Abstract
The term “Achilles Heel” has often been used in relation to potentially vulnerable target molecules for anticancer drugs within cancer cells. Its generalized application to an increasing range of diverse possibilities, however, detracts from the uniquely valuable way it describes the wild-type aerobic glycolysis target which persists within the immortal cancer cell phenotype. Here it is proposed that the persistence of the PKM2 isoform of the normal non mutant pyruvate kinase gene which induces aerobic glycolysis in a cancer cell whose phenotype has otherwise been immortalized by oncogenic somatic mutation provides a close analogous comparison with the immortal body of Achilles and his mortal vulnerable heel. Exploitation of the mechanism believed to explain the way aerobic glycolysis helps avoid oncogene induced apoptosis has led to the development of ONKONEK, a first in class therapeutic agent employing the new therapeutic paradigm of selective cancer cell limotherapy (SCCL) which starves cancer cells rather than attacking their cell division and is not expected to be restricted by the limitations of current chemotherapy.
The new therapeutic paradigm of selective cancer cell limotherapy (SCCL) starves cancer and, in particular, down-regulates PKM2 activity during cytokinesis in mitosis addicted cancer cells. Selective cancer cell limotherapy, by sparing normal cells provides a potential global cancer treatment which could potentially be delivered repeatedly until every cancer cell is destroyed.
Keywords
Aerobic glycolysis, Cancer, CDK4, Limotherapy, PKM2
Achilles Heel
The mythological story tells how the goddess Thetis, a sea nymph tried to immortalize her son, whose father Peleus was a mortal, by dipping him in the river Styx. Because she grasped him by the heel, the water did not touch that part of his body which remained a vulnerable target for the fatal arrow later shot by Paris during the Siege of Troy. The concept of the presence of a vulnerable “Achilles Heel” in an immortal cancer cell has been widely employed in the literature and has grown to describe a multiplicity of unrelated potentially vulnerable target sites in cancer that might be treated without damage to normal tissues [1–5]. In some cases, even multiple “heels” have been included in this encompassing title [6]. No clinically effective anticancer drug has yet, however, resulted from this paradigm. Most chemotherapy is restricted by cumulative toxicity because it permanently damages normal as well as cancer cells. Chemotherapy can only therefore generally be administered for a limited number of courses and cannot achieve enough repeated treatments to completely eradicate the majority of cancers.
Here it is proposed that the persistence of the PKM2 isoform of the normal non mutant pyruvate kinase gene which induces aerobic glycolysis in a cancer cell provides the closest analogous comparison with the immortal body of Achilles and his mortal vulnerable heel.
Aerobic Glycolysis
Heinrich Friederich Warburg was the first to identify disordered carbohydrate metabolism as a significant characteristic of cancer cells nearly a century ago. Since that time there has been a progressive interest in what subsequently came to be described as aerobic glycolysis [7], where cancer cells use only the glycolytic part of the carbohydrate catabolism pathway without the contribution of mitochondrial oxidative phosphorylation despite the abundant availability of oxygen. The mechanism underlying aerobic glycolysis is now known to be due to the presence within cancer cells of the PKM2 isoform of the non-mutated pyruvate kinase gene brought about by a switch of expression of exon 9 to exon 10, rather than the PKM1 isoform characteristic of normal adult mammalian cells [8]. Aerobic glycolysis is ubiquitously found in all human cancer cells in which it has been sought including myeloma, pediatric and pancreatic tumors [9–11].
The idea that normal cellular processes might play an important role in the etiology of cancer has rarely been considered [12]. The suggestion that a cause of cancer might lie in carbohydrate metabolism as suggested by Warburg did not meet with general scientific approval for many years and the ubiquitous presence of aerobic glycolysis throughout cancer still resists a convincing explanation [13]. The discovery of aerobic glycolysis was long considered to be a metabolic quirk of transformed cells, but recent studies in stem cells have challenged this model revealing that many normal stem cell populations also exhibit aerobic glycolysis [14,15]. Selectively killing cancer cells cannot therefore only rely on targeting aerobic glycolysis but also needs to operate in the context of the fundamental difference in mitotic cell division between cancer cells and normal stem cells, which is itself dependent on the key hallmarks of cancer: self-sufficiency in growth signals and limitless replicative potential [16]. These together account for the continuously repeated cycles of relentless cell division characteristic of all cancers, here termed mitotic addiction.
When Does Aerobic Glycolysis Arise in a Cancer Cell?
Recently it has been proposed that the reason for the presence of aerobic glycolysis in a cancer cell is because it is a preneoplastic change which is already present in the normal cell before it became transformed into a cancer cell by an oncogenic mutation [17,18].
Rather than being a consequence of malignant transformation, as previously thought, aerobic glycolysis in fact precedes malignant transformation (Figure1). This proposition is supported by reports that markers of aerobic glycolysis have recently been found in premalignant tissues [19,20].
