Abstract
Macrophages are members of the innate immune system; that originate from monocyte cells from the myeloid stem cells. In response to the tissue environment, monocytes differentiate into two subtypes of macrophages, M1, or M2. The M1 or classically activated macrophages (CAM) aggravate immune responses by releasing reactive oxygen species (ROS), and pro-inflammatory cytokines. The alternatively activated or M2 macrophages (AAM), are involved in the process of tissue repair by suppressing immune responses. The other type of macrophages are called tissue-resident macrophages and they originate from the primitive myeloid precursor cells of the yolk sac. Different types of macrophages affect the immune system through various pathways. Rheumatic diseases are inflammatory; disorders with an auto-immune basis that usually involve the musculoskeletal system, lungs, kidneys, and eyes. In rheumatoid arthritis (RA), the main affected organs are joints, bones, and cartilage; which cause joint swelling, tenderness, redness, decreased range of motion, and morning stiffness. The roles of macrophages in RA include the production of cytokines, activation of autoreactive B- and T-cells, and impairment of tissue repair in joints. The main purpose of this review article is to further discuss the underlying role of macrophages in inflammatory articular changes in RA patients.
Keywords
Macrophage, Rheumatoid arthritis, Synovial inflammation
Introduction
Macrophages are one of the important cellular components of the immune system, which can affect the immune responses by either suppressing or aggravating them. As the monocyte cells arise from myeloid stem cells residing in the red bone marrow, they circulate throughout the body via blood vessels. The monocytes differentiate into macrophages as they enter different tissues of the body [1]. According to the immune modulators present in the tissue environment, different subtypes of macrophages such as M1, or M2 are differentiated [2]. For instance, activation of toll-like receptors (TLR) as well as the presence of interferon-gamma (IFNγ), and tumor necrosis factor-alpha (TNF-α) leads to M1 or classically activated macrophages (CAM) differentiation. These cells aggravate immune responses by the release of reactive oxygen species (ROS), and pro-inflammatory cytokines such as IL-1β, and IL-6. So, they eventually help the immune system to overcome non-self antigens like pathogens and cancerous cells [3,4].
The evolution of the immune system has led to the development of an alternative way to activation of macrophages. These cells are called alternatively activated or M2 macrophages (AAM), which are induced by the presence of IL-4, and IL-13, and signaling through the activation of signal transducer and activator of transcription 6 (STAT6). M2 macrophages are a part of type 2 immune responses involved in the process of tissue repair by inducing immune response suppression [5]. The different gene expressions of M1 and M2 macrophages are responsible for the various functions of these cells. For instance, IL-10, C-C motif chemokine ligand (CCL)17, and CCL22 levels are increased in M2 macrophages. However, CD80 and CD86 scavenger receptors are expressed on M1 macrophages, which induce innate and adaptive responses [4]. Primitive myeloid precursor cells arising from the yolk sac are another source of macrophages that reside in different tissues of the body such as the central nervous system (CNS), and liver [5] (Table 1).
|
|
M1 macrophages |
M2 macrophages |
|
Also called the |
Classically activated macrophages (CAM) |
Alternatively activated macrophages (AAM) |
|
Immune modulators present in the tissue environment |
|
|
|
Gene expression |
Highly expressed CD80 and CD86 scavenger receptors. |
Increased expression of IL-10 CCL17, and CCL22 |
|
Overall effects on the body |
|
|
|
TLR: Toll-Like Receptor; STAT6: Signal Transducer and Activator of Transcription 6; IFNγ: Interferon-Gamma; TNF-α: Tumor Necrosis Factor-alpha; IL: Interleukin; CD: Cluster of Differentiation; CCL: C-C motif chemokine Ligand; ROS: Reactive Oxygen Species |
||
Rheumatic diseases are inflammatory disorders that usually involve the musculoskeletal system, lungs, kidneys, and eyes [6]. A possible underlying cause of these disorders is the attack of the immune system on different tissues. For instance, the immune system might attack the bony and articular structures, as in rheumatoid arthritis (RA). Rheumatoid arthritis causes synovitis of joints, destruction of bones (osteoporosis), and cartilage around the articular surface, which leads to decreased articular space. Joint swelling, tenderness, redness, decreased range of motion, and morning stiffness can lead to the diagnosis of synovitis [7]. RA usually affects the small joints of the hands (proximal interphalangeal and metacarpophalangeal) and feet (metatarsophalangeal) [8]. Due to the important roles of both monocyte-derived and tissue residential macrophages in the inflammatory responses and tissue repair processes of affected joints, the main purpose of this review article is to discuss the underlying role of macrophages in these inflammatory articular changes in RA patients.
