Introduction
Lung cancer continues to be the leading cause of cancer deaths worldwide [1]. Approximately 85% of lung cancers are histologically classified as non-small cell lung cancer (NSCLC) of which adenocarcinoma is the most frequent subtype. KRAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) is one of the most commonly mutated genes in human cancers [2]. KRAS mutations are the most common oncogenic driver mutations in adenocarcinoma of the lung and can be found in up to a quarter of patients [3,4]. The vast majority (95%) are associated with a history of nicotine use. In NSCLC, KRAS mutations most often occur in codons 12 and 13 and with a lower frequency in codon 61 [4]. The predominant mutations are G12C, G12V and G12D [5]. The KRAS G12C mutation is present in approximately 13% of patients with NSCLC [2]. KRAS mutations do generally not overlap with other oncogenic mutations. KRAS mutant NSCLC has been reported to be associated with shorter median overall survival (OS) and lower two-year survival rates, however, there is conflicting data on the prognostic significance of KRAS mutations [6-8].
The RAS proteins regulate signal transduction by activating different effectors, thereby controlling various cellular functions. There are three genes related to human tumors in the RAS gene family: Harvey rat sarcoma viral oncogene (HRAS), KRAS and Neuroblastoma rat sarcoma viral oncogene (NRAS), which are located on chromosomes 11, 12, and 1, respectively [9]. Among them, KRAS by far represents the most important one. The KRAS protein is missing a “pocket” for smallmolecule binding rendering drug development more difficult for targeted treatment. For decades, numerous efforts were made to target KRAS and its downstream pathways. The bestknown downstream pathway is the mitogen activated protein kinase (MAPK)-pathway, which consists of the RAS-rapidly accelerated fibrosarcoma (RAF)-extracellular signal-regulated kinase (MEK)-ERK signaling cascade (Figure 1). However, when activated, KRAS relays on upstream signals from cell surface receptors to additional downstream pathways, including PI3K-AKT-mTOR, RALGDS-RAL and TIAM-RAC, that also control normal cell function and proliferation and add further complexity for drug development.
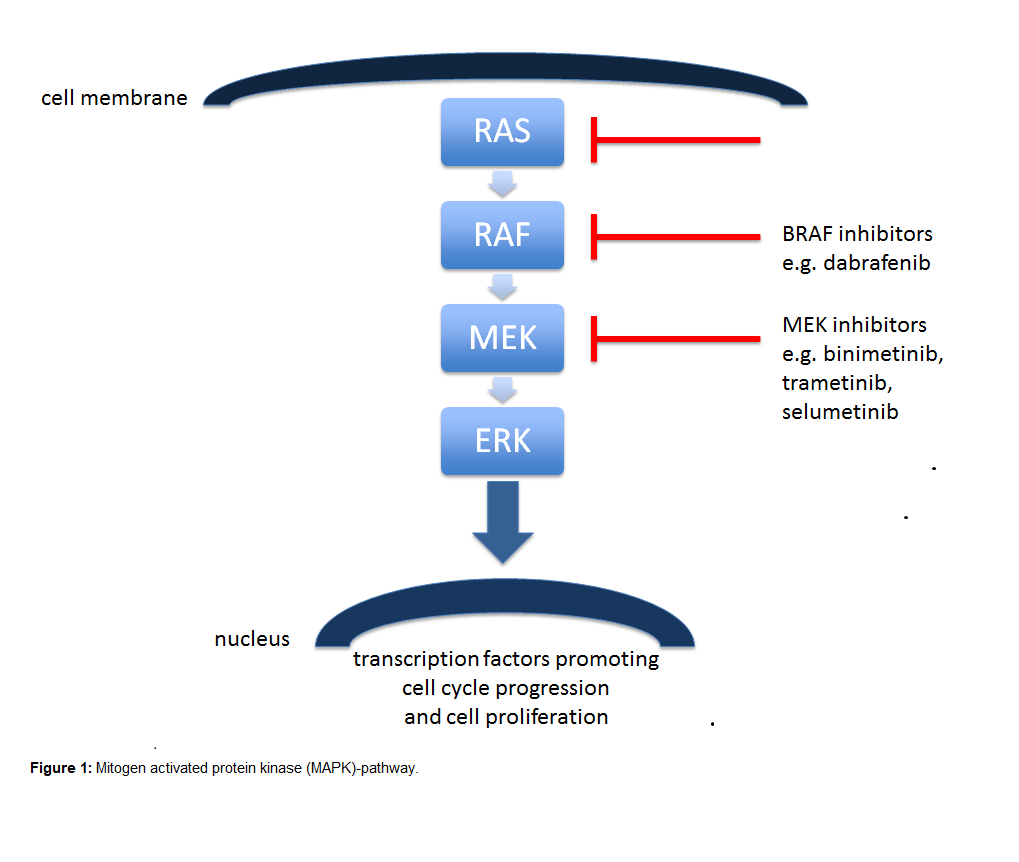
The first class of drugs directed to KRAS were the farnesyltransferase inhibitors (FTIs) tipifarnib, lonafarnib and second-generation salirasib. However, these trials were unsuccessful, in part due to the compensatory effect of the enzyme geranyltransferase, that among others processes KRAS to let it bind to cell membranes and to exert its activity. MEK is a serine/threonine kinase, a downstream signal of KRAS and BRAF. Activated RAF activates MEK, which activates ERK and other transcription factors promoting cell cycle progression and cell proliferation. MEK1/MEK2 inhibitors, selumetinib (AZD6244, ARRY-142886) and trametinib (GSK1120212, JTP- 74057) as monotherapy didn’t improve outcomes of patients with KRAS mutant NSCLC [10,11]. A potential reason may be that MEK inhibitors induce drug resistance by increased upstream signaling leading to ERK signaling activation [12].
