demonstrated that this is dependent on the levels of UCP2 expression in these cells [35]. UCP2 knockout mice exhibited enhanced serum levels of IL-1β, IL-6 and interferon-γ (IFN-γ) in response to listerial infection [35]. In this study, we showed that increased IL-1β expression in the livers of Mkp-2-/- (Figure 5A) that is consistent with reduced UCP2 expression. Other studies have demonstrated that UCP2 knockout displayed enhanced resistance to bacterial infection [35]. These results indicate that MKP-2 modulates the expression of pro- and anti-inflammatory cytokines by negatively regulating UCP2 protein expression in the liver.
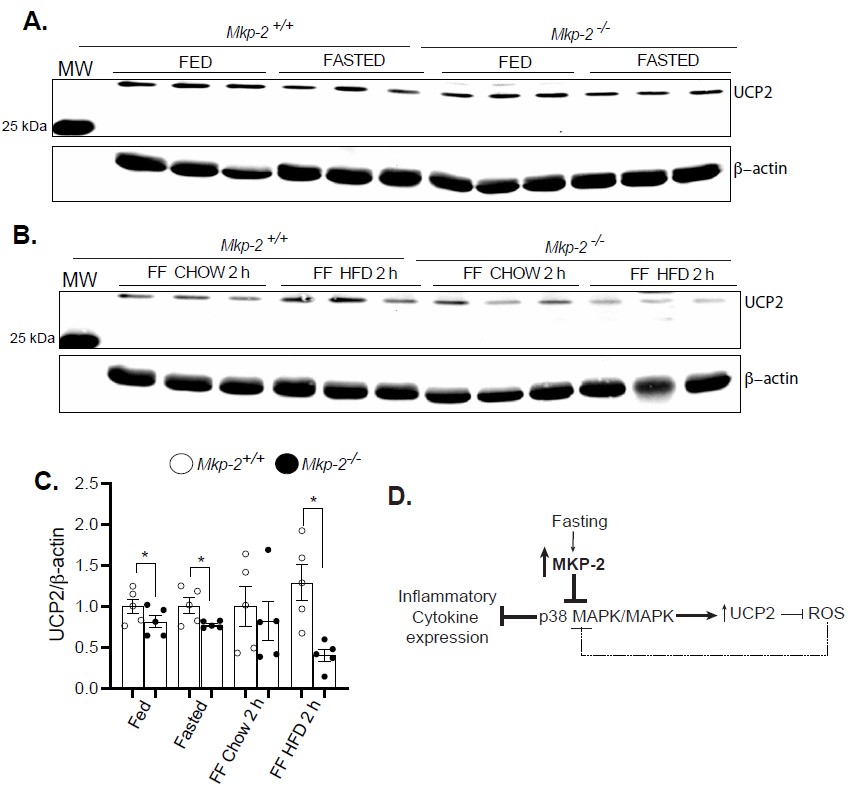
Figure 6. Reduced hepatic protein expression of UCP2 in response to refeeding a HFD in MKP-2-deficient mice after 48-h fast. Liver lysates from fasted and chow- and high-fat diet refed Mkp-2+/+ and Mkp-2-/- mice were analyzed by immunoblotting (A and B). Immunoblots were determined using fluorescent imaging and quantitated by densitometry for the levels of UCP2/β-actin (C). In the liver, fasting activates MKP-2 expression which serves to regulate p38 MAPK/UCP2-dependent pathways to control inflammatory cytokine expression. Activated p38 MAPK decreases UCP2 expression. As a feedback mechanism, UCP2 down-regulation enhances ROS-dependent p38 MAPK activation (D). Results represent n = 5 per genotype and data shown are the mean ± SEM; *; p <0.05, as determined by analysis of variance (ANOVA) with Bonferroni’s post-test for multiple comparisons. FF; Refed. Open bars, Mkp-2+/+ mice; closed bars, Mkp-2-/- mice.
