Abstract
Obesity has reached a global epidemic and it predisposes to the development of insulin resistance, type 2 diabetes and related metabolic diseases. Current interventions against obesity and/or type 2 diabetes such as calorie restriction, exercise, genetic manipulations or established pharmacological treatments have not been successful for many patients with obesity and/or type 2 diabetes. There is an urgent need for new strategies to treat insulin resistance, T2D and obesity. Increased activity of stress-responsive pathways has been linked to the pathogenesis of insulin resistance in obesity. In this commentary, we argue that chronic upregulation of MKP-1 in skeletal muscle is part of a stress response that contributes to the development of insulin resistance, T2D and obesity. Therefore, inhibition of MKP-1 in skeletal muscle is a potential strategy for the treatment of T2D and obesity. We highlight therapeutic strategies for potential targeting of MKP-1 in skeletal muscle for the treatment of metabolic diseases as well as other diseases of skeletal muscle.
Keywords
MAP kinase, MAP kinase phosphatase, Obesity, Insulin resistance
Introduction
Obesity is a major public health problem globally. It develops as a result of an imbalance between energy intake and expenditure, and predisposes to the development of multiple diseases including insulin resistance, type 2 diabetes (T2D), nonalcoholic fatty liver disease (NAFLD), cardiovascular disease and some types of cancers [1-4]. Skeletal muscle constitutes about 30-40% of total body mass in healthy humans. Skeletal muscle is the major tissue involved in the control of energy balance and glucose metabolism. The generation, maintenance, composition and repair of skeletal muscle is considered to be an important aspect of metabolic homeostasis [5,6]. Dysfunctional energy metabolism in skeletal muscle has been attributed to the development of insulin resistance, glucose intolerance, T2D and obesity [7,8]. Myokines, are muscle-derived secretory molecules that mediate crosstalk between skeletal muscle and other organs/systems including the liver, adipose tissue, bone, pancreas and cardiovascular system [9]. Thus, maintenance of healthy skeletal muscle impacts virtually all organ systems in the body. Disruption of skeletal muscle function leads to numerous types of diseases including muscular dystrophy, loss of muscle mass (atrophy), and muscular hypertrophy [10-12]. Skeletal muscle is the main source of animal protein for human consumption, and the growth and development of skeletal muscle directly impacts animal meat quality and quantity [13]. Exercise has been the main benefit used by space agencies in order to protect skeletal muscle health while in space for prolonged periods of time [14,15]. Because skeletal muscle plays such a major role in metabolism the benefits of exercise have been shown to promote the actions of insulin and improve overall metabolic indicators. Thus, targeting skeletal muscle in cases of correcting metabolic dysfunction such as T2D could be considered a rational strategy.
The signal transduction pathways that govern metabolic regulation in skeletal muscle are extremely complex. Here we will focus on the regulation of the mitogenactivated protein kinase (MAPK) pathway in skeletal muscle. The MAPKs are a family of serine/threonine kinases that have been shown to be directly involved in skeletal muscle metabolism. The MAPKs are activated by direct phosphorylation by their upstream MAPK kinases (MKKs). In contrast, the inactivation of the MAPKs is achieved by direct dephosphorylation on both regulatory threonine and tyrosine residues by the MAPK phosphatases (MKPs) [16]. The MKPs are a sub-family of enzymes known as dual-specificity phosphatases (DUSPs) that belong to a larger superfamily known as protein tyrosine phosphatases (PTPs). The archetypal MKP, MKP-1 has been studied extensively in a variety of cellular and biological systems. MKP-1 is abundantly expressed in skeletal muscle and is a critical negative regulator of p38 MAPK, c-Jun NH2 terminal kinase (JNK) and to a lesser extent, ERK activities [12,17,18]. We previously showed that MKP-1 is an important regulator of MAPK-dependent regulation of lipid homeostasis, energy metabolism, and mitochondrial biogenesis [16,18]. Work from this group using skeletal muscle-specific deletion of MKP-1 uncovered an important role of MKP-1 in this tissue. We demonstrated a major contribution of skeletal muscle MKP-1 in the regulation of glucose metabolism and energy homeostasis [18]. One of the most interesting observations of this work was the observation that MKP-1 is increased in its level of expression in skeletal muscle of obese humans [18]. These results, along with others that will be discussed here suggested that MKP-1 forms part of an important stress response that leads to reduced energy expenditure in skeletal muscle thereby contributing to weight gain. A paradigm consistent with the idea that stress positively correlates with obesity. Furthermore, it was discovered that MKP-1 participates through MAPK in crosstalk mechanisms with other tissues such as the liver to promote abnormalities in glucose metabolism and hepatic steatosis associated with metabolic disease [18]. Although MKP-1 expression has not been well studied in human obesity there is one study that reported increased MKP-1 expression in adipose tissue and macrophages in obese humans [19]. Our data is the first study that reported increased expression of MKP-1 in skeletal muscle of obese humans with concomitant dephosphorylation of p38 MAPK [18]. These findings are of significance because it implies that targeting skeletal muscle MKP-1 could be beneficial to the treatment of insulin resistance, T2D and obesity.
