Abstract
MAGIs (membrane-associated guanylate-kinases (MAGUK) inverted) are apical scaffolds conserved across evolution, which regulate cellular junctions. Low expression of MAGIs has been associated with tumorigenesis in a wide variety of cancers. This “tumor-suppressive” function of MAGIs has stimulated many studies to better understand the processes they control, and how their misregulation could contribute to cancer progression. In this mini review, we will describe and discuss the recent advances concerning the role of MAGIs, and propose a potential framework to explain the link between the junctional role of MAGIs and the variety of signaling pathways found altered upon MAGIs loss in cancer cells. We argue that through the diversity of MAGIs’ partners at the cell junctions, their impact on actin dynamics regulation, and the cellular contexts (other scaffolds or other actin regulators such as the AMOTs, NF2, etc.), the loss of MAGIs impacts different pathways, such as the PTEN/Akt, β-catenin, Hippo/YAP, or p38 pathways, to fuel tumorigenicity.
Keywords
MAGI, Cell junctions, Actin tension, ROCK, PTEN/Akt, Wnt/β-catenin, Hippo/YAP, p38 stress signaling pathways
MAGI Scaffolds and Cellular Junctions
The vertebrate epithelial tight junction-associated protein MAGIs belong to a family of membrane-associated guanylatekinases (MAGUK) containing MAGI-1, MAGI-2 and MAGI-3. Invertebrate genomes encode for only one MAGI orthologue, named MAGI-1 in Caenorhabditis elegans and Magi in Drosophila melanogaster. MAGI scaffolds are recruited to Tight Junctions (TJs) and Adherens Junctions (AJs) thereby connecting them with the actin cytoskeleton through a scaffolding function [1-3]. For instance, in mammalian epithelial cells, MAGI1 colocalizes with ZO-1 and α-4-actinin at apical TJs [2] and with E-cadherin and β-catenin at the basolateral AJs [4]. Similar junctional localizations along the lateral membranes are also found in invertebrate epithelial cells [5,6].
Due to the lack of redundancy, invertebrate models offer a unique opportunity to probe the function of MAGI scaffolds through loss-of-function approaches. In C. elegans, MAGI- 1 was shown to interact genetically with the cadherin/ catenin complex to define junction domains. Removing MAGI-1 induced a loss of the overall robustness of cell-cell adhesion, suggesting that MAGI-1 functions as an organizer that is able to ensure a correct segregation of the different cell adhesion complexes into distinct domains along the lateral plasma membrane [7]. In Drosophila, Magi localizes at the AJs, overlapping with AJs regulators and apical polarity determinants Par3/Bazooka and aPKC [8]. Using loss-offunction approaches, our lab showed that Magi, although dispensable for early development, is required in the developing eye in particular during junctions remodeling. We further showed that Magi, through its physical interaction with RASSF8, controls ASPP and Par3/Bazooka localization at AJs. We have then proposed that, by controlling the proper localization of Bazooka to remodeling junctions, Magi and the RASSF8-ASPP complex promoted the recruitment or stabilization of E-cadherin complexes at junction sites to regulate the integrity of the cell junctions ([6] and reviewed by Coopman et Djiane [9]). In these different studies, the main defects identified were at intercellular junctions, impacting morphogenesis and developmental apoptosis [6,7], supporting that the main function of MAGI scaffolds, consistent with their localizations, lies in junctional regulation. The defects were subtle suggesting that other complexes must act in parallel to MAGI scaffolds to regulate junctions.
Lynch et al. reported physical interactions between MAGI- 1, AFD-1/afadin, and SAX-7/L1CAM, part of a functional interactome that includes components of the core Cadherin/ Catenin complex, suggesting that MAGI-1 helps to partition and maintain a stable and spatially ordered apical junction during morphogenesis [5]. A recent study using elegant genetically engineered mouse EpH4 epithelial cells extended reports made in Drosophila, showing that MAGIs recruit the Par-3 regulator ASPP2 and control its localization to apical junctional complexes (AJC) in part through RASSF proteins. This MAGI/RASSF/ASPP complex modulates Par-3/aPKC recruitment and junction morphology: loss of MAGIs results in highly heterogeneous junctions across the epithelial sheet resulting in cells with very different apical sizes [10]. Mechanistically, the authors then showed that MAGI complexes control Rho-kinase ROCK localization and antagonize ROCKdriven contractility placing MAGIs in the mutual antagonism between Par-3/aPKC and ROCK in the maintenance of the homeostasis of intercellular tension [10]. Work from our lab in luminal breast cancer cell lines further supported a role for MAGI1 in the fine regulation of AJs in human breast cancer cell lines: MAGI1 deficient cells accumulated AJs resident proteins such as E-cadherin which extended basolaterally. MAGI1 knockdown also led to the accumulation of the AJs protein and actin cytoskeleton regulator AMOTL2. The link between MAGI scaffolds and actin dynamics was finally further supported by the observation that in MAGI1 deficient cells, cells were stiffer as measured by atomic force microscopy and subjected to higher ROCK activity [11].