Role of Macrophages in Synovitis of RA
In the human body, synovial joints (diarthrosis) connect different bones and because of their special structure, they allow high ranges of motion at the joint site. The normal synovial membranes covering the inner side of the synovial joints are thin and composed of lining and sublining layers [9]. Barrier-forming macrophages (type A synoviocytes) and fibroblast-like synoviocytes (FLS, type B synoviocytes) usually reside in the lining layer, regulating and protecting the functioning structure of the joint. However, the neural and vascular components of the synovium are located in the connective tissue of the sublining layer [10]. It was demonstrated that interstitial macrophages are derived from non-monocyte lineage and are present in the sublining layer of the synovial membrane. This group of macrophages differentiates into barrier-forming macrophages and, along with synovial fibroblasts forms the lining layer. Overall, synovial tissue macrophages are classified into two groups based on their origin, tissue-resident macrophages and infiltrating macrophages (derived from monocytes in the blood circulation) (Figure 1).
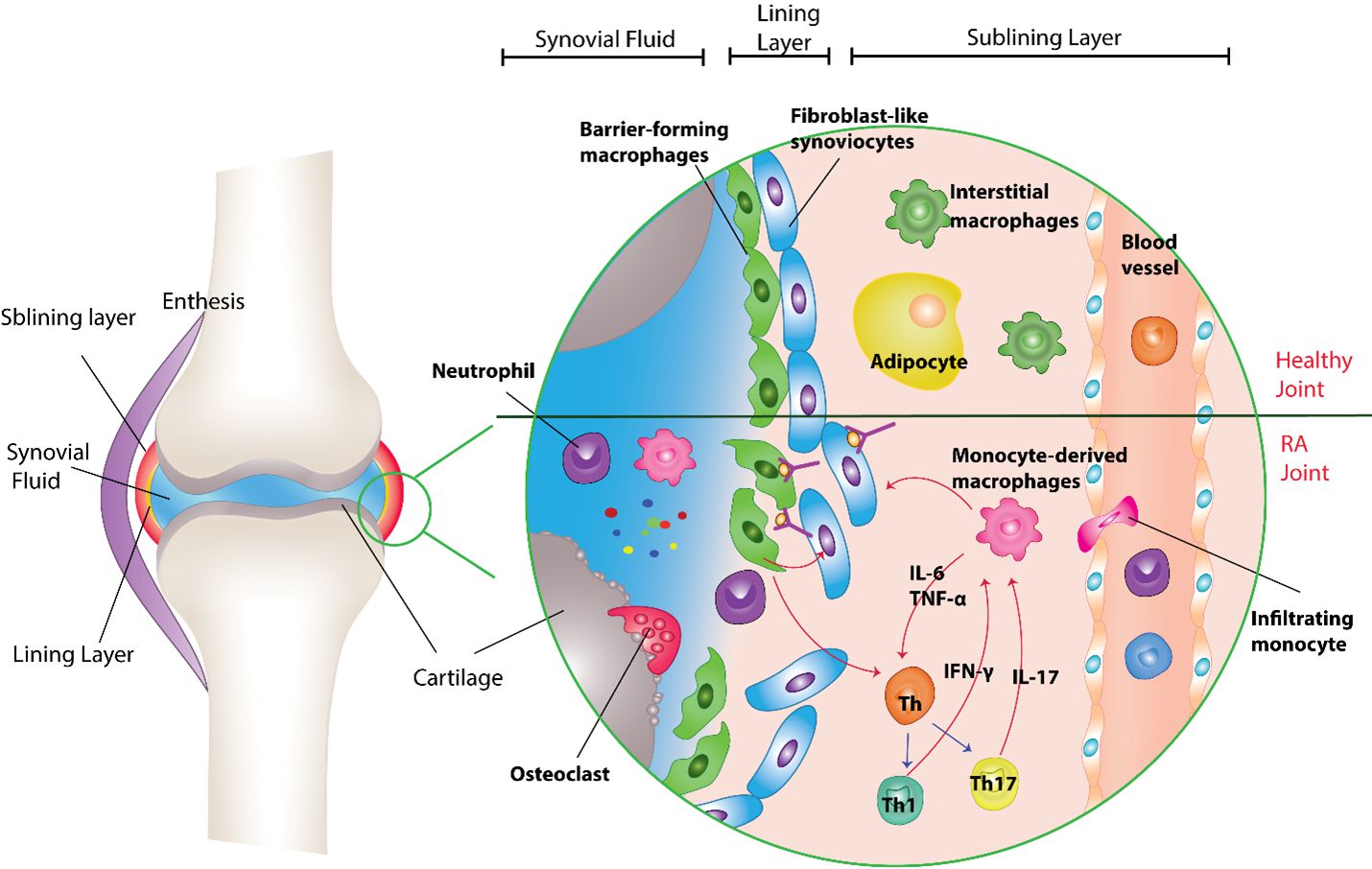
Figure 1. The roles of monocyte-derived and tissue-resident macrophages in healthy and RA joint. The normal synovial membranes covering the inner side of synovial joints are thin and composed of the lining and sublining layers. Interstitial macrophages are derived from non-monocyte lineage present in the sublining layer of the synovial membrane. They are differentiated into barrier-forming macrophages and along with synovial fibroblasts form the lining layer. The barrier-forming macrophages, protect the synovium from inflammatory attacks by reducing the infiltration of immune cells through tight junctions, suppressing inflammation, and digesting infiltrated neutrophils. In RA patients, produced immune complexes (ICs) are deposited on the synovial layers. The deposition of these complexes leads to the barrier-forming macrophage activation and destruction