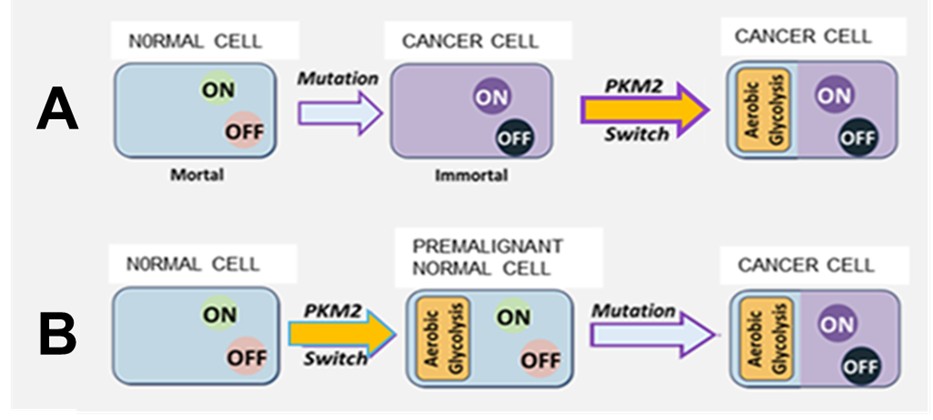
The persistence of the PKM2 isoform switch through succeeding generations of the newly created cancer provides an explanation for its presence in all cancer cells. Figure 1 compares the canonical somatic mutation theory of cancer (A) and the Achilles Heel modification (B), adoption of which implies that the cancer cell should be considered a hybrid mortal/immortal cell like the mythical Achilles with a mortal aerobic glycolysis “heel” in an otherwise immortal “body”.
The widely held earlier belief that each of the many different types of cancer cell, caused by different types of oncogenic somatic mutation arising in a wide range of different normal cells, suddenly “decides” to effect an identical change in the isoform expression of the same single normal gene, pyruvate kinase, (the suggested reasons being different from case to case) in every cancer cell, seems an unlikely explanation for the ubiquitous presence of aerobic glycolysis throughout cancer.
Oncogenic Mutations Require Normal CDK4 and PKM2 Interaction in Order to Achieve Immortalization with Avoidance of Apoptosis
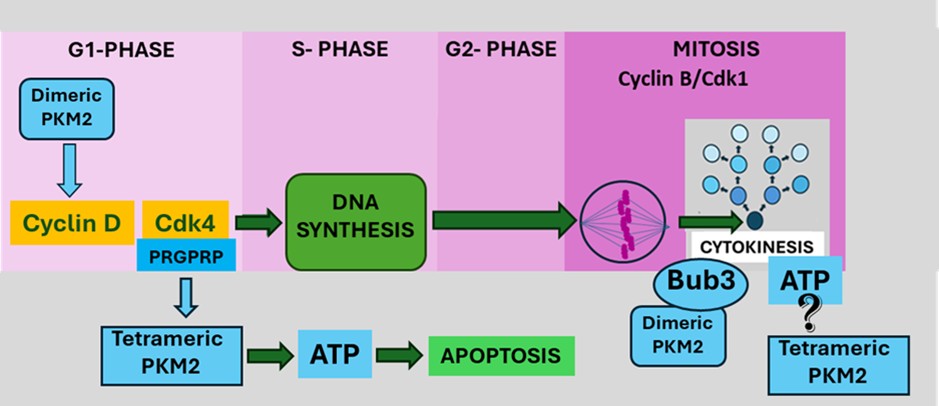
In advanced cancers reasons provided for escape from apoptosis by cancer cells have been suggested to be increasing or decreasing expression of anti- or proapoptotic genes, respectively or stabilizing or de-stabilizing anti- or pro-apoptotic proteins, respectively [21]. At the moment of initial malignant transformation of a normal cell to a cancer cell a simpler method of avoiding apoptosis would be needed. Apoptosis is an energy dependent process. ATP availability determines whether necrosis or apoptosis is a cell’s final fate [22]. It has now been proposed that at the moment of malignant transformation, in addition to activating relentless cell division through the canonical Cdk4/CyclinD-pRb-E2F pathway, In the G1 phase of the cell cycle (Figure 4) Cdk4 levels rise and interact with tetrameric PKM2 causing a transient lowering of intracellular ATP that is sufficient to prevent apoptosis but not sufficiently low to cause necrosis.
Pyruvate Kinase M2
The aerobic glycolysis phenotype in cancer cells is a consequence of the isotypic switch in expression of the pyruvate kinase gene to express the PKM2 isotype by expression of exon 10 rather than the PKM1 isotype expressed by exon 9. This results in a different amino acid sequence between amino acid sites 379–411 [23]. PKM2 can exist as a dimer with multiple interaction sites and functions or may form a tetramer which controls the transformation from phosphoenolpyruvate to pyruvate with the release of one molecule of ATP [24] (Figure 2A). The pyruvate is then converted into lactic acid by the enzyme lactate dehydrogenase.