Discussion
Previous work demonstrated that MKP-2 is upregulated in major insulin-responsive tissues in response to excess nutrients and plays an important role in liver metabolism [26]. Here, we evaluated the effect of MKP-2 deficiency on hepatic metabolic and inflammatory responses using fasting-refeeding paradigm. In this study fasting induced a significant increase in the expression of MKP-2 mRNA and protein, while refeeding with chow diet significantly attenuated the expression compared with HFD. After fasting, hepatic p38 MAPK phosphorylation was downregulated, which is consistent with increased Mkp-2 expression. Few other studies have demonstrated elevated ERK and p38 MAPK phosphorylation in the paraventricular nucleus and increased ERK phosphorylation in the hypothalamic arcuate nucleus of fasted wild type mice. Similar to this, in rat hepatocytes and macrophages, fasting led to ERK and JNK activation. Together, these findings show that fasting stress causes the expression of MKP-2 in the liver as well as selective activation and/or downregulation of p38 MAPK, ERK, and JNK in various tissues and cell types.
Although the immune system responds to insults by injury or infection, there are studies that suggest excess nutrients stimulate the immune system in metabolically active tissues such as the liver as part of the physiological response [7,36,37]. Also, it has been shown that MKP-2 is a negative regulator of JNK and p38 MAPK in macrophages and that it inhibits the expression of proinflammatory cytokines in response to LPS [38]. Our studies demonstrate that fasting activates MKP-2 expression that plays an important role in modulating the innate immune response by regulating the hepatic gene expression of inflammatory cytokines including IL-1β (Figure 5A). Interestingly, Mkp-2-/- mice exhibited reduced protein levels of UCP2 in the liver, which prompts us to discover a pathway by which MKP-2 negatively regulates IL-1β by antagonizing UCP2 in response to fasting and refeeding. Refeeding with either chow or HFD upregulated hepatic IL-1β expression in Mkp-2-/- mice. In addition, some studies have reported that fatty acids induce UCP2 expression [39,40] and here we showed reduced accumulation of fat in the fasted livers of Mkp-2-/- mice that is associated with decreased UCP2 protein expression. The mechanisms of how UCP2 increases IL-1β expression remain unclear. In this study we demonstrated for the first time that downregulation of UCP2 protein expression upregulated IL-1β levels in the liver suggesting that MKP-2 in part mediates the effects of UCP2 on IL-1β expression. Refeeding with an HFD-induced increased hepatic TLR2 expression in Mkp-2+/+ mice in comparison with Mkp- 2-/- mice refed with a chow diet. Table 1 showed that HFD contained higher cornstarch compared with chow diet in Table 2. This is consistent with a previous study that showed enhanced hepatic TLR2 in response to dietary carbohydrate [41]. However, this was attenuated in MKP-2 deficient mice. Similarly, TLR2 knockout mice exhibited reduced hepatic CXCL1 and CXCL10 [41] and this is consistent with decreased CXCL1 and CXCL10 in MKP-2 deficient mice [31]. It has been shown that activation of p38 MAPK in macrophages in response to LPS decreased UCP2 expression and increased production of mitochondrial ROS [42]. In a model of taxol-induced mitochondrial stress in human melanoma cells, another study showed enhanced p38 MAPK activation-mediated increase in mitochondrial ROS production [43]. Our data suggest a model in which fasting activates MKP-2 expression in the liver, which controls the expression of inflammatory cytokines via p38 MAPK/UCP2-dependent pathways (Figure 6D).