Role of MKP-1 in Biological Processes and Disease Development
Contribution of skeletal muscle MKP-1 in obesity and insulin resistance
MAPK signaling is necessary for the maintenance of skeletal muscle mass, regenerative myogenesis and muscle atrophy [12,20,21] and promotion of obesity-induced insulin resistance [22]. Overexpression of MKP-1 inhibits the expression and activity of PGC1a, a master regulator of mitochondrial biogenesis and energy expenditure, by impairing p38 MAPK-mediated PGC1a phosphorylation [17,18] (Figure 1). Further studies showed impaired muscle regeneration, reduced body weight, muscle mass, muscle cross-sectional area and exacerbated myopathy in MKP-1- deficient mice [12].
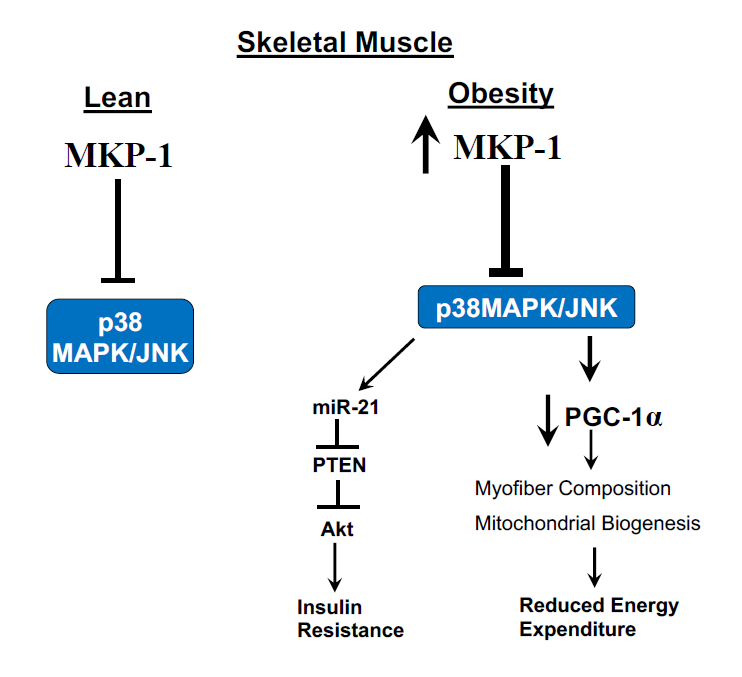
Figure 1: Model for MKP-1 regulation of MAPK pathway in obese skeletal muscle. MKP-1 regulate the activities of p38 MAPK/JNK pathway. In obesity, MKP-1 is upregulated, which inhibit p38 MAPK thereby impairing PCG1α activity leading to decreased Mitochondrial function and reduced energy expenditure. Overexpression of MKP-1 also leads to development of insulin resistance by controlling the activation of p38 MAPK/JNK through the miR-21/PTEN/Akt pathway.
We showed that MKP-1 when deleted specifically in skeletal muscle relieves the inhibition of both JNK and p38 MAPK. Remarkably, instead of enhanced JNK/p38 MAPK activity driving obesity and insulin resistance as previous studies suggested [23], these mice are resistant to diet-induced obesity and are insulin sensitive [18]. Significantly, MKP-1 is upregulated in high-fat diet feeding in mice [17] and in obese in human subjects [18]. These data suggest that MKP-1 upregulation contributes to the development of obesity and insulin resistance by antagonizing the JNK/p38 MAPK signaling module. We found that enhanced p38 MAPK/JNK activities increased miR-21 expression in skeletal muscle lacking MKP-1, leading to reduced PTEN expression, thereby upregulating Akt activity. These results indicate that inhibition of MKP- 1 in skeletal muscle could promote insulin sensitivity in part by MKP-1 mediated upregulation of Akt through a MAPK/miR-21/PTEN pathway (Figure 1). Further work is needed to substantiate the human data and definitively prove whether these findings in mice are recapitulated in humans. Here, using tissue-specific approaches the interpretation that the negative effects of skeletal muscle MKP-1 ultimately prevail in the development of obesity and insulin resistance has been established. This work shifts the entire idea that progression of obesity and insulin resistance as it pertains to JNK/p38 MAPK is simply a consequence of enhanced activity of these MAPKs. These findings raise the possibility that targeting MKP-1 in skeletal muscle may provide therapeutic potential for the treatment of obesity, insulin resistance and T2D.