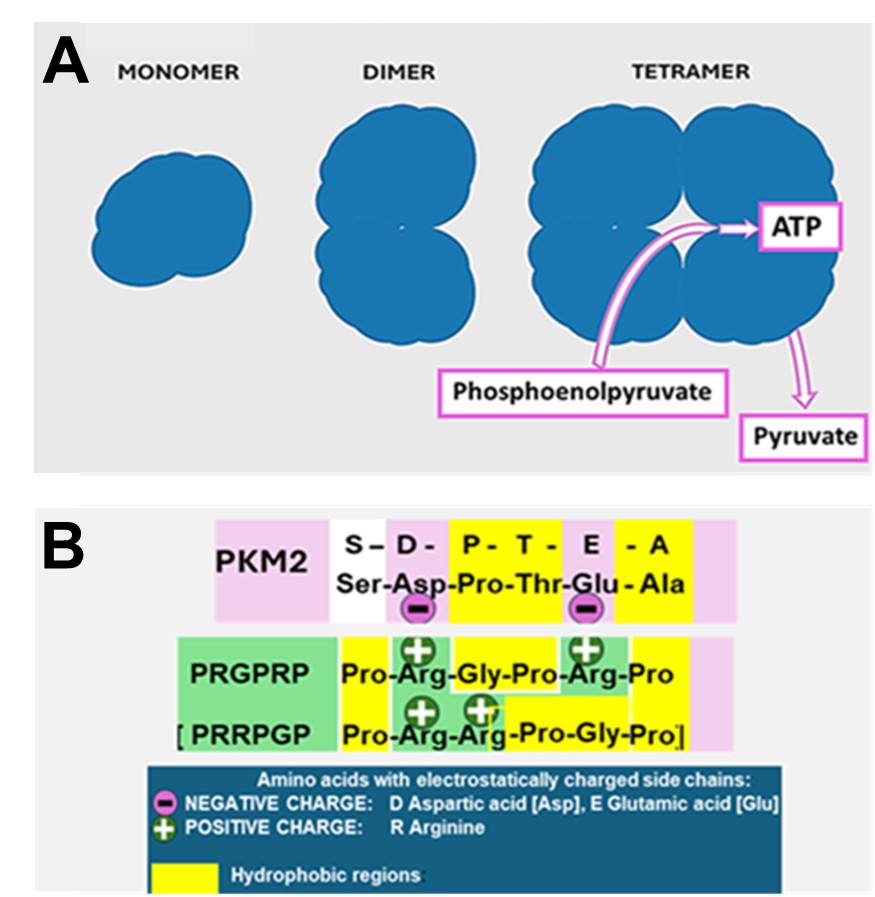
Dimeric PKM2
Whereas PKM2 exerts its metabolic glycolytic function in the cytosol predominantly in its tetrameric form, the PKM2-specific exon 10 encodes a nuclear localization signal facilitating dimeric PKM2 import into the nucleus [24]. Nuclear translocation requires post-translational changes such as activation of ERK1/2 by EGRF signaling which causes ERK1/2 to bind to Ileu429/Leu431 and phosphorylate PKM2 at Ser37 [25]. The dimer transfers into the nucleus in the G1 phase of the cell cycle where it becomes involved in many signaling interactions including binding to phosphorylated-Tyr333 of β-catenin and phosphorylation of PKM2 at Thr454 which facilitates the transcriptional co-activation of HIF-1α and β-catenin [26] ultimately resulting in upregulation of cyclin D [27]. The dimeric form of PKM2 not only passively avoids participating in glycolysis but using PEP as a phosphate donor acts as a signaling molecule which, catalyzes tyrosine phosphorylation of STAT3 [28].
Tetrameric PKM2
The tetrameric form of PKM2 (Figures 2A and 2B) is responsible for control of the rate-limiting step in glycolysis that shifts glucose metabolism from the normal respiratory chain to lactate production [29]. In contrast to the multiple signaling interactions of its dimeric form, the role of tetrameric of PKM2 is the generation of ATP derived from the transformation of phosphoenolpyruvate to pyruvate. Unlike the complex picture of modulation and activation of the PKM2 dimer, less information is available about the control of PKM2 tetramerization and its energy producing role except that PKM2 activators such as fructose-1-6-biphosphate (FBP) promote allosteric changes which influence active tetramer formation [30]. This activity is abrogated by a site directed mutation of PKM2 at Cys-423 within the tetramerization zone. In summary, PKM2 dimer can be regarded as an active protein kinase signaling moiety and PKM2 tetramer is an active pyruvate kinase providing intracellular energy in the form of ATP [31].