The rationale for performing the SAKK 19/16 trial was the unmet need to treat patients with KRAS mutated lung cancer [13]. The trial was developed based on preclinical and clinical evidence, that MEK-Inhibition plus chemotherapy may improve outcomes [14-17], although there have also been disappointing results such as in the SELECT-1 trial, which showed no benefit for the addition of selumetinib to docetaxel as second-line therapy for advanced KRAS mutant NSCLC [10]. At time of start of the SAKK 19/16 trial, a cisplatin combination was recommended as first-line treatment in advanced and metastatic NSCLC based on a meta-analysis by Ardizzoni et al. [18]. Pemetrexed and cisplatin were therefore chosen as chemotherapy backbone in combination with binimetinib (MEK162). Binimetinib is an oral inhibitor of the MAPK kinases MEK1 and MEK2. The aim of the SAKK 19/16 trial was to determine the recommended phase 2 dose of binimetinib and assess early anti-tumor activity. Cisplatin, pemetrexed and binimetinib were administered for 4 cycles followed by a binimetinib and pemetrexed maintenance until disease progression (PD) or unacceptable toxicity. The protocol suggested maintenance pemetrexed as standard treatment given a randomized phase III trial previously has demonstrated a survival benefit [19]. For binimetinib an intermittent schedule (d1-14 q21 days) was chosen because MEK inhibitors have shown to be more active as pulsatile instead of continuous treatment in preclinical studies [20]. Eligible patients had treatment naive, metastatic or locally advanced NSCLC unsuitable for curative treatment and a KRAS mutation (codon 12, 13 or 61). Eighteen patients were enrolled. The primary endpoint were dose-limiting toxicities (DLT), and two different dose levels of binimetinib (30 and 45 mg BID) were explored. Secondary endpoints included adverse events (AE) and objective response rate (ORR), progression-free survival (PFS) and OS. DLT did not occur in the nine evaluable patients. Thus, 45mg binimetinib BID was declared to be the maximum tolerated dose (MTD). In 9 out of 10 patients treated at the MTD at least one serious adverse event (SAE) occurred. The most common treatment-related grade 3 (G3) AE of all 16 evaluable patients for safety were lung infection (25%), fatigue (19%) and anemia (19%). Of the 10 patients who received binimetinib at the MTD fatigue (30%), nausea (20%), anemia (20%), hypertension (20%) and lung infection (20%) were the most common G3 AEs. The ORR was 29% (95% CI, 35-87) in all 14 evaluable patients and 33% (95% CI, 7-70) in the nine patients treated at the MTD. PFS and OS in the MTD group were 5.7 months (95% CI, 1.1-14.0) and 6.5 months (95% CI, 1.8–not reached).
Although DLT was not observed, the high rates of SAEs and the significant proportion of patients not evaluable for DLT indicate tolerability issues, that were most likely mainly chemotherapy-related. As the patients with KRAS mutations generally have a significant history of nicotine use resulting in relevant comorbidities and impaired performance status, retrospectively, carboplatin likely might have been a better tolerable choice.
In summary, the addition of binimetinib to a cisplatin-based first-line chemotherapy seems to be feasible with no DLT at a dose of 45 mg binimetinib BID, although the high rates of SAEs should be taken into account. However, no sign of improved anti-tumor activity of the treatment combination of cisplatin, pemetrexed and binimetinib as first-line therapy in KRAS mutated metastatic NSCLC was observed.