of joint structures and the attraction of immune cells to the site of inflammation. Therefore, the monocytes along with other immune cells infiltrate the synovial tissues from peripheral blood. Blood monocytes are differentiated from the monocyte-derived macrophages and participate in tissue inflammation. IFN-γ and IL-17 secreted from Th 1 and Th17 are the cytokines responsible for the stimulation of macrophages to release other cytokines like IL-6, and TNF-α, which leads to the initiation of the inflammation process. These cytokines released from the macrophages, in turn, activate the differentiation of T cell lymphocytes into Th1 and Th17 subtypes. It is found that macrophages can also regulate FLS function and behavior in a feedback loop.
Tissue-resident macrophages
Tissue residential macrophages, which are derived from a non-monocyte lineage along with synovial fibroblasts, form the lining layer of the synovial cavity. Because of their location in the synovium, barrier-forming macrophages same as M2-type macrophages, protect the synovium from inflammatory attacks by reducing the infiltration of immune cells through tight junctions, suppressing inflammation, and digesting infiltrated neutrophils. Due to the differences in gene expression of macrophages induced by their surrounding immunologic stimuli, different types of macrophages have diverse surface markers and roles in immune processes. For instance, the tissue-resident macrophages located in the synovial lining have CD68, T-cell immunoglobulin and mucin domain-containing 4 (TIMD-4), and MER proto-oncogene, tyrosine kinase (MerTK) markers. They also express CD163 and CD206 receptors, which are mainly expressed by M2 type of macrophage [11,12]. The main role of MerKT activation is to stimulate signaling pathways that induce phagocytosis of non-functional cells by macrophages [13]. It is also found that MerKT signaling in macrophages increases the production of a group of molecules called specialized pro-resolving mediators (SPM), which leads to tissue repair and reduces inflammation [14].