An Arginine Rich Amino Acid Sequence from the C’-terminal Non-kinase Region of CDK4 Downregulates ATP
An isolated PRGPRP construct in a cyclic amphiphilic cassette was shown to cause a fall in intracellular ATP and the global death of a wide range of human cancer cell lines by necrosis [19,25]. Normal diploid human keratinocytes exposed to PRGPRP underwent cell cycle arrest but did not die. The fall in cancer cell ATP was accompanied by inhibition of lactate dehydrogenase (a downstream indicator of PKM2 activity). Time lapse photomicrography [in appendix] shows that human lung cancer cells went through only a single division and then died as a result of exposure to THR 53, a construct containing the PRGPRP warhead in a cyclic amphiphilic vector of Ala/Leu/Lys repeats. The depletion of energy by PRGPRP appears to be critical for cancer cells to survive cell division. A modification of the PRGPRP warhead creating an increase in lipophilicity by the substitution of the warhead prolines by unnatural amino acids and incorporation into a ArgArg/TrpTrp repeat cassette to yield a previously described potential therapeutic agent, HILR-056 [19] has been renamed ONKONEK and has recently entered preclinical animal trials.
THR53 Docks at the End of the PKM2 Tetramerization Interface
THR53, a cyclic amphiphilic derivative of the Cdk4 C’-terminal hexapeptide Pro-Arg-Gly-Pro-Arg-Pro [32], was found to inhibit lactate dehydrogenase at dose which caused a fall in ATP accompanied by selective cancer cell necrosis. A comparison of the linear amino acid sequences of PKM1 and PKM2 was therefore undertaken in a search for a putative binding site for PRGPRP [17]. This revealed a Ser-Asp-Pro-Thr-Glu-Ala site unique to PKM2 lying at one end of the tetramerization interface, as a potential ionic interactive site for PRGPRP. It is proposed this interaction allosterically down-modulates ATP production.
Structural chemical modelling by computer modelling [previously unpublished data] is consistent with the possibility of a PRGPRP warhead carried in an Ala-Lys-Leu- repeat amphiphilic cassette (THR53) docking at the predicted SDPTEA site on the A, B, C, and D subsites of tetrameric PKM2 (Figure 3). Figure 3 depicts the simulated plausible interaction site for PRGPRP in a THR 53 cassette lying in the A subunit of tetrameric PKM2. This interaction is repeated on subunits B, C, and D of the symmetrical molecule. The interaction (depicted in detail in Figure 2B) is ionic not covalent and is thus its maintenance is dependent on the continuous presence of PRGPRP and leaves no permanent molecular damage. Conventional chemotherapy irreversibly damages normal as well as cancer tissues which permits it to only be administered for a limited number of treatments which are, in general, insufficient to eradicate the majority of cancers. Because SCCL agents are likely to have a transient non-damaging effect on normal cells they can potentially be given repeatedly without being limited by normal tissue toxicity, until every cancer cell is eradicated.
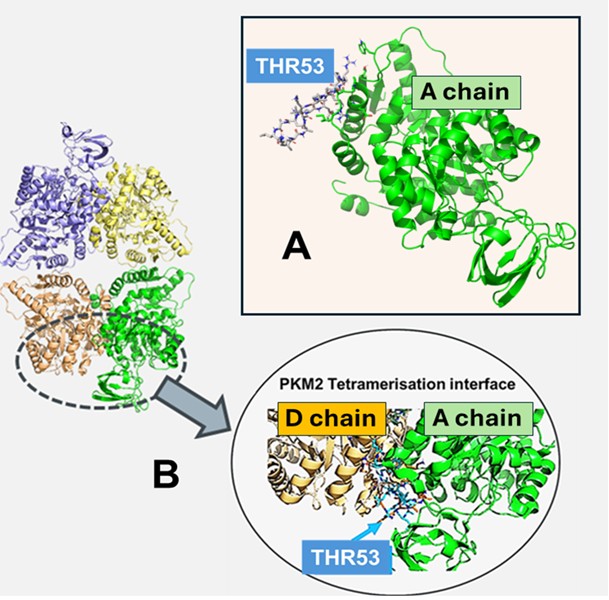
Figure 3. Simulated docking of PRGPRP warhead in a cyclic amphiphilic cassette (THR 53) with tetrameric PKM2. The THR 53 peptide  was built using Rosetta. Simulation with enhanced sampling was used to obtain stable solution structures; Docking simulations were performed with and without amino acid restraint. A. Binding to A chain with restraint. B. Binding to A chain overlayed with D chain.
was built using Rosetta. Simulation with enhanced sampling was used to obtain stable solution structures; Docking simulations were performed with and without amino acid restraint. A. Binding to A chain with restraint. B. Binding to A chain overlayed with D chain.
Cytokinesis
Cytokinesis is the final stage of cell division during which separation of the two daughter cells created by the mitotic process occurs [33,34]. Myosin II is the principal motor for cytokinesis using the energy derived from the hydrolysis of ATP to move along actin filaments, causing contraction of the contractile ring and ultimately splitting the cell into two daughter cells. PKM2 [35], but not PKM1 [36] has been reported to play a critical regulatory role in cytokinesis by binding to and phosphorylating the spindle checkpoint Bub3 at Y207 required for the Bub3-Bub1 complex to interact with Blinkin [37]. PKM2 also interacts with the light chain of myosin II (MCL2) [38,39].