The junctional functions of MAGIs have also been extensively studied in podocytes, specialized epithelial cells of the Bowman’s capsule in the kidneys. In mice models, Magi1 or Magi2 loss of functions are associated with tighter slit diaphragm and thus filtration problems. At the cellular level, MAGIs interact physically with several transmembrane junctional proteins such as JAM-4 or Nephrin and loss of Magi1 or Magi2 results in abnormal localization of different junctional proteins and in particular in the accumulation of Claudin 5 [12-15] suggesting that the filtration problems are a consequence of altered junctions. In cultured podocytes, Magi1 has been shown to promote Rap1 signaling (activated Rap1 levels) in particular during junctional challenge with calcium switch assay [15]. Rap1 is a small G-protein implicated in the regulation of many biological processes (e.g. junctions remodeling) and more particularly in the regulation of actin cytoskeleton dynamics through its main inhibitory effect on RhoA and ROCK (via Araf3 or Arhgap29 [16,17]).
In vertebrates, besides epithelial cells, MAGIs have also been associated with the regulation of endothelial cell junctions. In human umbilical vein endothelial (HUVECs) and human arterial endothelial (HAECs) cells, MAGI1 regulates the activation of RAP1 that is responsible for the maintenance and the maturation of AJs. MAGI1 localized to cell-cell contacts forming a complex with vascular E-cadherin (VE-cadherin) and β-catenin and through its binding partner PDZ-GEF1 (also known as RAPGEF2), promotes RAP1 [18]. Similarly, to what could be seen in epithelial cells, this activation of RAP1 signaling in endothelial cells could thus be predicted to inhibit ROCK activity since studies showed that Rap1 signaling in endothelial cells inhibits RhoA signaling via Arhgap29 [16]. Studies in zebrafish also demonstrated a key role of AMOTL2 and its binding partner MAGI1 in bridging cadherin complexes and the actin cytoskeleton tension, ultimately controlling the morphogenesis of vessels [19]. Thus, in endothelial cells, similarly to what was described for epithelial cells, junctional MAGI scaffolds appear linked to small-G-protein activity regulation, actin cytoskeleton remodeling and potentially to ROCK activity. The vascular endothelial functions of MAGI1 were extensively depicted and discussed in a complete and recent review written by Wörthmuller et al. [20].
Alday-Parejo et al. recently identified MAGI1 as a novel component of focal adhesions (FAs) in endothelial cells. MAGI1 co-localized with paxillin at mature focal adhesion and actin stress fibers and regulated their dynamics [21]. The loss of MAGI1 induced a phosphorylation of paxillin consistent with increased focal adhesion turnover. In this context, overexpressed MAGI1 enhanced integrin-mediated endothelial cell adhesion to extracellular matrix proteins and promoted RhoA and Rac1 activation. At the cellular level, using HUVECs cells in vitro, the effects of MAGI1 translated in increased adhesion and decreased pseudo-capillary formation in vitro and angiogenesis in vivo [21]. RhoA is one of the main activators of ROCK activity, and it would be interesting to see whether in these conditions MAGI1 promotes higher ROCK activity. However, it should be noted that the link reported here between MAGI1 levels and ROCK activity in endothelial cells would be opposite to that seen in epithelial cells [10,11] or from what could have been predicted from the antagonism between Rap1 and RhoA signaling [16]. These opposing effects might reflect the complex interactions between Rap1 and RhoA signaling, in the context of different junctional compartments (AJs vs FAs). This new role of MAGI scaffolds at FAs in endothelial cells remains to be studied in other cell types and in particular epithelial cells.
Thus, works in different cell types and organisms firmly establish a core function of MAGI scaffolds in fine-tuning the link between cellular junctions and actin cytoskeleton dynamics (Figure 1). The fine details of this regulation remain poorly understood and the high number of potential actors, together with the existence of parallel scaffolds performing similar and redundant functions (e.g. ZO proteins [10]), suggests that the effects of MAGIs might be context specific. Nevertheless, studies so far point towards a potential common output in the regulation of small G-protein signaling and ROCK activity.