|
Ingredient |
kcal/gm |
grams/kg |
kcal/kg |
|
Casein |
3.58 |
200 |
716.00 |
|
L-Cystine |
4 |
3 |
12 |
|
Sucrose |
3.6 |
68.8 |
275.20 |
|
Cornstarch |
3.6 |
125 |
475.00 |
|
Dyetrose |
3.8 |
125.00 |
475.00 |
|
Soybean Oil |
9 |
25 |
225.00 |
|
t-Butylhydroquinone |
0 |
0.005 |
0 |
|
Cellulose |
0 |
50 |
0 |
|
Lard |
9 |
245 |
2205.00 |
|
Vitamin Mix #300050 |
3.92 |
10 |
39.20 |
|
Choline Bitartrate |
0 |
2 |
0 |
|
Salt Mix #210088 |
1.6 |
10 |
16.00 |
|
Dicalcium Phosphate |
0 |
13 |
0.00 |
|
Calcium Carbonate |
0 |
5.5 |
0.00 |
|
Potassium Citrate H20 |
0 |
16.5 |
0.00 |
|
|
|
773.805 |
3963.400 |
|
Ingredient |
kcal/gm |
grams/kg |
kcal/kg |
|
Casein |
3.58 |
200 |
716 |
|
L-Cystine |
4 |
3 |
12 |
|
Sucrose |
4 |
350 |
1400 |
|
Cornstarch |
3.6 |
315 |
1134 |
|
Dyetrose |
3.8 |
35 |
133 |
|
Soybean Oil |
9 |
25 |
225 |
|
t-Butylhydroquinone |
0 |
0.005 |
0 |
|
Lard |
9 |
20 |
180 |
|
Cellulose |
0 |
50 |
0 |
|
Mineral Mix #210088 |
1.6 |
10 |
16 |
|
Dicalcium Phosphate |
0 |
13 |
0 |
|
Calcium Carbonate |
0 |
5.5 |
0 |
|
Potassium Citrate H2O |
0 |
16.5 |
0 |
|
Vitamin Mix # 300050 |
3.92 |
10 |
39.2 |
|
Choline Bitartrate |
0 |
2 |
0 |
|
|
|
1055.005 |
3855.2 |
Metabolic syndrome is closely associated with the development of hepatic steatosis [3,44]. The dysregulation of fatty acid metabolism by the liver is mainly responsible for the accumulation of fat in the liver. Our data demonstrate that MKP-2-deficient mice were protected from fasting-induced hepatic steatosis. Furthermore, reduced liver damage was observed in fasted MKP-2-deficient mice as evident by lower serum ALT and AST levels. Our results reveal that changes in hepatic fat metabolism that resulted in resistance to hepatic steatosis in MKP- 2-deficient mice impact glucose uptake in the liver. Mkp-2-/- mice exhibited reduced GLUT2 hepatic gene expression, a key glucose transporter in hepatocytes. This suggests that the reduced accumulation of fat in the liver stimulated decreased glucose uptake through reduced GLUT2 expression thereby limiting glucose as substrate for processes such as lipogenesis.
More importantly, Mkp-2-/- mice exhibited reduced adipose expression of key lipolytic genes LIPIE, ATGL and PLIN1. The decreased lipolysis in fasted Mkp-2-/- mice results in reduced hepatic absorption of fatty acids, which explains protection in hepatic steatosis. All of the research done points to the possibility that blocking MKP-2 could offer a fresh approach to treating metabolic disease. Anti-sense strategies for targeted inhibition of hepatic MKP-2 in obese patients may be useful in treating hepatic steatosis. Whether MKP-2 is overexpressed in human type 2 diabetes is still unknown. We showed recently that hepatic MKP-2 protein expression levels are upregulated in obese/NASH humans [26], indicating that there is a relationship between MKP-2 expression and fat mass. Furthermore, it is unclear which tissue serves as a major conduit for MKP-2 impact on metabolism. Metabolic analysis of MKP-2 tissue-specific deleted mice will be necessary for future work.
Conclusions
Our findings showed that the hepatic inflammatory response to fasting is altered by MKP-2 deficiency. These results lend credence to the idea that upregulation of MKP-2 is a physiologically relevant response and may help the liver better utilize hepatic lipids while fasting. According to these findings, MKP-2 expression is increased in response to alterations in nutritional cues, such as fasting and an abundance of nutrients. Upregulation of MKP-2 while fasting would enhance lipolysis and promote the development of fatty liver disease. This would offer therapeutic intervention strategies for the management of fatty liver disease.
Acknowledgements
A.L. is supported by UAH Faculty Startup and New Faculty Research Funding Program. K.M is supported by UTEP Faculty Startup Award.
Disclosures
No potential conflicts of interest relevant to this article were reported.
Author Contributions
A.L. conceived, designed, and wrote the manuscript. S.J, G.W., J.S., I.S, N.G., C.S., M.W., and U.P. performed experiments. S.J. and A.L. analyzed data. K.M. edited, designed, and revised the manuscript.
References
2. Andolfi C, Fisichella PM. Epidemiology of obesity and associated comorbidities. Journal of Laparoendoscopic & Advanced Surgical Techniques. 2018 Aug 1;28(8):919-24.