Skeletal muscle MKP-1 and its contribution in liver diseases
There is a growing body of literature that focuses on the role of skeletal muscle in the development of liver disease. Studies have shown impaired skeletal muscle function in NAFLD patients [24] and in chronic liver diseases including cirrhosis [25,26]. Whether the damage of skeletal muscle tissue is causal, a provoking factor or an effect of the chronic liver disease remains to be established. Also, the mechanism by which skeletal muscle affects liver diseases or whether alterations in skeletal muscle function contribute to the progression of liver disease is not known. We previously showed that mice lacking MKP-1 in the liver (MKP1-LKO) results in altered expression of hormones and cytokines along with resistance to hepatic steatosis [27]. Additionally, MKP1-LKO mice exhibit reduced mitochondrial function suggesting that deleting MKP-1 in the liver might inhibit skeletal muscle function possibly as a result of crosstalk that occurs between the liver and skeletal muscle tissue through a hepatic MKP-1-dependent pathway. Conversely, showed resistance to hepatic steatosis in mice lacking expression of MKP-1 in skeletal muscle. One potential mechanism for this crosstalk is the effect of myokines secreted from the skeletal muscle that mediate hepatic lipid metabolism. The exact molecular mechanism for this relationship has yet to be investigated.
Skeletal muscle MKP-1 in mitochondrial function, sarcopenia and aging
Sarcopenia is defined by the gradual and extensive loss of skeletal muscle mass, strength and function [28]. It is a common complication observed in about 70% of liver cirrhosis patients [29,30] and it affects almost all elderly people [31]. The underlying mechanisms for the development of sarcopenia and the effective treatment for sarcopenia have not been discovered. Exercise is known to slow the progression of sarcopenia where it partially improves mitochondrial biogenesis and protein turnover. Sarcopenia is associated with increased obesity in the elderly. A decline in the proportion of type I myofibers, which are mitochondria rich, is observed in obese patients and the proportion of type I myofibers positively correlates with overall metabolic health [7-11,32]. Thus, the predominant view reflects the notion that the levels of oxidative myofiber composition negatively correlates with the development of metabolic syndrome. Indeed, this is consistent with the concept proposed here suggesting that skeletal muscle MKP-1 overexpression promotes insulin resistance and obesity by reducing oxidative myofiber composition (Figure 1).
The stress-responsive MAPKs control processes such as insulin signaling, glucose homeostasis, fatty acid metabolism and energy expenditure [33,34]. Mice with skeletal muscle-specific deletion of JNK1 are unaffected by diet-induced obesity but are insulin sensitive [35]. p38 MAPK has been shown to stimulate glucose uptake in muscle cells [36] and constitutive p38 MAPK activation in skeletal muscle promotes mitochondrial biogenesis [37]. Although it is realized that metabolic stressors such as inflammation and nutrient excess activate both p38 MAPK and JNK [38,39], the results of these studies reflect the individual actions of these MAPKs on metabolism. The studies described here on MKP-1 in skeletal muscle represent the integrated response of antagonizing these stress-responsive MAPKs on metabolism which has revealed the importance of MKP-1 as a crucial regulator in the progression of metabolic disease.
One of the main reasons for the actions of MKP-1/MAPK in skeletal muscle metabolism relates to the fact that this pathway regulates energy expenditure through controlling the composition of oxidative and glycolytic muscle fibers. By examining the composition of skeletal muscle myofibers we found that there was an increase in the proportion of slow oxidative myofibers and reduction in the proportion of fast glycolytic myofibers in skeletal muscle derived from mice lacking MKP-1 expression in this tissue [18]. Thus, our results imply that changes in myofiber type composition could contribute to the enhanced oxidative capacity and reduced glycolytic capacity of skeletal muscles in mice lacking skeletal muscle MKP-1. Mitochondrial oxidation, ATP synthesis, and myofiber type composition contribute to the control of skeletal muscle endurance [40,41]. Skeletal muscle-specific MKP-1-deficient mice were observed to exhibit significantly increased levels of endurance which, further supports the interpretation that the oxidative myofiber composition of these skeletal muscles is improved. These observations provide an explanation for the increased levels of mitochondrial efficiency and increased whole-body energy expenditure in skeletal muscle-specific MKP-1-deficient mice and thus, resistance to diet-induced obesity. These results suggest that skeletal muscle MKP-1 through a MAPK dependent pathway(s) modulates mitochondrial biogenesis and subsequently influences whole body energetics. Impaired mitochondrial biogenesis and protein turnover has been reported to promote the development of sarcopenia [30]. Therefore, overexpression of skeletal muscle MKP-1 would be anticipated to promote age- and diet-induced stresses on mitochondrial dysfunction and contribute to the progression of sarcopenia.