The signaling function of PKM2 involved in the control of cytokinesis at the Bub3 site is likely to be carried out by dimeric PKM2, but as yet no mechanism has been provided to explain the source of the energy required for the cleavage required to separate the mitotic parent cell into two new daughter cells. It is proposed that in addition to its dimeric conformation as a regulatory signaling molecule, PKM2 at this site can also assume its tetrameric motor conformation. Tetrameric PKM2 would provide the ATP required to carry out the physical process of separation of the two daughter cells that completes cytokinesis. Inhibition of ATP production by tetrameric PKM2 by a PRGPRP bearing molecule such as THR53 at this critical final phase of cancer cell division would result in death. This would provide an explanation for the morphological appearance of THR-53 treated H460 Human Non-small cell lung cancer cells immediately after failed mitoses as seen on time lapse video photomicrography (APPENDIX video: Right Hand Panel).
The Limitations of Targeting Somatic Mutations
Present targets used for the design of anticancer treatments are the result of somatic mutations which cause the relative overexpression or activation by modifying structural alterations of normal cellular molecules, such as signal transduction factors, cell surface receptors and those neoantigens recognized by the immune system. By contrast PKM2 expression rather than PKM1 expression provides a unique phenotype with an absolute rather than a relative difference between cancer and normal in which reliable targets for uniquely selective anticancer agents can be sought.
Notably, because expression of the PKM2 isoform is not the result of mutation it should provide a stable target uninfluenced by the heterogeneity and genetic instability that is characteristic of mutation induced targets. Somatic mutations are stochastic events, and as such, their distribution and penetrance will be random across any field of cancer cells.
Observations of tissue sections of typical cancers stained immunohistochemically for expression of HER2 for example, show a random pattern of fluorescence intensity from cell to cell [40]. Some cells show high expression of the HER2 receptor, a few show little or no expression and the majority lie in between.
Such uneven penetrance means that a therapeutic drug targeted at a specific oncogene is likely to kill cells with the highest penetrance leaving those with lower or no oncogene expression to survive and repopulate to give rise to a resistant cancer (Figure 5). A similar argument applies to the neoantigens induced by mutations, against which the body mounts an immune response. It is not surprising, therefore, that widespread therapeutic induced acquired resistance is sufficiently common in oncology to merit the title of a hallmark of cancer.
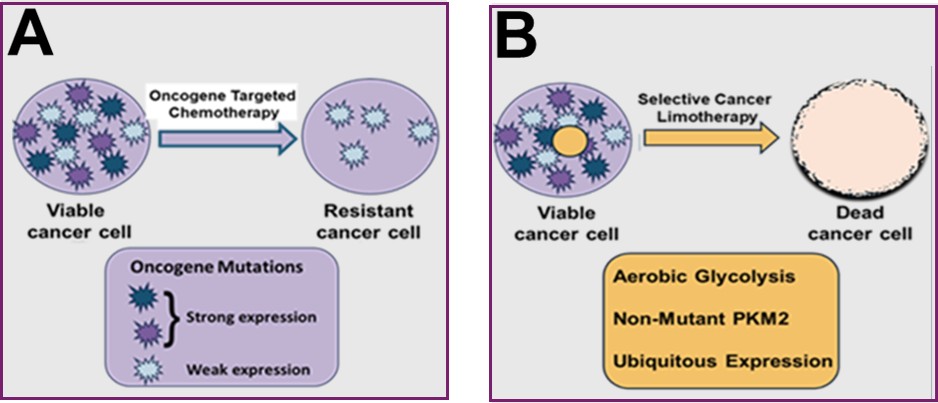
Spontaneous Cancer Cell Death
Unrestrained cell division is widely acknowledged to be the key hallmark of cancer leading to the general belief that cancer cells are immortal. The fundamental assay of unrestrained cancer cell division is a clonogenic assay which measures the ability of a cancer cell to go through repeated divisions creating daughter cells each of which are capable of further unrestrained divisions [41]. Despite the development of optimal tissue culture conditions for human cancer cell lines since the middle of the last century and the improved ease with which the human cancer cell lines themselves have adapted to in vitro growth, there still remains a wide variation in the percentage of single human cancer cells which give rise to colonies in clonogenic assays. This variability is reflected in the “plating efficiency”, a value obtained for an individual cell line reflecting the percentage likelihood of clone formation from a single cell.