3. Lawan A, Bennett AM. Mitogen-activated protein kinase regulation in hepatic metabolism. Trends in Endocrinology & Metabolism. 2017 Dec 1;28(12):868-78.
4. Beyerlein A, Toschke AM, Schaffrath Rosario A, von Kries R. Risk factors for obesity: further evidence for stronger effects on overweight children and adolescents compared to normal-weight subjects. PloS one. 2011 Jan 20;6(1):e15739.
5. Ard J. Obesity in the US: what is the best role for primary care?. BMJ. 2015 Feb 5;350:g7846.
6. Ludwig DS, Sørensen TI. An integrated model of obesity pathogenesis that revisits causal direction. Nature Reviews Endocrinology. 2022 May;18(5):261-2.
7. Margină D, Ungurianu A, Purdel C, Tsoukalas D, Sarandi E, Thanasoula M, et al. Chronic inflammation in the context of everyday life: dietary changes as mitigating factors. International Journal of Environmental Research and Public Health. 2020 Jun;17(11):4135.
8. Westenberger G, Sellers J, Fernando S, Junkins S, Han SM, Min K, et al. Function of mitogen-activated protein kinases in hepatic inflammation. Journal of Cellular Signaling. 2021;2(3):172-80.
9. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. Journal of hepatology. 2019 Jan 1;70(1):151-71.
10. Trefts E, Gannon M, Wasserman DH. The liver. Current Biology. 2017 Nov 6;27(21):R1147-R1151.
11. Peng WC, Logan CY, Fish M, Anbarchian T, Aguisanda F, Álvarez-Varela A, et al. Inflammatory cytokine TNFα promotes the long-term expansion of primary hepatocytes in 3D culture. Cell. 2018 Nov 29;175(6):1607-19.
12. Yang YM, Kim SY, Seki E. Inflammation and Liver Cancer: Molecular Mechanisms and Therapeutic Targets. Seminars in Liver Disease. 2019 Feb;39(1):26-42.
13. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nature Reviews Molecular Cell Biology. 2021 May;22(5):346-66.
14. Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2007 Aug 1;1773(8):1358-75.
15. Li W, Yang GL, Zhu Q, Zhong XH, Nie YC, Li XH, et al. TLR4 promotes liver inflammation by activating the JNK pathway. European Review for Medical & Pharmacological Sciences. 2019 Sep 1;23(17):7655-62.
16. Lawan A, Shi H, Gatzke F, Bennett AM. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cellular and Molecular Life Sciences. 2013 Jan;70:223-37.
17. Hillgartner FB, Salati LM, Goodridge AG. Physiological and molecular mechanisms involved in nutritional regulation of fatty acid synthesis. Physiological Reviews. 1995 Jan 1;75(1):47-76.
18. DeBose-Boyd RA. Significance and regulation of lipid metabolism. Seminars in Cell & Developmental Biology. 2018 Sep;81:97.
19. Towers AE, Oelschlager ML, Juda MB, Jain S, Gainey SJ, Freund GG. HFD refeeding in mice after fasting impairs learning by activating caspase-1 in the brain. Metabolism. 2020 Jan 1;102:153989.
20. Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, et al. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. The Journal of Clinical Investigation. 1998 Jan 1;101(1):1-9.
21. Shimomura I, Shimano H, Korn BS, Bashmakov Y, Horton JD. Nuclear sterol regulatory element-binding proteins activate genes responsible for the entire program of unsaturated fatty acid biosynthesis in transgenic mouse liver. Journal of Biological Chemistry. 1998 Dec 25;273(52):35299-306.
22. Lawan A, Zhang L, Gatzke F, Min K, Jurczak MJ, Al-Mutairi M, et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Molecular and Cellular Biology. 2015; 35(1):26-40.
23. Acosta-Rodríguez VA, de Groot MH, Rijo-Ferreira F, Green CB, Takahashi JS. Mice under caloric restriction self-impose a temporal restriction of food intake as revealed by an automated feeder system. Cell Metabolism. 2017 Jul 5;26(1):267-77.
24. Baeck C, Tacke F. Balance of inflammatory pathways and interplay of immune cells in the liver during homeostasis and injury. EXCLI journal. 2014;13:67-81.
25. Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013 Nov 14;39(5):806-18.