The role of the MerTK, CD206, and CD136-positive macrophages in gaining remission in RA patients was studied by Alivernini et al. The results of their study showed that patients with remission RA have significantly higher levels of MerTK+CD206+ and CD206+CD163+ macrophages, compared with the RA patients who are either resistant to medications or did not receive any medications. The analysis of disease activity in RA patients showed that higher levels of MerTK+CD206+, CD206+CD163+, and MerTK+CD163+ macrophages were observed in RA patients with lower disease activity. The result of macrophage activation by lipo-polysaccharides (LPS) showed that the release of IL-6, IL-1β, TNF-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), CCL2, and CCL3 was significantly diminished in MerTK+ cells of remission RA patients, compared to the MerTK- cells of active RA patients. However, there was no significant difference in the secretion of IL-10, IL-8, IL-13, IFNα2, and IL-12 [15].
Macrophages can also affect the pattern of gene expression in synovial fibroblasts. For instance, MerTK+CD206+ macrophages from RA patients in remission state might protect the structure of the joint by reducing inflammation and causing a higher expression of collagen, and TGF-β genes in FLS. However, the MerTK- CD206- macrophages may cause severe damage to joint structures by stimulating fibroblasts to significantly increase the expression of various genes, such as IL6, CCL2, CCL20, CXCL1, 8, MMP1, 3, and RANKL. Indeed, MerTK+ macrophages of RA patients in remission state secreted significantly higher amounts of resolvin D1, than MerTK+ macrophages of patients with active RA [15]. Resolvin D1 has anti-inflammatory effects, including reduction of IL-1B, IL-6, TNF-α, and clinical symptoms of arthritis in mice models. Indeed, resolvin D1 inhibits the formation of new blood vessels and synovial fibroblasts, which in turn protects joints from inflammation and destruction. Therefore, the lower serum level of resolvin D1 in RA patients is one of the possible causes of joint damage in this disease [16].
Infiltrating macrophages
Autoimmune inflammation of synovial joints is a characteristic feature of rheumatoid arthritis [17]. The basis of the immunopathogenesis of rheumatoid arthritis is not clearly revealed yet. However, it is known that both genetic and environmental elements play substantial roles in the development of auto-reactive immune cells [18]. These auto-reactive cells recognize self-antigens and produce antibodies against them. The auto-antibodies produced in RA include rheumatoid factor (RF), anti-citrullinated protein antibody (ACPA), and anti-carbamylated protein antibody (anti-CarP). The final result of the activation of self-reactive cells and antibody formation is the deposition of immune complexes (ICs) in various tissues, including the synovial layers [19].
The deposition of these complexes can further stimulate the immune system via the complement pathway, which leads to the activation of barrier-forming macrophages, and destruction of joint structures, and the attraction of immune cells to the site of inflammation [20,21]. Indeed, the dysfunctional tight junctions between the cells of the lining layer of the synovial membrane and the increased number of blood vessels, due to the presence of vascular endothelial growth factor (VEGF) in the sublining layer, can itself increase the deposition of immune complexes in a positive feedback loop [22]. The cellular components of the inflammation process circulate throughout the body via the blood. Immunological signaling molecules are responsible for the attraction of inflammatory cells to synovial joints [23]. These lead to residential macrophages, synovial fibroblasts, and bone cells (osteoclasts) activation and infiltration of neutrophils, monocytes, B cells, and Th1 cells into the joints and Th17 lymphocyte polarization.