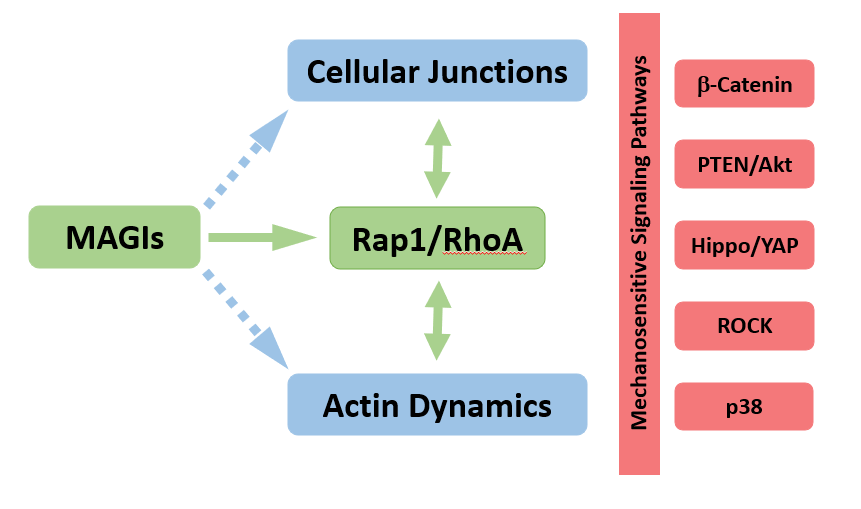
Figure 1: Model for MAGIs scaffolds function. While the effects on cellular junctions and actin dynamics represent a « core » function, their implications in terms of signaling pathways’ regulation are dependent on context (cell type, mutations), accounting for the diversity of signaling pathways misregulated in cancer cells with impaired MAGIs.
Besides their obvious adhesive role, cellular junctions also represent signalization hubs where signaling pathway components accumulate and cluster to form signalosomes. Junctional signalosomes can be modulated not only by classic ligand/receptor interactions but also by adhesive cues and cytoskeletal inputs, allowing integration of a wide array of signals. Pathways sensitive to mechanical cues or cellular architecture are thus particularly important to consider here in the context of processes controlled by MAGIs. In different cancer cell lines, junctional MAGI scaffolds have indeed been shown to control several signaling pathways. Although the pathways identified are diverse (for a recent and exhaustive review see [22]), it is possible that the underlying cause for signaling pathway modulation by MAGIs could be linked to their role as actin dynamics’ regulators (Figure 1).
MAGIs and the PTEN/Akt and β-Catenin Pathways
In several cancer cell lines, such as liver, colon, or renal cancer cell lines, MAGIs have been proposed to promote PTEN activity and thus inhibit the activity of the pro-tumoral kinase AKT. The first evidence of a MAGI1/PTEN signalosome came from the direct binding between MAGIs and PTEN, shown initially by yeast two-hybrid assay. The overexpression of MAGI1 in MDCK and 293 cells potentiated the interaction of PTEN with junctional complexes and promoted E-cadherin-dependent cell-cell aggregation [20,23-26].
A similar involvement of a PTEN/MAGI1 axis was also suggested in renal cell carcinoma, in which MAGI1 behaved as a tumor suppressor, inhibiting the invasion and migration of cells through a stabilization of the PTEN/MAGI1/β-catenin complex in order to inhibit the β-catenin signaling pathway [27]. In human T cells, MAGI1 inhibited Akt activity through its interaction with PTEN and MEK1 suggesting that a decreased expression of MAGI1 contributes to the development of several types of T-cell leukemia through the stimulation of both Akt and MEK pathways [28]. While the link between MAGIs and increased PTEN activity (and thus low Akt activity) is well established in the literature, one intriguing possibility would be that the strength of that link is under the control of junctions and actin dynamics. Indeed, PTEN activity is controlled by many post-translational modifications including phosphorylation by ROCK (reviewed in [29]) which appears mainly as an activator of PTEN activity. Moreover, complex cross talks exist between PTEN and junction dynamics [30,31]. In that respect, loss of MAGIs, which promotes increased ROCK activity [10,11], would not always result in lower PTEN activity through the compensatory activity of ROCK.