MKP-1 in Cardioprotection
Heart disease and its associated complications including heart failure, atrial fibrillation and myocardial ischemia and infarction are leading causes of death and disability globally [42,43]. The molecular basis for the development and progression of cardiac diseases are not completely clear. Reperfusion is critical in rescuing the ischemic myocardium from infarction and cardiomyocyte death [43,44]. Insulin protects cardiac myocytes [45,46] while oxidative stress [44] has been implicated in ischemia-reperfusion induced cardiomyocyte apoptosis [44]. In cardiac myocytes, insulin resistance affects the cytoprotective effects of insulin that is mediated by induction of MKP-1 expression [47]. In a rat model of ischemia-reperfusion, dexamethasone-induced cardiomyocyte protection has been shown to be mediated by upregulation of MKP-1 expression [48]. In contrast, MKP-1 down-regulation by parathyroid hormone-related peptide has been shown to be cardioprotective [51]. It has been demonstrated that cardiac myocytes derived from MKP-1 knockout mice were protected from oxidative injury [49]. These studies demonstrate that the cardioprotective effects of MKP-1 are controversial. However, inhibition of MKP-1 protects against oxidative stress-induced myocytes apoptosis and modulation of the activity of MKP-1 during myocardial ischemia-reperfusion might be beneficial for cardiac function.
MKP-1 as a Potential Therapeutic Target
Rationale
There is significant interest in the role and implication of phosphatases in metabolic diseases. The ability to selectively target these signaling pathways holds tremendous therapeutic potential for the treatment of obesity and insulin resistance. Many studies have shown that MKP-1 may have important functions in health and disease [18,50-52]. Some of the roles for MKP-1 in obesity, energy homeostasis, insulin resistance, hepatic steatosis, diabetes and cardioprotection have been uncovered [17,27,50,53]. In the following section we will discuss the potential for targeting of MKP-1 as a potential strategy for the treatment of insulin resistance, T2D and obesity.
Inhibition of MKP-1 in muscle
Several DUSPs have been implicated in human diseases including cancer, neurological and muscle disorders, metabolic disorders, inflammatory and cardiovascular diseases [52]. To date a combination of structural, biochemical, and genetic data unequivocally supports the conclusion that DUSPs exhibit overwhelming specificity towards the MAPKs [54,55]. MKP-1 is the prototypical member of this family of enzymes. Despite the fact MKP-1 has been extensively studied compared with other members of this family of enzymes, only recently has the crystal structure of the human MKP-1 catalytic domain been reported [56]. The absence of information about the crystal structure of this enzyme has hindered development of inhibitors to target MKP-1. Inhibitors of MKP-1 have been identified through the efforts of highthroughput screens [57]. The fact that the PTP domain of MKPs are all highly similar presents a challenge towards the development of potent and specific MKP-1 inhibitors. Since the crystal structure of MKP-1 has been solved, structure-based drug design is now possible.
Considering the contribution of MKP-1 in metabolic homeostasis, the ability to selectively target MKP-1 could be of great therapeutic potential for the treatment of metabolic diseases. In the last two decades kinases have appeared as a major class of druggable targets for the treatment of cancers and other diseases. Many drugs targeted against protein-tyrosine kinases (PTKs) [58] have had a significant impact on the treatment of various cancers [59]. However, many challenges exist with drugs targeted against PTKs for example, certain cancers treated with PTK inhibitors succumb to either intrinsic or acquired resistance to such treatments. Therefore, other targets and approaches are needed. Investigators both in industry and academia are working to find small molecule therapeutics targeting PTPs. However, these efforts to generate small molecules targeting the active site of PTPs have encountered challenges because of the polar nature of the active site [60,61]. New approaches such as allosteric inhibitors are being developed to target PTPs [58]. One such effort is in the development of allosteric inhibitors for PTP1B for the treatment of diabetes and obesity [62]. This strategy avoids formation of the active conformation of the enzyme by obstructing mobility of the catalytic center. Similarly, small molecule allosteric inhibitors of MKP-1 would be highly attractive target for therapeutic intervention in metabolic diseases including, insulin resistance, T2D and obesity.