MEK Inhibition in KRAS Mutant NSCLC – Quo vadis?
In a more recent phase I study by Fung et al., binimetinib was investigated in combination with carboplatin and pemetrexed as chemotherapy backbone in patients with stage IV nonsquamous NSCLC [21]. No prior chemotherapy was allowed. Unlike in the SAKK 19/16 trial, patients with KRAS wild type, sensitizing EGFR mutations or ALK-fusions or high PD-L1 levels were eligible if previously treated with the respective tyrosine kinase inhibitors (TKIs) or immune checkpoint inhibitors. In this study 6 cycles of binimetinib, carboplatin and pemetrexed were administered followed by a maintenance therapy with pemetrexed/binimetinib until PD. Whereas binimetinib in the SAKK 19/16 trial was administered for 14 days in a 21-day cycle, in the study by Fung et al. binimetinib was administered for days 1-5 in cycle 1 followed by chemotherapy on day 8 and binimetinib from day 8-26. In the following cycles (21 daycycles) binimetinib was given continuously BID except for a washout period 2 days prior to the next chemotherapy (except for dose level -1, in which binimetinib was administered on days 1-14 of each cycle starting from cycle 2). Aim of the study was to determine the recommended phase 2 dose and to assess safety. With their schedule of binimetinib, two patients reached a DLT event at 45 mg BID (elevated alanine aminotransferase (ALT) for longer than 7 days and G3 ocular toxicity) resulting in a recommended dose of 30mg binimetinib BID. AEs of any grade occurred in all 13 enrolled patients with eight patients developing a G3/4 AE. In total, 12/13 patients were evaluated for efficacy with an ORR of 50% (95% CI, 21.1-78.9) by investigator assessment and of 33.4% by independent review (95% CI, 9.9-65.1). The median PFS was 4.5 months (95% CI, 2.6-NA). Seven patients enrolled in this study harbored a KRAS mutation, among those the ORR was 57.1% by investigator assessment and 28.6% by independent review. Although disease control rate (DCR) was 100% in patients harboring a KRAS mutation and ORR were slightly higher in KRAS mutated patients no meaningful conclusion can be drawn from this small sample size with regards to activity of the combination. In addition, no information on the PFS in the KRAS mutated subgroup was provided. Despite the DLTs, the study by Fung et al. showed a slightly more manageable toxicity profile compared to the SAKK 19/16 trial, which used a cisplatin combination (G3 or higher AE rate of 61.5% versus 66.7% in the SAKK 19/16 trial). The ORR of the two studies is comparable and lack a clear signal of additional activity of binimetinib in this situation that is in line with previous studies evaluating other MEK inhibitors with platinum-doublet chemotherapy [22-24].
Although KRAS is the oncogene most frequently mutated in human cancer, targeting the RAS-RAF-MEK-ERK pathway remains a challenge. Most recently, the focus of drug development laid on the codon variant in the KRAS protein G12C accounting for 39% of KRAS mutations in NSCLC, followed by G12V (21%) and G12D (17%) [5]. The incidence of KRASG12C is therefore similar to the one of EGFR mutations in a Western European population. Recently, sotorasib, a covalentKRASG12C inhibitor locking KRAS in its inactive GDP-bound state by irreversibly binding to the switch II pocket [25], was evaluated in the CodeBreaK 100 study in patients with previously treated advanced or metastatic KRASG12C mutated cancer (n=129) [26]. The ORR for patients with NSCLC (59/129 patients) was 32.2% (95% CI, 20.62-45.64) and the DCR 88.1% (95% CI, 77.07-95.09). AEs of G3 or higher were reported in 52.7% of all patients (68/129 patients). These results led to accelerated approval of sotorasib for advanced and metastatic NSCLC by the FDA in May 2021. Sotorasib is currently investigated further in several ongoing studies. Adagrasib is another KRASG12C inhibitor currently in clinical development. It is a potent inhibitor that irreversibly and selectively binds KRASG12C and locks it in its inactive state. Seventy-nine pretreated patients with NSCLC were enrolled in the KRYSTAL-1 trial and received adagrasib 600mg BID. Of 51 patients evaluable for efficacy 23 patients (45%) showed a partial remission and 26 patients had stable disease [27]. Sotorasib and adagrasib have also recently shown early signs of activity in KRASG12C mutated colorectal cancer in combination with panitumumab and cetuximab, respectively [28,29]. However, the results of the prespecified analysis of the CodeBreaK100 trial for previously treated KRASG12C mutant colorectal cancers showed a modest anti-tumor activity and manageable safety but did not reach the benchmark with an ORR of 9.7% (6/62 patients, 95% CI, 3.6-19.9) [30]. Given these recent developments with selective KRASG12C inhibitors demonstrating activity in a further subset of KRAS mutated NSCLC patients as well as the emergence of immunotherapy (which has significant activity particularly in the subset of KRAS mutated NSCLC) [31], the two first-line trials investigating binimetinib plus chemotherapy discussed above have to be viewed in this new context and the question arises: Quo vadis MEK inhibition in KRAS mutated NSCLC?