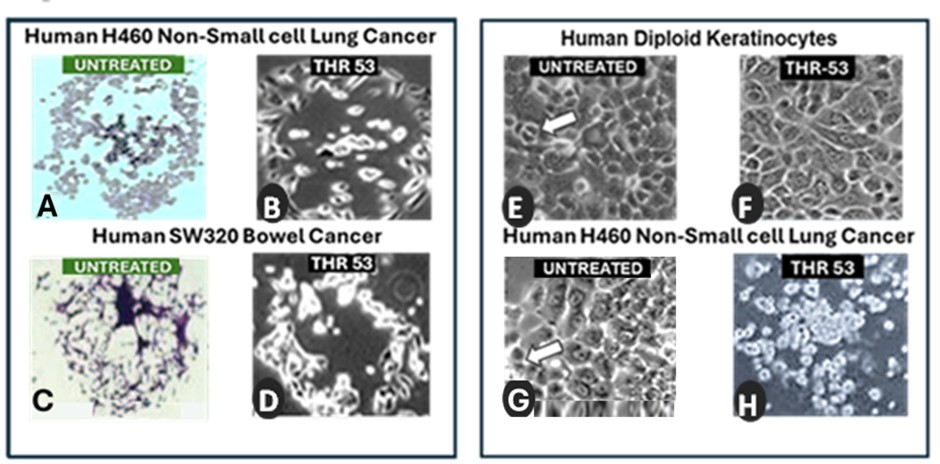
Serial photo microscopic studies of different human cancer stem cell lines growing under optimal growth conditions in tissue culture clonogenic assays reveal considerable differences in the percentage of cells able to successfully accomplishes the requisite number of consecutive cell divisions to form an intact colony of over fifty daughter cells. It is therefore necessary to normalize the results of therapeutic clonogenic assays to the relative plating efficiency of each cell line when comparing different treatments. The morphological appearances of the dying colonies in hematoxylin and eosin light microscopy images of untreated clonogenic assays (Figures 6A and 6C) are strikingly similar in appearance to phase contrast images of human cancer cell lines in which exposure to the ONKONEK analogue, THR53, has induced a drop in intracellular ATP causing necrotic death (Figures 6B and 6D) [32]. The plating efficiency of control untreated human cancer cells in clonogenic assays across a range of different untreated cell lines has been measured as being between 25% and 71.7% with a mean of 50.1%. It appears, therefore, that around half of human cancer cells undergo spontaneous cancer cell death by necrosis because they run out of energy when attempting to form colonies by repeated divisions of cancer stem cells. This spontaneous attrition of energy in a cancer undergoes further depletion following treatment with an anti-metabolic agent such as THR-53 or ONKONEK, that targets aerobic glycolysis causing even further ATP depletion providing an explanation of the similar morphological appearances between spontaneously dying untreated cells and cells exposed to THR-53 seen in H460 Non-small cell lung cancer (Figures 6A and 6B) and SW320 colorectal cancer (Figures 6C and 6D). Cancer cells should thus not be viewed as super-cells which outperform normal cells so much as damaged cells prone to spontaneous cell death by necrosis which makes them vulnerable to anti-metabolic agent attack by agents targeting aerobic glycolysis. The cancer cells die because, driven by mitotic addiction, they cannot avoid entering cytokinesis although they have insufficient ATP to complete it successfully.
Mitotic Addiction
The term oncogene addiction describes the process in which cancers with genetic, epigenetic, or chromosomal irregularities become dependent on one or several genes for maintenance and survival. As a result, cancer cells rely on continuous signaling from these oncogenes which make them desirable targets for new chemotherapeutic agents [42]. Cancer cell autonomous proliferation, however, is not only dependent on addiction to a relatively small group of dominant oncogenes, but also relies on many other phenotypic factors including inactivation of suppressor genes and the non-mutation-induced isotypic expression of normal PKM2 responsible for aerobic glycolysis. The principal difference between cancer and normal cells, therefore is that, further to addiction to oncogenes, cancer cells are addicted to mitosis itself whose critical final step of cytokinesis (Figure 4) is suggested here to require ATP generated by tetrameric PKM2 located at the dimeric PKM2 binding site to Bub3 and myosin II. In this way an anti-metabolic therapeutic attack on aerobic glycolysis selectively kills cancer cells by inhibiting mitotic cell division.
Mitoses are a prominent feature of typical human tumors growing in tissue culture, often seen as doublets under phase microscopy (Figure 6G arrow), which having replicated their genetic code detach from the substratum whilst undergoing the physical process of fission to yield two new daughter cells each containing a new copy of the original genome. Repopulating stem cells in normal tissues seen in short term culture of normal human diploid keratinocytes show mitoses of similar, though more regular, appearance (Figure 6E arrow). ATP cannot be stored easily within cells [43] and human keratinocyte stem cells exposed to a PRGPRP congener undergo cell cycle arrest showing a flattened appearance with no evidence of mitoses (Figure 6F). Normal stem cells do not therefore proceed into mitosis and so do not become vulnerable to killing by PKM2 inhibitors during cytokinesis. Human in vitro cancer cell lines such as H460 Non-small cell lung cancer, exposed to homologues of the wild-type Cdk4 C’-terminal hexapeptide PRGPRP are unable because of their mutated signaling systems to stop dividing and proceed into mitosis terminating in a cytokinesis lacking sufficient ATP for successful completion.
Thus, although normal stem cells and cancer cells both exhibit aerobic glycolysis, anti-metabolic agents containing warheads such as PRGPRP, targeted at down-regulating PKM2 are able to selectively kill cancer cells by depriving them of the energy needed to complete a successful mitosis whilst only causing a pause in the cell cycle in normal stem cells. Attacking aerobic glycolysis thus appears to be a subtle way of attacking the unique tendency of the cancer as opposed to the normal cell, to undergo unrestrained mitotic division.