There are other MKP inhibitors that have been developed but are poor [57]. However, recent work on the development of MKP-5 inhibitors has opened the door to novel strategies of allosteric modulation of the MKPs, in general, that can potentially be applied to MKP-1 [63]. The first co-crystal structure of a small molecule inhibitor with MKP-5 revealed an unknown allosteric site that resides within the catalytic domain of MKP-5. This allosteric site is conserved in many other MKPs, including MKP-1. It was demonstrated that key residues within the MKP-5 allosteric site are critical for defining compound binding and mutation of these residues in MKP-5 to those found in MKP-1 abrogates the ability of the compound to inhibit MKP-5. The catalytic domains of the DUSPs are between 36 and 57% identical to that of MKP-5 suggesting that sufficient differences exist in which specificity can be achieved. These results pave the way for the development of a new class of MKP-specific allosteric inhibitors. Given the renewed interest in the development of allosteric PTP inhibitors and its emerging success in other PTPs such as SHP-2 and PTP-1B, the development of MKP-1 allosteric inhibitors is an exciting therapeutic strategy.
In unstressed conditions MKP-1 is expressed at relatively low levels. However, in obese states and type 2 diabetes, MKP-1 is upregulated. Skeletal muscle MKP- 1 is overexpressed in obese humans [18]. Thus, MKP-1 would be a good target for therapeutic intervention only in those diseased tissues where its expression is aberrantly increased. It is important to perform a systematic survey of human tissue-specific MKP-1 gene expression and splicing to unravel new opportunities for therapeutic target identification and evaluation. Development of a metabolic disease-targeted tissue-specific promoter system would be a desirable tool. This approach is already in use in the treatment of certain types of cancers [64]. To target skeletal muscle MKP-1 to treat metabolic diseases, there is need to design a dual promoter technology in which a skeletal muscle-specific transcription system under the control of a human alpha skeletal actin promoter antisense-based therapeutics against MKP-1. Recent studies have demonstrated tissue-specific oligonucleotide delivery that utilizes both viral and nonviral delivery vectors [65]. Strategies such as blocking expression of MKP-1 in skeletal muscle with antisense oligonucleotides could be beneficial in improving insulin sensitivity and prevent the development of obesity. Using the antisensebased therapeutics against PTP1B has shown efficacy in clinical trials [66,67].
Considering the fact that studies on cardioprotective effects of MKP-1 [45,47-49] including stress-responsive MAPK [68] are controversial, more studies are needed to elucidate how modulation of MKP-1 could be beneficial in the treatment of cardiac diseases. Future work will require cardioprotective analysis of MKP-1 in tissue-specific mouse models.
The encouraging message here is that inhibition of MKP- 1 would be expected to provide a positive therapeutic value in the area of treating myocardial injury resulting from ischemic-reperfusion insults.
Challenges
One of the challenges towards inhibiting MKPs are concerns about toxicity and/or adverse side-effects because it means removing a widely expressed MAPK antagonist. However, this is unlikely to be a major cause of concern since whole body deletion of MKP-1 [17,50] or specifically MKP-1 deletion in skeletal muscle [18] does not result in overt effects in unstressed animals. Furthermore, specific inhibition of MKP-1 causes distinct upregulation of the nuclear pool of MAPKs. Major side effects due solely to increased nuclear MAPK activation are anticipated to be greatly lessened because of the restricted effects on the nuclear pool of MAPKs. Inhibition of MKP-1 pharmacologically in skeletal muscle, possibly through the development of drugs that can be targeted selectively to this tissue, will result in the activation of the nuclear pool of MAPKs causing upregulation of genes that promote energy expenditure without affecting the cytosolic pool of MAPKs that interfere with insulin sensitivity. Collectively, these mechanisms due to their spatiotemporally restricted effects, could afford highly favorable and tolerable longterm side-effects. The validity of these ideas will need to be rigorously tested in an assortment of mouse models of metabolic disease.
Conclusion
After over a decade of study of the physiological function of MKP-1 in the regulation of metabolic homeostasis in mice and more recently in humans, these data collectively point to the notion that chronic upregulation of MKP-1 in skeletal muscle is part of a stress response mediated by p38 MAPK and JNK activities and this may play an important contributing role in the development of insulin resistance, T2D and obesity. Despite MKP-1 being a challenging, target there are potential strategies that if successfully executed could lead to inhibiting MKP-1 as a treatment of metabolic disease.
Acknowledgements
A.M.B. is supported by NIH grants P01-DK57751, R01 HL134166 and R01 AR66003. A.L. is supported by UAH Faculty Startup and New Faculty Research Funding Program.
Conflict of Interest
No potential conflicts of interest relevant to this article were reported.
Author Contributions
A.L. wrote the manuscript and A.M.B. reviewed and edited the manuscript.
References
2. Wu HW, Backman D, Kizer KW. Restructuring a state nutrition education and obesity prevention program: implications of a local health department model for SNAPEd. Journal of Public Health Management and Practice. 2017 Jan 1;23(1):e28-36.
3. Arroyo-Johnson C, Mincey KD. Obesity epidemiology worldwide. Gastroenterology Clinics. 2016 Dec 1;45(4):571-9.
4. Donohoe F, Wilkinson M, Baxter E, Brennan DJ. Mitogen-Activated Protein Kinase (MAPK) and obesityrelated cancer. International Journal of Molecular Sciences. 2020 Jan;21(4):1241.