Therefore, monocytes along with other immune cells infiltrate the synovial tissues from the peripheral blood. Blood monocytes differentiate into monocyte-derived macrophages and participate in tissue inflammation. One of the major cells responsible for the inflammatory changes in joints of RA patients is suggested to be these infiltrating macrophages. Indeed, the surface markers of these infiltrating macrophages derived from monocytes include CD14, myeloid-related protein (MRP)8, and 14 [11,12]. Other changes in the synovium of RA patients include thickening of the synovial membrane due to the increased number of macrophages and fibroblasts, increased synthesis of connective tissue components by fibroblasts, and formation of new blood vessels in the sublining layer will occur subsequently [10,22].
Macrophages and Synovial Inflammation
There are many possible ways in which macrophages can contribute to joint damage in RA. Infiltrating macrophages communicate with other cells of the immune system through signaling molecules and direct cellular contact [24]. The contact of macrophages with T cells through CD40/CD40L, LFA1/ICAM1 (leukocyte function-associated antigen 1/intracellular adhesion molecule 1), OX40 (CD143)/OX40L (CD252), and CCL20/CCR6 is one of the stimuli for macrophage activation [25,26]. Indeed, IFN-γ and IL-17 secreted from Th1 and Th17 are the cytokines responsible for the stimulation of macrophages to release other cytokines like IL-6 and TNF-α, which leads to the initiation of the inflammation process [27]. The other stimuli for macrophage activation include the immune complexes of auto-antibodies such as RF and ACPA, with their molecular ligands like citrullinated proteins. These complexes bind to TLR4 (Toll-like receptor 4), MyD88 (myeloid differentiation primary response 88), and FcγR (Fc gamma receptor) on macrophages' surface, which in turn stimulates the release of inflammatory cytokines from macrophages [28]. These cytokines released from macrophages activate the differentiation of T cell lymphocytes into Th1 and Th17 subtypes [25]. So, the interactions between macrophages and T cells in the pathogenesis of RA are two-way roads (Figure 1).
Haringman et al. studied the role of macrophages in the clinical symptoms of RA by assessing synovial samples of 88 patients with active RA for CD68+ macrophage cells. The activity of the disease was determined by calculating the “28 joint count Disease Activity Score (DAS28)” for each patient. Of 70 patients who received medication for RA, 11 patients showed a good response to the medication. Indeed, these patients showed a significantly higher reduction in the number of macrophages in the synovial sub-lining layer, compared with the patients with a moderate response (n=35) and those who did not respond to the medication (n=42). Indeed, the analysis of changes in disease activity (DAS28) and the number of macrophages present in the sub-lining layer after receiving the medications showed a notable positive correlation [29].
The increased production of various molecules by macrophages is found in synovial fluid samples of patients with RA [30]. Liu et al. examined the synovial membrane and synovial fluid samples of 23 RA patients for the presence of macrophages and mRNA or proteins of matrix metalloproteinase (MMP)-3, 9, and 12. The results of their study showed a higher expression of MMP-3, 9, and 12 in the synovial sample of RA patients, compared with samples of 29 patients with osteoarthritis. The presence of macrophages was detected in all RA synovial samples. However, in the sample of OA patients, the number of macrophages was low. Further examination showed that macrophages seem to be responsible for MMP-12 enzyme production, as there was a correlation between the presence of macrophages and MMP-12 in RA samples [31]. The role of MMPs in the RA pathogenesis process is to enzymatically destroy various particles in the extracellular matrices of articular surfaces (for instance, collagen and gelatin) [32]. Indeed, the destruction of joint components produces new self-antigens, which in turn leads to the formation of new auto-antibodies against them. This process causes the chronic autoimmune basis of the disease [25].
Cytokines and Chemokines
As mentioned earlier, the communication of macrophages with other immune cells occurs through cytokines and chemokines released by macrophages. In this section, we will review the different chemicals released from macrophages and their particular roles in recruiting various aspects of the immune processes of RA.