As suggested earlier, the effect of MAGIs on PTEN/Akt signaling was frequently concomitant with an action on canonical β-catenin signaling, a pathway frequently activated in tumor cells when cadherin complexes disassemble. Furthermore, Akt prevents GSK-3β activity, thus promoting β-catenin stabilization and signaling [32]. In brain tumors, low MAGI1 expression correlated with cancer progression in gliomas [25]. The mechanism of action of MAGI1 in glioma cells indicated a function via both Wnt/β-catenin and PTEN/ Akt pathways. More precisely, the knockdown of MAGI1 induced a decrease in E-cadherin protein level along with an increase in N-cadherin and vimentin indicating the induction of an EMT process. In addition, silencing MAGI1 significantly upregulated β-catenin and cyclin D1 levels in glioma cell lines with both PTEN expression reduction and significant increase in the levels of phospho-Akt [25]. A second study in glioma cells showed a potent tumor suppressor action of MAGI1 mainly through the PTEN/Akt pathway [33]. Similar PTEN/Akt and β-catenin signaling alterations have been proposed in response to MAGI1 knockdown in colon [4] and luminal breast cancer cells [34].
MAGIs and the Hippo/YAP Pathway
The Hippo pathway is a key regulator of cell growth, organ size and tissue homeostasis in response to diverse stimuli including cell density or actin cytoskeleton tension. Intense research identified several branches of the Hippo/YAP pathway. The “canonical” Hippo pathway is centered around the kinases Hippo/MST and Warts/LATS which ultimately phosphorylate and prevent YAP/TAZ to enter the nucleus and turn on the transcription of target genes (mainly proliferative and anti-apoptotic genes in cancer cells). But YAP/TAZ nucleocytoplasmic shuttling is also under the control of alternative pathways such as Src, actin cytoskeleton tension or cytoplasmic trapping by several proteins including AMOTs [35,36]. Several connections between MAGIs and YAP regulation have been reported.
First, MAGIs can physically interact with YAP regulators. Proteome-wide efforts have systematically mapped the proteins interacting with Hippo pathway components. This identified direct binding between LATS2 and MAGI1 [37]. Furthermore, Chastre et al. reported that MAGI1 directly interacted with TRIP6, a protein known to bind to LATS1/2 [3]. As mentioned previously, MAGI scaffolds also bind directly to AMOTs, apical scaffolds playing important roles in the regulation of cadherin-based junctions and their linkage to the actin cytoskeleton [38]. AMOTs are also negative regulators of YAP acting by sequestering it in the cytoplasm (reviewed in [35]). In particular, in luminal breast cancer cells, we have shown that AMOTL2 binds to MAGI1 through its PPxY motifs. In MAGI1 depleted cells, we described a dramatic accumulation of AMOTL2 and an exclusion of YAP from the nucleus [11]. This nuclear exclusion of YAP is consistent with previous studies showing that in epithelial cells with high levels of E-cadherin (as seen in MAGI1-depleted cells), β-catenin and NF2 act to exclude YAP from the nucleus [39]. Importantly we showed that AMOTL2 is required for the increased tumorigenicity of MAGI1-depleted luminal breast cancer cells, demonstrating that AMOTL2 is a critical mediator in this context. However, because YAP was excluded from the nucleus in MAGI1 deficient cells, the increased tumorigenicity of MAGI1 depleted cells could not be attributed to the “canonical” oncogenic role of YAP.
Second, it has also been reported that MAGI1 can control the expression of LATS1/2 at the mRNA level in endothelial cells. Abe et al. showed that MAGI1 post-translational modifications, such as S741 phosphorylation and K931 de-SUMOylation, led to endothelial cell activation [40]. Investigating further the relationship between MAGI1 and the Hippo pathway, the authors recently showed that MAGI1 depletion increased both mRNA and protein expression of LATS1/2, leading to inhibition of YAP/TAZ activity, thus participating in endothelial cell monolayer permeability [41].