5. Kim G, Kim JH. Impact of Skeletal Muscle Mass on Metabolic Health. Endocrinology and Metabolism. 2020 Mar 1;35(1):1-6.
6. Tallis J, James RS, Seebacher F. The effects of obesity on skeletal muscle contractile function. Journal of Experimental Biology. 2018 Jul 1;221(13).
7. Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. New England Journal of Medicine. 2004 Feb 12;350(7):664-71.
8. Stuart CA, McCurry MP, Marino A, South MA, Howell ME, Layne AS, et al. Slow-twitch fiber proportion in skeletal muscle correlates with insulin responsiveness. The Journal of Clinical Endocrinology & Metabolism. 2013 May 1;98(5):2027-36.
9. Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nature Reviews Endocrinology. 2012 Aug;8(8):457-65.
10. Zierath JR, Hawley JA. Skeletal muscle fiber type: influence on contractile and metabolic properties. PLoS Biol. 2004 Oct 12;2(10):e348.
11. Oberbach A, Bossenz Y, Lehmann S, Niebauer J, Adams V, Paschke R, et al. Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with type 2 diabetes. Diabetes Care. 2006 Apr 1;29(4):895-900.
12. Shi H, Boadu E, Mercan F, Le AM, Flach RJ, Zhang L, et al. MAP kinase phosphatase-1 deficiency impairs skeletal muscle regeneration and exacerbates muscular dystrophy. The FASEB Journal. 2010 Aug;24(8):2985-97.
13. Wang S, Jin J, Xu Z, Zuo B. Functions and regulatory mechanisms of lncRNAs in skeletal myogenesis, muscle disease and meat production. Cells. 2019 Sep;8(9):1107.
14. Capri M, Morsiani C, Santoro A, Moriggi M, Conte M, Martucci M, et al. Recovery from 6-month spaceflight at the International Space Station: muscle-related stress into a proinflammatory setting. The FASEB Journal. 2019 Apr;33(4):5168-80.
15. Rittweger J, Albracht K, Flück M, Ruoss S, Brocca L, Longa E, et al. Sarcolab pilot study into skeletal muscle’s adaptation to long-term spaceflight. npj Microgravity. 2018 Sep 17;4(1):1-9.
16. Lawan A, Bennett AM. MAP kinase phosphatases emerge as new players in metabolic regulation. Protein Tyrosine Phosphatase Control of Metabolism. Bence KK, Ed. New York, Springer. 2013:221.
17. Roth RJ, Le AM, Zhang L, Kahn M, Samuel VT, Shulman GI, et al. MAPK phosphatase-1 facilitates the loss of oxidative myofibers associated with obesity in mice. The Journal of Clinical Investigation. 2009 Dec 1;119(12):3817- 29.
18. Lawan A, Min K, Zhang L, Canfran-Duque A, Jurczak MJ, Camporez JP, et al. Skeletal muscle–specific deletion of MKP-1 reveals a p38 MAPK/JNK/Akt signaling node that regulates obesity-induced insulin resistance. Diabetes. 2018 Apr 1;67(4):624-35.
19. Khadir A, Tiss A, Abubaker J, Abu-Farha M, Al-Khairi I, Cherian P, et al. MAP kinase phosphatase DUSP1 is overexpressed in obese humans and modulated by physical exercise. American Journal of Physiology-Endocrinology and Metabolism. 2015 Jan 1;308(1):E71-83.
20. Xie SJ, Li JH, Chen HF, Tan YY, Liu SR, Zhang Y, et al. Inhibition of the JNK/MAPK signaling pathway by myogenesis-associated miRNAs is required for skeletal muscle development. Cell Death & Differentiation. 2018 Sep;25(9):1581-97.
21. Shi H, Scheffler JM, Pleitner JM, Zeng C, Park S, Hannon KM, et al. Modulation of skeletal muscle fiber type by mitogen-activated protein kinase signaling. The FASEB Journal. 2008 Aug;22(8):2990-3000.
22. Sabio G, Davis RJ. cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends in Biochemical Sciences. 2010 Sep 1;35(9):490-6.
23. Brown AE, Palsgaard J, Borup R, Avery P, Gunn DA, De Meyts P, et al. p38 MAPK activation upregulates proinflammatory pathways in skeletal muscle cells from insulin-resistant type 2 diabetic patients. American Journal of Physiology-Endocrinology and Metabolism. 2015 Jan 1;308(1):E63-70.