26. Fernando S, Sellers J, Smith S, Bhogoju S, Junkins S, Welch M, et al. Metabolic Impact of MKP-2 Upregulation in Obesity Promotes Insulin Resistance and Fatty Liver Disease. Nutrients. 2022 Jun 15;14(12):2475.
27. Chen J, Vitetta L. Gut microbiota metabolites in NAFLD pathogenesis and therapeutic implications. International Journal of Molecular Sciences. 2020 Jul 23;21(15):5214.
28. Tomaz LM, Barbosa MR, Farahnak Z, Lagoeiro CG, Magosso NS, Lavoie JM, et al. GLUT2 proteins and PPARγ transcripts levels are increased in liver of ovariectomized rats: reversal effects of resistance training. Journal of Exercise Nutrition & Biochemistry. 2016 Jun;20(2):51-7.
29. Jensen MD, Haymond MW, Gerich JE, Cryer PE, Miles JM. Lipolysis during fasting. Decreased suppression by insulin and increased stimulation by epinephrine. The Journal of Clinical Investigation. 1987 Jan 1;79(1):207-13.
30. Recazens E, Mouisel E, Langin D. Hormone-sensitive lipase: sixty years later. Progress in Lipid Research. 2021 Apr 1;82:101084.
31. Donath MY. When metabolism met immunology. Nature Immunology. 2013 May;14(5):421-2.
32. Kuo LH, Tsai PJ, Jiang MJ, Chuang YL, Yu L, Lai KT, et al. Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia. 2011 Jan;54:168-79.
33. Boro M, Balaji KN. CXCL1 and CXCL2 regulate NLRP3 inflammasome activation via G-protein–coupled receptor CXCR2. The Journal of Immunology. 2017 Sep 1;199(5):1660-71.
34. Emre Y, Nübel T. Uncoupling protein UCP2: when mitochondrial activity meets immunity. FEBS letters. 2010 Apr 16;584(8):1437-42.
35. Rousset S, Emre Y, Join-Lambert O, Hurtaud C, Ricquier D, Cassard-Doulcier AM. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine. 2006 Aug 1;35(3-4):135-42.
36. Kodra AL, Mucida D. To eat or not to eat: type 2 immunity controls food avoidance behavior. Trends in Immunology. 2023 Aug 15; 44(9):665-7.
37. de Carvalho Ribeiro M, Szabo G. Role of the inflammasome in liver disease. Annual Review of Pathology: Mechanisms of Disease. 2022 Jan 24;17:345-65.
38. Al-Mutairi MS, Cadalbert LC, McGachy HA, Shweash M, Schroeder J, Kurnik M, et al. MAP kinase phosphatase-2 plays a critical role in response to infection by Leishmania mexicana. PLoS pathogens. 2010 Nov 11;6(11):e1001192.
39. Hauton D, Evans R. Utilisation of fatty acid and triacylglycerol by rat macrophages: the effect of endotoxin. Cellular Physiology and Biochemistry. 2002 Nov 20;12(5-6):293-304.
40. Newsholme P, Procopio J, Lima MM, Pithon‐Curi TC, Curi R. Glutamine and glutamate—their central role in cell metabolism and function. Cell Biochemistry and Function. 2003 Mar;21(1):1-9.
41. Oarada M, Takahashi-Nakaguchi A, Abe T, Nikawa T, Miki T, Gonoi T. Refeeding with glucose rather than fructose elicits greater hepatic inflammatory gene expression in mice. Nutrition. 2015 May 1;31(5):757-65.
42. Emre Y, Hurtaud C, Nübel T, Criscuolo F, Ricquier D, Cassard-Doulcier AM. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochemical Journal. 2007 Mar 1;402(2):271-8.
43. Selimovic D, Hassan M, Haikel Y, Hengge UR. Taxol-induced mitochondrial stress in melanoma cells is mediated by activation of c-Jun N-terminal kinase (JNK) and p38 pathways via uncoupling protein 2. Cellular Signalling. 2008 Feb 1;20(2):311-22.
44. Jensen-Cody SO, Potthoff MJ. Hepatokines and metabolism: Deciphering communication from the liver. Molecular Metabolism. 2021 Feb 1;44:101138.