There are numerous approaches ongoing targeting various components of the RAS-RAF-MEK-ERK pathway: HL-085, a novel ATP non-competitive MEK inhibitor is explored in a phase I trial (ClinicalTrials.gov Identifier: NCT03990077). In addition, pulse MEK inhibition combined with CTLA-4 blockade may prolong the survival time of KRAS mutant tumors in mice, possibly due to T-cell activation and increased CTLA-4 expression resulting from pulse therapy. Furthermore, the combination of a MEK- and RAF-inhibitor may also be promising as currently investigated in a phase I trial of LXH254 and trametinib as well as belvarafenib combined with cobimetinib (ClinicalTrials. gov Identifier: NCT02974725 and NCT03284502). Finally, VS- 6766 (RO5126766), a novel targeted drug that inhibits both MEK and RAF, is being evaluated in a phase II clinical study (ClinicalTrials.gov Identifier: NCT04620330). SHP2 plays a crucial role in KRAS mutation-driven tumors. It is involved in the downstream signal transduction of various growth factors and cytokines. The combination of SHP2 with MEK inhibitors resulted in a synergistic effect to control tumor growth in KRAS mutant NSCLC xenograft models [32]. A single drug phase I study and a clinical trial in combination with pembrolizumab are ongoing (ClinicalTrials.gov Identifier: NCT03634982, ClinicalTrials.gov Identifier: NCT04418661).
Since the inhibition of the MAPK-pathway activates the PI3K-pathway, reducing KRAS mutated cell sensitivity to MEK inhibitors, an approach to target the PI3K-AKT-mTOR- and RAF-MEK-ERK-pathways simultaneously could be promising, but potentially results in increased toxicity. Likewise, if in KRAS mutant NSCLC MEK is inhibited, STAT3 is activated via fibroblast growth factor receptor and Janus kinase, thus combined inhibition of MEK and Janus kinase has shown to result in tumor regression [33]. Finally, as ERK is the final kinase in the MAPK-pathway, the resistance of KRAS mutated tumors to RAF or MEK inhibitors is frequently caused by ERK feedback activation. Combined inhibition of ERK may be another strategy to prevent drug resistance. Currently, ERK inhibitors such as JSI-1187-01 and ASN007 are in phase I clinical trials (ClinicalTrials.gov Identifier: NCT04418167 and NCT03415126), respectively.
In summary, MEK inhibitors could still play a role in the future in the treatment of KRAS mutated advanced NSCLC patients and may find their place as combination partners with other targeted agents given the complexity of the RAS-RAF-MEKERK- pathway and/or immunotherapy. Results of ongoing respective trials are eagerly awaited.
References
2. Consortium AP. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017;7(8):818-31.
3. Sequist LV, Heist RS, Shaw AT, Fidias P, Rosovsky R, Temel JS, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22(12):2616-24.
4. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba, II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998-2006.
5. Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smokingrelated KRAS-mutant cancers. Clin Cancer Res. 2012;18(22):6169-77.
6. El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, et al. Characteristics and Outcomes of Patients With Metastatic KRASMutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol. 2019;14(5):876-89.
7. Graziano SL, Gamble GP, Newman NB, Abbott LZ, Rooney M, Mookherjee S, et al. Prognostic significance of K-ras codon 12 mutations in patients with resected stage I and II non-small-cell lung cancer. J Clin Oncol. 1999;17(2):668-75.
8. Schiller JH, Adak S, Feins RH, Keller SM, Fry WA, Livingston RB, et al. Lack of prognostic significance of p53 and K-ras mutations in primary resected non-small-cell lung cancer on E4592: a Laboratory Ancillary Study on an Eastern Cooperative Oncology Group Prospective Randomized Trial of Postoperative Adjuvant Therapy. J Clin Oncol. 2001;19(2):448-57.
9. Burns TF, Borghaei H, Ramalingam SS, Mok TS, Peters S. Targeting KRAS-Mutant Non-Small-Cell Lung Cancer: One Mutation at a Time, With a Focus on KRAS G12C Mutations. J Clin Oncol. 2020;38(35):4208-18.
10. Janne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crino L, et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA. 2017;317(18):1844-53.