Conclusion
Cancer cells can be best understood as hybrids, composed of a somatic mutation-induced immortal phenotype and a normal aerobic glycolysis phenotype. Chemotherapy has been designed in the past to attack cell division by causing irreparable damage to the molecules involved in DNA synthesis and, where it is still frequently used, can only be given for a limited number of courses because of accumulating damage in normal cells. Around 568 different potential targets have now been described as resulting from somatic mutations affecting the on and off signals controlling cell division and creating the immortal cancer phenotype [44]. Rapid advances in DNA sequencing in cancer have led to the potential approach of personalized therapy by selectively targeting specific mutations responsible for increased cell division in individual patients with less damage to normal cells. These mutated targets suffer, however, from uneven heterogenous phenotypic expression and target disappearance due to genetic instability which are likely to lead to drug resistance. Similar constraints apply to the neoantigens on the surface of cancer cells which are targeted by the body’s own immune system. Further therapeutic advances in cancer would benefit from druggable non-mutant stable targets. It has been suggested that the PKM2 isoform of the pyruvate kinase gene responsible for aerobic glycolysis, which is found ubiquitously throughout malignant cells, could provide such a target, making it the true “Achilles Heel” of cancer. Spontaneous cell death in cancer cells but not normal cells make them already more vulnerable than normal cells to energy depletion which would provide targeting aerobic glycolysis in general with a selective advantage. More specifically targeting ATP production by PKM2 deprives cancer cells driven into cytokinesis by mitotic addiction of the required energy to complete division into two daughter cells.
The whole person dietary approach of limotherapy, has not infrequently been proposed as a way of treating cancer [45] but it applies to the whole soma of an intact individual, both cancer and normal and does not comply with the desired dictum “The tumor should shrink faster than the patient”. Here a new therapy is described that applies limotherapy exclusively to the cancer cells but not to adjacent or distant normal cells within the body. Non-nucleate mature erythrocytes which like cancer cells depend on aerobic glycolysis should not be affected by this treatment because they use the PKR enzyme which does not contain the same SDPTEA target as PKM2.
The discovery that the unique hexapeptide sequence PRGPRP in an external loop of the C’-terminus of Cdk4 modulates the production of ATP by PKM2 has led to the development of ONKONEK [18,32], a first in class example of the new therapeutic paradigm of selective cancer cell limotherapy, a global cancer therapeutic approach which does not damage normal cells but depletes ATP in cancer cells. The putative site of action of PRGPRP congeners at the final cytokinetic step of mitosis provides a role for aerobic glycolysis in the context of the cell cycle and an explanation for why antimetabolic therapy can lead to the death of dividing cancer cells. Because the new therapeutic approach can potentially kill individual cancer cells within the body by intermittent starvation rather than by generalized irreparable molecular damage it has the potential to be administered repeatedly for as long as is necessary until the very last cancer cell is eradicated.
Acknowledgements
I am very grateful to Dr. Leonie Windeln and Professor Jonathan Essex for the computer simulated docking plausibility studies of THR53 with tetrameric PKM2 selected examples of which are shown in Figure 3.
Funding
Personal.
Ethics
No ethical approval required.
Clinical Trials
None.
Data Availability
All data is in earlier literature and in patent applications except new unpublished structural chemistry data in Figure 3.
References
2. Dodson M, Zhang DD. The pyrimidinosome is cancer's Achilles' heel. Nat Cell Biol. 2023 Jun;25(6):798–9.
3. Fan Y, Xue H, Li Z, Huo M, Gao H, Guan X. Exploiting the Achilles' heel of cancer: disrupting glutamine metabolism for effective cancer treatment. Front Pharmacol. 2024 Mar 6;15:1345522.
4. Iulianna T, Kuldeep N, Eric F. The Achilles' heel of cancer: targeting tumors via lysosome-induced immunogenic cell death. Cell Death Dis. 2022 May 30;13(5):509.
5. Berico P, Coin F. Is TFIIH the new Achilles heel of cancer cells? Transcription. 2018;9(1):47–51.
6. Gahramanov V, Vizeacoumar FS, Morales AM, Bonham K, Sakharkar MK, Vizeacoumar FJ, et al. Cancer Cell’s Seven Achilles Heels: Considerations for design of anti-cancer drug combinations. bioRxiv. 2023 Jan 3:2023-01.
7. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004 Nov;4(11):891–9.
8. Israelsen WJ, Vander Heiden MG. Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol. 2015 Jul;43:43–51.
9. Gavriatopoulou M, Paschou SA, Ntanasis-Stathopoulos I, Dimopoulos MA. Metabolic Disorders in Multiple Myeloma. Int J Mol Sci. 2021 Oct 22;22(21):11430.
10. Rellinger EJ, Craig BT, Alvarez AL, Dusek HL, Kim KW, Qiao J, et al. FX11 inhibits aerobic glycolysis and growth of neuroblastoma cells. Surgery. 2017 Mar;161(3):747–52.