Finally, MAGIs also emerge as relaying or sensing mechanical inputs, for instance in endothelial cells, where MAGI1 colocalizes with VE-cadherin at the level of cellular junctions under static and laminar flow conditions [42]. In this study, the authors then showed that MAGI1 expression was increased under fluid shear stress and its overexpression led to phosphorylation of PKA and eNOS in vitro and in vivo. The control of YAP activity by the actin cytoskeleton is well established in various cell types ranging from cultured fibroblasts to Drosophila wing imaginal discs epithelial cells (Reviewed in [43]). The control of Hippo/YAP by MAGIs could thus also be mediated by MAGIs’ control of actin dynamics. Of particular interest here again is the regulation of ROCK activity by MAGIs shown in different contexts [10,11]. Indeed, in mesenchymal cells (such as human mesenchymal stem cells), in response to stiff extracellular matrix, the actin tension driven in part by ROCK activity promotes YAP nuclear translocation [44]. Moreover, hyperactivation of RhoA/ROCK1/ actomyosin signaling through RhoA transfected MCF10A cells was recently shown to be both necessary and sufficient to drive oncogenic TEAD/YAP transcription [45]. However, the link between loss of MAGIs, higher ROCK activity, and thus YAP nuclear translocation, could not be observed in our study on luminal breast cancer cells depleted of MAGI1, where increased ROCK activity was accompanied by YAP retention in the cytoplasm [11]. These conflicting results suggest that the control of YAP localization and signaling by MAGIs might be controlled by several opposing mechanisms (ROCK on one hand but increased AMOTL2 and E-Cadherin on the other hand), ensuring a balanced response. Alternatively, it could also indicate that depending on the pool of actin, and where ROCK activity is triggered, different outputs on YAP localization might be achieved. More studies are thus needed to better understand the link between MAGI scaffolds and the control of Hippo/YAP signaling.
MAGIs and the ER Stress Pathway
In a recent study, Abe et al. nicely demonstrated in vivo that endothelial cell activation (NF-κB pathway activation) by various proinflammatory stimuli such as disturbed flow, TNF-α, or Thrombin (Thb), required MAGI1 and Rap1 signaling [40]. Furthermore, they demonstrated that MAGI1 was required during Thb-induced ER stress and proposed that MAGI1, through its binding to ATF6, supports ATF6 nuclear translocation where it promotes ER-stress response such as XBP-1 transcription [40]. Thus, this study supports a key role for the MAGI1/Rap1 module in supporting NF- κB signaling, even though the involvement of RhoA/ROCK remains to be explored. It also points towards an effect on ATF6 mediated ER-stress response. Interestingly, a separate in vivo study recently revealed that ROCK activation in mammary tumors selectively activates PERK, a kinase known to induce one branch of ER stress-induced unfolded protein response (UPR) [46]. PERK activation led to the expression and secretion of CRELD2 that instructed CAFs to enhance tumor growth. In this activated ROCK model, depleting CRELD2 suppressed tumor progression thus demonstrating that the paracrine ROCKPERK- ATF4-CRELD2 axis promotes breast cancer progression [46]. Given our results and that of others [10,11], suggesting increased ROCK activity upon MAGI1 loss, it would thus be interesting to investigate whether in epithelial cells the loss of MAGIs would trigger, via ROCK, PERK-ATF4 ER-stress response. Since in endothelial cells the presence of MAGI1 appears required for ATF6 ER-stress response [40], studies aiming at deciphering these seemingly opposite roles of MAGI1 on ERstress are needed.
MAGIs and the p38 Stress Pathway
In luminal breast cancer cell lines, we have shown that the loss of MAGI1 promoted the activation of the p38 stress signaling pathway. Importantly, p38 activity was required for several cancerous traits such as increased cell growth and survival in low attachment conditions upon MAGI1 depletion. We further showed that this elevated p38 activity required both AMOTL2 and ROCK activity suggesting that in response to MAGI1 loss, elevated junctional proteins and junction dysfunction, together with actin cytoskeletal tension could activate the p38 stress pathway to fuel tumorigenicity [11]. p38 signaling is typically activated in response to cellular stress, such as osmotic shock [47]. We thus propose a model where E-cadherin and AMOTL2 enrichment, together with increased ROCK activity and actin tension, result in compressive forces that could represent a new stress signal activating p38. Indeed, amongst the genes regulated transcriptionally by p38, SOX9, a gene mostly responsive to osmotic shock, was the most robustly activated upon MAGI1 knockdown [11].
Although the p38 signaling pathway has been primarily implicated as a tumor suppressor, recent studies demonstrated that it could also act as tumor promoting pathway depending on both cancer type and stage. Given the core function of MAGI scaffolds on junctions and actin dynamics, as well as reports linking MAGI loss and increased ROCK activity, it would be important to re-evaluate the role of the p38 pathway, and potentially other stress-response pathways controlled by ROCK, in cancer types in which loss of MAGIs represents a bad prognosis factor.