11. Blumenschein GR Jr., Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC). Ann Oncol. 2015;26(5):894-901.
12. Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: The role of MEK. Cancer Treat Rev. 2017;53:61-9.
13. Froesch P, Mark M, Rothschild SI, Li Q, Godar G, Rusterholz C, et al. Binimetinib, pemetrexed and cisplatin, followed by maintenance of binimetinib and pemetrexed in patients with advanced non-small cell lung cancer (NSCLC) and KRAS mutations. The phase 1B SAKK 19/16 trial. Lung Cancer. 2021;156:91-9.
14. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebocontrolled, phase 2 study. Lancet Oncol. 2013;14(1):38-47.
15. Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, et al. AZD6244 (ARRY-142886), a potent inhibitor of mitogenactivated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/ pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6(8):2209-19.
16. Gandara DR, Hiret S, Blumenschein GR, Barlesi F, Delord J-P, Madelaine J, et al. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with docetaxel in KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): A phase I/Ib trial.American Society of Clinical Oncology; 2013.
17. Kelly K, Mazieres J, Leighl NB, Barlesi F, Zalcman G, Gordon MS, et al. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with pemetrexed for KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): A phase I/Ib trial. American Society of Clinical Oncology; 2013.
18. Ardizzoni A, Boni L, Tiseo M, Fossella FV, Schiller JH, Paesmans M, et al. Cisplatin- versus carboplatin-based chemotherapy in firstline treatment of advanced non-small-cell lung cancer: an individual patient data meta-analysis. J Natl Cancer Inst. 2007;99(11):847-57.
19. Paz-Ares LG, de Marinis F, Dediu M, Thomas M, Pujol JL, Bidoli P, et al. PARAMOUNT: Final overall survival results of the phase III study of maintenance pemetrexed versus placebo immediately after induction treatment with pemetrexed plus cisplatin for advanced nonsquamous non-small-cell lung cancer. J Clin Oncol. 2013;31(23):2895-902.
20. Choi H, Deng J, Li S, Silk T, Dong L, Brea EJ, et al. Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer. Cell Rep. 2019;27(3):806-19 e5.
21. Fung AS, Graham DM, Chen EX, Stockley TL, Zhang T, Le LW, et al. A phase I study of binimetinib (MEK 162), a MEK inhibitor, plus carboplatin and pemetrexed chemotherapy in non-squamous nonsmall cell lung cancer. Lung Cancer. 2021;157:21-9.
22. Melosky B, Bradbury P, Tu D, Florescu M, Reiman A, Nicholas G, et al. Selumetinib in patients receiving standard pemetrexed and platinum-based chemotherapy for advanced or metastatic KRAS wildtype or unknown non-squamous non-small cell lung cancer: A randomized, multicenter, phase II study. Canadian Cancer Trials Group (CCTG) IND.219. Lung Cancer. 2019;133:48-55.
23. Greystoke A, Steele N, Arkenau HT, Blackhall F, Md Haris N, Lindsay CR, et al. SELECT-3: a phase I study of selumetinib in combination with platinum-doublet chemotherapy for advanced NSCLC in the first-line setting. Br J Cancer. 2017;117(7):938-46.
24. Goffin JR, Nicholas G, Mates M, Tu D, Chen E, Laurie SA, et al. Canadian Cancer Trials Group (CCTG) IND215: A phase Ib study of Selumetinib in patients with untreated advanced or metastatic NSCLC who are receiving standard chemotherapy regimens. Invest New Drugs. 2019;37(3):498-506.
25. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217-23.
26. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383(13):1207-17.
27. Jänne P, Rybkin II, Spira A, Riely G, Papadopoulos K, Sabari J, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in advanced/ metastatic non–small-cell lung cancer (NSCLC) harboring KRAS G12C mutation. European Journal of Cancer. 2020;138:S1-S2.
28. Weiss J, Yaeger R, Johnson M, Spira A, Klempner S, Barve M, et al. LBA6 KRYSTAL-1: Adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Annals of Oncology. 2021;32:S1294.
29. Fakih M, Falchook G, Hong D, Yaeger R, Chan E, Mather O, et al. 434P CodeBreaK 101 subprotocol H: Phase Ib study evaluating combination of sotorasib (Soto), a KRASG12C inhibitor, and panitumumab (PMab), an EGFR inhibitor, in advanced KRAS p. G12Cmutated colorectal cancer (CRC). Annals of Oncology. 2021;32:S551.
30. Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, et al. Sotorasib for previously treated colorectal cancers with KRASG12C mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. The Lancet Oncology. 2021.
31. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30(8):1321-8.
32. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med. 2018;24(7):954-60.
33. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26(2):207-21.