24. Petta S, Ciminnisi S, Di Marco V, Cabibi D, Camma C, Licata A, et al. Sarcopenia is associated with severe liver fibrosis in patients with non-alcoholic fatty liver disease. Alimentary Pharmacology & Therapeutics. 2017 Feb;45(4):510-8.
25. Yu R, Shi Q, Liu L, Chen L. Relationship of sarcopenia with steatohepatitis and advanced liver fibrosis in nonalcoholic fatty liver disease: a meta-analysis. BMC Gastroenterology. 2018 Dec;18(1):1-6.
26. Tandon P, Ney M, Irwin I, Ma MM, Gramlich L, Bain VG, et al. Severe muscle depletion in patients on the liver transplant wait list: its prevalence and independent prognostic value. Liver Transplantation. 2012 Oct;18(10):1209-16.
27. Lawan A, Zhang L, Gatzke F, Min K, Jurczak MJ, Al- Mutairi M, et al. Hepatic mitogen-activated protein kinase phosphatase 1 selectively regulates glucose metabolism and energy homeostasis. Molecular and Cellular Biology. 2015 Jan 1;35(1):26-40.
28. Roth Flach RJ, Bennett AM. Mitogen-activated protein kinase phosphatase-1–a potential therapeutic target in metabolic disease. Expert Opinion on Therapeutic Targets. 2010 Dec 1;14(12):1323-32.
29. Ebadi M, Bhanji RA, Mazurak VC, Montano-Loza AJ. Sarcopenia in cirrhosis: From pathogenesis to interventions. Journal of Gastroenterology. 2019 Aug 7:1- 5.
30. Coen PM, Musci RV, Hinkley JM, Miller BF. Mitochondria as a target for mitigating sarcopenia. Frontiers in Physiology. 2019 Jan 10;9:1883.
31. Bhanji RA, Montano-Loza AJ, Watt KD. Sarcopenia in cirrhosis: Looking beyond the skeletal muscle loss to see the systemic disease. Hepatology. 2019 Dec;70(6):2193- 203.
32. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiological Reviews. 2011 Oct;91(4):1447-531.
33. Pal M, Febbraio MA, Lancaster GI. The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and insulin resistance. The Journal of Physiology. 2016 Jan 15;594(2):267-79.
34. Manieri E, Sabio G. Stress kinases in the modulation of metabolism and energy balance. Journal of Molecular Endocrinology. 2015 Oct;55(2):R11.
35. Sabio G, Kennedy NJ, Cavanagh-Kyros J, Jung DY, Ko HJ, Ong H, et al. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Molecular and Cellular Biology. 2010 Jan 1;30(1):106-15.
36. Xi X, Han J, Zhang JZ. Stimulation of glucose transport by AMP-activated protein kinase via activation of p38 mitogen-activated protein kinase. Journal of Biological Chemistry. 2001 Nov 2;276(44):41029-34.
37. Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, et al. Exercise stimulates Pgc-1a transcription in skeletal muscle through activation of the p38 MAPK pathway. Journal of Biological Chemistry. 2005 May 20;280(20):19587-93.
38. Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Molecular Metabolism. 2017 Feb 1;6(2):174-84.
39. Nandipati KC, Subramanian S, Agrawal DK. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Molecular and Cellular Biochemistry. 2017 Feb 1;426(1- 2):27-45.
40. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. The FEBS Journal. 2015 Feb;282(4):647-72.
41. Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocrine Reviews. 2003 Feb;24(1):78-90.
42. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Executive summary: heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation. 2015 Jan 27;131(4):434-41.
43. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circulation Research. 2015 Feb 13;116(4):674-99.
44. De Hert S, Moerman A. Myocardial injury and protection related to cardiopulmonary bypass. Best Practice & Research Clinical Anaesthesiology. 2015 Jun 1;29(2):137-49.
45. Webster I, Smith A, Lochner A, Huisamen B. The role of MKP-1 in insulin-induced cardioprotection. Cardiovascular Drugs and Therapy. 2017 Jun 1;31(3):247- 54.
46. Aikawa R, Nawano M, Gu Y, Katagiri H, Asano T, Zhu W, et al. Insulin prevents cardiomyocytes from oxidative stress–induced apoptosis through activation of PI3 kinase/ Akt. Circulation. 2000 Dec 5;102(23):2873-9.
47. Morisco C, Marrone C, Trimarco V, Crispo S, Monti MG, Sadoshima J, et al. Insulin resistance affects the cytoprotective effect of insulin in cardiomyocytes through an impairment of MAPK phosphatase-1 expression. Cardiovascular Research. 2007 Dec 1;76(3):453-64.
48. Fan WJ, Genade S, Genis A, Huisamen B, Lochner A. Dexamethasone-induced cardioprotection: a role for the phosphatase MKP-1?. Life Sciences. 2009 Jun 5;84(23- 24):838-46.