Conclusion
Low MAGIs expression levels have been shown to promote tumorigenesis in a wide variety of cancers. Besides their key role in cellular junctions biology, the loss of MAGIs in cancerous cells has been shown to regulate several key signaling pathways. Due to their nature as scaffold proteins, the diversity of their binding partners and the partly redundant functions undertaken by other junctional scaffolds as well as the complex interactions between the small G-proteins, Rap1 and RhoA, the regulation of signaling pathways by MAGIs appears to be context specific (Figure 1). Furthermore, the different processes controlled by MAGIs may have sometimes opposite effects on the signaling pathways, epitomizing the complex and intricate feedbacks occurring at junctions ensuring cellular homeostasis. Further studies should help uncover how such homeostatic responses are perverted in cancer cells, resulting in increased tumorigenicity when MAGIs scaffolds are lost.
Funding
Research in the laboratory is supported by the French National Agency ANR ANR-18-CE14-0041, Fondation de France and Ligue Régionale Contre le Cancer.
Competing and Declaration of Interests
The authors have approved the manuscript and agreed with the submission. None of the authors has conflict of interest to declare.
Abbreviations
AFD1: Afadin1; AJs: Adherens Junctions; AMOT: Angiomotin; aPKC: Atypical Protein Kinase C; ASPP: Apoptosis Stimulating Proteins of p53; CAFs: Cancer Associated Fibroblasts; CRELD2: Cysteine Rich With EGF Like Domains 2; EMT: Epithelial to Mesenchymal Transition; ER: Endoplasmic Reticulum; GSK3: Glycogen Synthase Kinase 3; HAECs: Human Aortic Endothelial Cells; HUVECs: Human Umbilical Vein Endothelial Cells; LAST1/2: Large Tumor Suppressor Kinase 1/2; L1CAM: L1 Cell Adhesion Molecule; MAGI: Membrane-Associated Guanylate- Kinases (MAGUK) Inverted; MEK1: MAP/ERK Kinase1; NF2: Neurofibromatose de type 2; PDZ-GEF1: PDZ-Guanine Nucleotide Exchange Factor; PERK: Protein Kinase RNA-like ER kinase; PTEN: Phosphatase and Tensin homolog; RASSF8: Ras Association Domain Family Member 8; RhoA: Ras homolog family member A; ROCK: Rho-associated protein kinase; SOX9: SRY-box transcription factor 9; TEAD: TEA Domain family member; TJs: Tight Junctions; TNF-α: Tumor Necrosis Factor alpha; TRIP6: Thyroid Hormone Receptor Interactor 6; UPR: Unfolded Protein Response; YAP: Yes-Associated Protein; ZO1: Zonula Occludens-1
References
2. Patrie KM, Drescher AJ, Welihinda A, Mundel P, Margolis B. Interaction of two actin-binding proteins, synaptopodin and α-actinin-4, with the tight junction protein MAGI-1. Journal of Biological Chemistry. 2002 Aug 16;277(33):30183-90.
3. Chastre E, Abdessamad M, Kruglov A, Bruyneel E, Bracke M, Di Gioia Y, et al. TRIP6, a novel molecular partner of the MAGI-1 scaffolding molecule, promotes invasiveness. The FASEB Journal. 2009 Mar;23(3):916-28.
4. Zaric J, Joseph JM, Tercier S, Sengstag T, Ponsonnet L, Delorenzi M, et al. Identification of MAGI1 as a tumor-suppressor protein induced by cyclooxygenase-2 inhibitors in colorectal cancer cells. Oncogene. 2012 Jan;31(1):48-59.
5. Lynch AM, Grana T, Cox-Paulson E, Couthier A, Cameron M, Chin-Sang I, et al. A genome-wide functional screen shows MAGI-1 is an L1CAM-dependent stabilizer of apical junctions in C. elegans. Current Biology. 2012 Oct 23;22(20):1891-9.
6. Zaessinger S, Zhou Y, Bray SJ, Tapon N, Djiane A. Drosophila MAGI interacts with RASSF8 to regulate E-Cadherin-based adherens junctions in the developing eye. Development. 2015 Mar 15;142(6):1102-12.
7. Stetak A, Hajnal A. The C. elegans MAGI-1 protein is a novel component of cell junctions that is required for junctional compartmentalization. Developmental Biology. 2011 Feb 1;350(1):24-31.
8. Padash Barmchi M, Samarasekera G, Gilbert M, Auld VJ, Zhang B. Magi is associated with the Par complex and functions antagonistically with Bazooka to regulate the apical polarity complex. PLoS One. 2016 Apr 13;11(4):e0153259.