11. Yan L, Raj P, Yao W, Ying H. Glucose Metabolism in Pancreatic Cancer. Cancers (Basel). 2019 Sep 29;11(10):1460.
12. Warenius HM. Are critical normal gene products in cancer cells the real therapeutic targets? Anticancer Res. 2002 Sep-Oct;22(5):2651–5
13. Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016 Mar;41(3):211–8.
14. DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab. 2020 Feb;2(2):127–9.
15. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009 May 22;324(5930):1029–33.
16. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57–70.
17. Warenius HM. The essential molecular requirements for the transformation of normal cells into established cancer cells, with implications for a novel anti-cancer agent. Cancer Rep (Hoboken). 2023 Aug;6(8):e1844.
18. Warenius HM. Selectively depleting the energy of cancer cells: A new therapeutic paradigm. J Cancer Biol. 2024 Feb 5;5(1):1–10.
19. Chen X, Yi C, Yang MJ, Sun X, Liu X, Ma H, et al. Metabolomics study reveals the potential evidence of metabolic reprogramming towards the Warburg effect in precancerous lesions. J Cancer. 2021 Jan 10;12(5):1563–74.
20. Cruz MD, Ledbetter S, Chowdhury S, Tiwari AK, Momi N, Wali RK, et al. Metabolic reprogramming of the premalignant colonic mucosa is an early event in carcinogenesis. Oncotarget. 2017 Mar 28;8(13):20543–57.
21. Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013 Dec;23(12):620–33.
22. Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997 May 15;57(10):1835–40.
23. Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, function and expression and regulation. Cell Biosci. 2019 Jun 26;9:52.
24. Chen X, Chen S, Yu D. Protein kinase function of pyruvate kinase M2 and cancer. Cancer Cell Int. 2020 Oct 29;20(1):523.
25. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol. 2012 Dec;14(12):1295–304.
26. Zhou C, Chen T, Xie Z, Qin Y, Ou Y, Zhang J, et al. RACK1 forms a complex with FGFR1 and PKM2, and stimulates the growth and migration of squamous lung cancer cells. Mol Carcinog. 2017 Nov;56(11):2391–9.
27. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011 Dec 1;480(7375):118–22.
28. Zhou Z, Li M, Zhang L, Zhao H, Şahin Ö, Chen J, et al. Oncogenic Kinase-Induced PKM2 Tyrosine 105 Phosphorylation Converts Nononcogenic PKM2 to a Tumor Promoter and Induces Cancer Stem-like Cells. Cancer Res. 2018 May 1;78(9):2248–61.
29. Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012 Oct;8(10):839–47.
30. Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure. 1998 Feb 15;6(2):195–210.
31. Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol Cell. 2012 Mar 9;45(5):598–609.
32. Warenius HM, Kilburn JD, Essex JW, Maurer RI, Blaydes JP, Agarwala U, et al. Selective anticancer activity of a hexapeptide with sequence homology to a non-kinase domain of Cyclin Dependent Kinase 4. Mol Cancer. 2011 Jun 13;10:72.
33. Pollard TD. Mechanics of cytokinesis in eukaryotes. Curr Opin Cell Biol. 2010 Feb;22(1):50–6.
34. Green RA, Paluch E, Oegema K. Cytokinesis in animal cells. Annu Rev Cell Dev Biol. 2012;28:29–58.
35. Ohba S, Tang Y, Johannessen TA, Mukherjee J. PKM2 Interacts With the Cdk1-CyclinB Complex to Facilitate Cell Cycle Progression in Gliomas. Front Oncol. 2022 Mar 22;12:844861.
36. Lang Y. Research Progress on the Role of PKM2 in the Cell Cycle of Tumor Cells and Immune Cells. Advances in Life Science for Adolescents. 2025 May 28;1(1).
37. Jiang Y, Li X, Yang W, Hawke DH, Zheng Y, Xia Y, et al. PKM2 regulates chromosome segregation and mitosis progression of tumor cells. Mol Cell. 2014 Jan 9;53(1):75–87.
38. Jiang Y, Wang Y, Wang T, Hawke DH, Zheng Y, Li X, et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat Commun. 2014 Nov 21;5:5566.
39. Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol. 2005 Jul;15(7):371–7.
40. Potts SJ, Krueger JS, Landis ND, Eberhard DA, Young GD, Schmechel SC, et al. Evaluating tumor heterogeneity in immunohistochemistry-stained breast cancer tissue. Lab Invest. 2012 Sep;92(9):1342–57.
41. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–9.
42. Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008 May 1;68(9):3077–80.
43. Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, Bononi A, et al. ATP synthesis and storage. Purinergic Signal. 2012 Sep;8(3):343–57.
44. Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C, et al. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020 Oct;20(10):555–72.
45. Safdie FM, Dorff T, Quinn D, Fontana L, Wei M, Lee C, et al. Fasting and cancer treatment in humans: A case series report. Aging (Albany NY). 2009 Dec 31;1(12):988–1007.