49. Datta NS, Chukkapalli S, Vengalil N, Zhan E, Przyklenk K, Lasley R, et al. Parathyroid hormone-related peptide protects cardiomyocytes from oxidative stressinduced cell death: First evidence of a novel endocrine– cardiovascular interaction. Biochemical and Biophysical Research Communications. 2015 Dec 4;468(1-2):202-7.
50. Wu JJ, Roth RJ, Anderson EJ, Hong EG, Lee MK, Choi CS, et al. Mice lacking MAP kinase phosphatase-1 have enhanced MAP kinase activity and resistance to dietinduced obesity. Cell Metabolism. 2006 Jul 1;4(1):61-73.
51. Broome DT, Datta NS. Mitogen-activated protein kinase phosphatase-1: function and regulation in bone and related tissues. Connective Tissue Research. 2016 May 3;57(3):175-89.
52. Seternes OM, Kidger AM, Keyse SM. Dual-specificity MAP kinase phosphatases in health and disease. Biochimica Et Biophysica Acta (BBA)-Molecular Cell Research. 2019 Jan 1;1866(1):124-43.
53. Lawan A, Shi H, Gatzke F, Bennett AM. Diversity and specificity of the mitogen-activated protein kinase phosphatase-1 functions. Cellular and Molecular Life Sciences. 2013 Jan 1;70(2):223-37.
54. Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) Shaping the outcome of MAP kinase signalling. The FEBS Journal. 2013 Jan;280(2):489-504.
55. Tonks NK. Protein phosphatases: From molecules to networks: Introduction. The FEBS Journal. 2013 Jan;280(2):323.
56. Gumpena R, Lountos GT, Raran-Kurussi S, Tropea JE, Cherry S, Waugh DS, et al. Crystal structure of the human dual specificity phosphatase 1 catalytic domain. Protein Science. 2018 Feb;27(2):561-7.
57. R Doddareddy M, Rawling T, J Ammit A. Targeting mitogen-activated protein kinase phosphatase-1 (MKP- 1): structure-based design of MKP-1 inhibitors and upregulators. Current Medicinal Chemistry. 2012 Jan 1;19(2):163-73.
58. Stanford SM, Bottini N. Targeting tyrosine phosphatases: time to end the stigma. Trends in Pharmacological Sciences. 2017 Jun 1;38(6):524-40.
59. Imasawa T, Kitamura H, Ohkawa R, Satoh Y, Miyashita A, Yatomi Y. Unbalanced expression of sphingosine 1-phosphate receptors in diabetic nephropathy. Experimental and Toxicologic Pathology. 2010 Jan 1;62(1):53-60.
60. Chen L, Pernazza D, Scott LM, Lawrence HR, Ren Y, Luo Y, et al. Inhibition of cellular Shp2 activity by a methyl ester analog of SPI-112. Biochemical Pharmacology. 2010 Sep 15;80(6):801-10.
61. Scott LM, Chen L, Daniel KG, Brooks WH, Guida WC, Lawrence HR, et al. Shp2 protein tyrosine phosphatase inhibitor activity of estramustine phosphate and its triterpenoid analogs. Bioorganic & Medicinal Chemistry Letters. 2011 Jan 15;21(2):730-3.
62. Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nature Chemical Biology. 2014 Jul;10(7):558-66.
63. Gannam ZT, Min K, Shillingford SR, Zhang L, Herrington J, Abriola L, et al. An allosteric site on MKP5 reveals a strategy for small-molecule inhibition. Science Signaling. 2020 Aug 25;13(646).
64. Blagosklonny MV. Tissue-selective therapy of cancer. British Journal of Cancer. 2003 Oct;89(7):1147-51.
65. Xia X, Pollock N, Zhou J, Rossi J. Tissue-Specific Delivery of Oligonucleotides. In:Oligonucleotide-Based Therapies. Humana, New York, NY. 2019; pp. 17-50.
66. Zinker BA, Rondinone CM, Trevillyan JM, Gum RJ, Clampit JE, Waring JF, et al. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proceedings of the National Academy of Sciences. 2002 Aug 20;99(17):11357-62.
67. Waring JF, Ciurlionis R, Clampit JE, Morgan S, Gum RJ, Jolly RA, et al. PTP1B antisense-treated mice show regulation of genes involved in lipogenesis in liver and fat. Molecular and Cellular Endocrinology. 2003 May 30;203(1-2):155-68.
68. Weinbrenner C, Liu GS, Cohen MV, Downey JM. Phosphorylation of tyrosine 182 of p38 mitogenactivated protein kinase correlates with the protection of preconditioning in the rabbit heart. Journal of Molecular and Cellular Cardiology. 1997 Sep 1;29(9):2383-91.