9. Coopman P, Djiane A. Adherens Junction and E-Cadherin complex regulation by epithelial polarity. Cellular and Molecular Life Sciences. 2016 Sep;73(18):3535-53.
10. Matsuzawa K, Ohga H, Shigetomi K, Shiiya T, Hirashima M, Ikenouchi J. MAGIs regulate aPKC to enable balanced distribution of intercellular tension for epithelial sheet homeostasis. Communications Biology. 2021 Mar 12;4(1):1-1.
11. Kantar D, Mur EB, Mancini M, Slaninova V, Salah YB, Costa L, et al. MAGI1 inhibits the AMOTL2/p38 stress pathway and prevents luminal breast tumorigenesis. Scientific Reports. 2021;11:5752.
12. Balbas MD, Burgess MR, Murali R, Wongvipat J, Skaggs BJ, Mundel P, et al. MAGI-2 scaffold protein is critical for kidney barrier function. Proceedings of the National Academy of Sciences. 2014 Oct 14;111(41):14876-81.
13. Yamada H, Shirata N, Makino S, Miyake T, Trejo JA, Yamamoto- Nonaka K, et al. MAGI-2 orchestrates the localization of backbone proteins in the slit diaphragm of podocytes. Kidney International. 2021 Feb 1;99(2):382-95.
14. Ihara KI, Asanuma K, Fukuda T, Ohwada S, Yoshida M, Nishimori K. MAGI-2 is critical for the formation and maintenance of the glomerular filtration barrier in mouse kidney. The American Journal of Pathology. 2014 Oct 1;184(10):2699-708.
15. Ni J, Bao S, Johnson RI, Zhu B, Li J, Vadaparampil J, et al. MAGI- 1 interacts with nephrin to maintain slit diaphragm structure through enhanced Rap1 activation in podocytes. Journal of Biological Chemistry. 2016 Nov 1;291(47):24406-17.
16. Post A, Pannekoek WJ, Ross SH, Verlaan I, Brouwer PM, Bos JL. Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proceedings of the National Academy of Sciences. 2013 Jul 9;110(28):11427-32.
17. Kooistra MR, Dubé N, Bos JL. Rap1: a key regulator in cell-cell junction formation. Journal of Cell Science. 2007 Jan 1;120(1):17- 22.
18. Sakurai A, Fukuhara S, Yamagishi A, Sako K, Kamioka Y, Masuda M, et al. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Molecular Biology of the Cell. 2006 Feb;17(2):966-76.
19. Hultin S, Zheng Y, Mojallal M, Vertuani S, Gentili C, Balland M, et al. AmotL2 links VE-cadherin to contractile actin fibres necessary for aortic lumen expansion. Nature Communications. 2014 May 7;5(1):1-3.
20. Wörthmüller J, Rüegg C. MAGI1, a scaffold protein with tumor suppressive and vascular functions. Cells. 2021 Jun 14;10(6):1494.
21. Alday-Parejo B, Ghimire K, Coquoz O, Albisetti GW, Tamò L, Zaric J, Stalin J, Rüegg C. MAGI1 localizes to mature focal adhesion and modulates endothelial cell adhesion, migration and angiogenesis. Cell Adhesion & Migration. 2021 Jan 1;15(1):126-39.
22. Kotelevets L, Chastre E. A New Story of the Three Magi: Scaffolding Proteins and lncRNA Suppressors of Cancer. Cancers.2021 Aug 24;13(17):4264.
23. Wu X, Hepner K, Castelino-Prabhu S, Do D, Kaye MB, Yuan XJ, et al. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proceedings of the National Academy of Sciences. 2000 Apr 11;97(8):4233-8.
24. Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, et al. Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. Journal of Biological Chemistry. 2000 Jul 14;275(28):21477-85.
25. Lu Y, Sun W, Zhang L, Li J. Silencing of MAGI1 promotes the proliferation and inhibits apoptosis of glioma cells via The Wnt/ β-catenin and PTEN/AKT signaling pathways. OncoTargets and Therapy. 2019;12:9639.
26. Kotelevets L, van Hengel J, Bruyneel E, Mareel M, Van Roy F, Chastre E. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. The FASEB Journal. 2005 Jan;19(1):115-7.
27. Wang W, Yang Y, Chen X, Shao S, Hu S, Zhang T. MAGI1 mediates tumor metastasis through c-Myb/miR-520h/MAGI1 signaling pathway in renal cell carcinoma. Apoptosis. 2019 Dec;24(11):837-48.
28. Kozakai T, Takahashi M, Higuchi M, Hara T, Saito K, et al. MAGI-1 expression is decreased in several types of human T-cell leukemia cell lines, including adult T-cell leukemia. International Journal of Hematology. 2018 Mar;107(3):337-44.
29. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature Reviews Molecular Cell Biology. 2012 May;13(5):283-96.
30. Fournier MV, Fata JE, Martin KJ, Yaswen P, Bissell MJ. Interaction of E-cadherin and PTEN regulates morphogenesis and growth arrest in human mammary epithelial cells. Cancer Research. 2009 May 15;69(10):4545-52.
31. Bardet PL, Guirao B, Paoletti C, Serman F, Léopold V, Bosveld F, et al. PTEN controls junction lengthening and stability during cell rearrangement in epithelial tissue. Developmental Cell. 2013 Jun 10;25(5):534-46.
32. Albrecht LV, Tejeda-Muñoz N, De Robertis EM. Cell biology of canonical Wnt signaling. Annual Review of Cell and Developmental Biology 2021 Oct 6;37:369-89.
33. Li ZY, Li XH, Tian GW, Zhang DY, Gao H, Wang ZY. MAGI1 inhibits the proliferation, migration and invasion of glioma cells. OncoTargets and Therapy. 2019;12:11281.
34. Alday-Parejo B, Richard F, Wörthmüller J, Rau T, Galván JA, Desmedt C, Santamaria-Martinez A, Rüegg C. MAGI1, a new potential tumor suppressor gene in estrogen receptor positive breast cancer. Cancers. 2020 Jan 16;12(1):223.
35. Maugeri-Saccà M, De Maria R. The Hippo pathway in normal development and cancer. Pharmacology & Therapeutics. 2018 Jun 1;186:60-72.
36. Charras G, Yap AS. Tensile forces and mechanotransduction at cell–cell junctions. Current Biology. 2018 Apr 23;28(8):R445-57.
37. Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Science Signaling. 2013 Nov 19;6(302):rs15.
38. Hildebrand S, Hultin S, Subramani A, Petropoulos S, Zhang Y, Cao X, et al. The E-cadherin/AmotL2 complex organizes actin filaments required for epithelial hexagonal packing and blastocyst hatching. Scientific Reports. 2017 Aug 25;7(1):1-7.
39. Furukawa KT, Yamashita K, Sakurai N, Ohno S. The epithelial circumferential actin belt regulates YAP/TAZ through nucleocytoplasmic shuttling of merlin. Cell Reports. 2017 Aug 8;20(6):1435-47.
40. Abe JI, Ko KA, Kotla S, Wang Y, Paez-Mayorga J, Shin IJ, et al. MAGI1 as a link between endothelial activation and ER stress drives atherosclerosis. JCI Insight. 2019 Apr 4;4(7):125570.
41. Abe RJ, Savage H, Imanishi M, Banerjee P, Kotla S, Paez- Mayorga J, et al. P90RSK-MAGI1 module controls endothelial permeability by post-translational modifications of MAGI1 and hippo pathway. Frontiers in Cardiovascular Medicine. 2020 Nov 13;7:542485.
42. Ghimire K, Zaric J, Alday-Parejo B, Seebach J, Bousquenaud M, Stalin J, et al. MAGI1 mediates eNOS activation and NO production in endothelial cells in response to fluid shear stress. Cells. 2019 Apr 27;8(5):388.
43. Gaspar P, Tapon N. Sensing the local environment: actin architecture and Hippo signalling. Current Opinion in Cell Biology. 2014 Dec 1;31:74-83.
44. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011 Jun;474(7350):179-83.
45. Esposito D, Pant I, Shen Y, Qiao RF, Yang X, Bai Y, et al. ROCK1 mechano-signaling dependency of human malignancies driven by TEAD/YAP activation. Nature Communications. 2022 Feb 4;13(1):1-4.
46. Boyle ST, Poltavets V, Kular J, Pyne NT, Sandow JJ, Lewis AC, et al. ROCK-mediated selective activation of PERK signalling causes fibroblast reprogramming and tumour progression through a CRELD2-dependent mechanism. Nature Cell Biology. 2020 Jul;22(7):882-95.
47. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nature Reviews Molecular Cell Biology. 2021 May;22(5):346-66.