To further understand the patterns of cytokine expression in RA patients, Yeo et al, analyzed the expression of cytokines in synovial biopsies from RA patients in two groups, the “early RA” and the “established RA” (the respective median disease durations were 6 and 38). In comparison to the control subjects, the patients with established RA had significantly elevated expression levels of CXCL7, 8, 9,13, CCL2, 8, 20, IL-1α, 1β, 2, 12, 21, 32, TNF-α, OX40L, and galactin3. The patients of the early RA group had higher expression of CXCL4 and CXCL7 compared with the control group. It is assumed that CXCL4 and CXCL7 are expressed by macrophages because the pattern of CXCL4 and CXCL7 expression is related to the presence of macrophages [33]. Indeed, these cytokines attract neutrophils to the inflamed joints [10]. These infiltrating neutrophils increase joint inflammation and destruction by secreting reactive oxygen species (ROS), causing oxidative stress, defensins, and tissue-damaging enzymes such as proteases [34,35].
IL-1, 6, 12, 18, 23, and TNF-α are other cytokines released by macrophages in the synovium of cases with RA [36-38] and are essential for the induction of joint damage [39]. It has been found that macrophages can regulate FLS function and behavior in a feedback loop. IL-1 and TNF-α, as products of macrophages, have autocrine and paracrine effects on FLS. They induce fibroblast proliferation and production of IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-8. [40]. They also stimulate synovial fibroblasts to synthesize matrix metalloproteinases (MMPs) and prostaglandins and contribute to synovitis and bone erosion [41].
Both tissue-resident and inflammatory cytokines can affect osteogenesis through the release of cytokines, including bone morphogenetic protein (BMP) 2 and 4, and TGF-β1 [42]. Activated macrophages also produce oncostatin M (OSM, a member of the IL-6 family), which induces osteogenic differentiation of mesenchymal stem cells and also stimulates the differentiation of osteoclasts [43,44]. Research has shown that OSM affects synovial fibroblasts in RA, induces RA-FLS to produce pro-inflammatory cytokines [45,46], and enhances their invasive ability. Besides, OSM increases the secretion of MMPs from RA-FLS and facilitates the formation of pannus in RA [47].
The overall effects of macrophage cytokines in RA include induction of new osteoclasts formation, connective tissue-damaging enzymes release (proteases, collagenases, and MMPs), and activation and differentiation of other cells of the immune system (neutrophils, NK cells, mast cells, T, and B lymphocytes) [39]. On the other hand, the secretion of immune-regulating cytokines such as IL-4, 5, 10, and 13 from B lymphocytes and regulatory T cells counteracts the osteodestructive signals of macrophages [48].
Macrophages and Bone Health
The rate of fractures in the vertebral column of patients with RA is nearly two times higher than that in the general population [49]. Indeed, a study showed that patients with RA have greater levels of bone turnover as the levels of bone formation (alkaline phosphatase), resorption (Type-I collagen cross-linked N-telopeptide), and matrix-related markers (pentosidine and homocysteine) were higher in these patients than in control subjects. Moreover, the bone mineral density of RA patients was lower than that of controls. Overall, the higher levels of bone destruction in RA patients may explain their higher fracture rates and more severe fractures [50]. The duration, disease activity, and disability caused by RA are risk factors predisposing patients to future fractures. In addition, the disability caused by RA and the duration of RA were both higher in patients with osteoporosis [51]. Indeed, the bone-destroying functions of osteoclasts produced from peripheral blood mononuclear cells (PBMCs) of RA patients were significantly higher in patients with elevated serum ESR levels [52]. These findings highlight the importance of understanding the effects of RA on bone health.
The renewal of bones in the body and their health relies on two processes, the destruction of bony material by osteoclasts and the synthesis of a new bony matrix by osteoblasts [53]. Osteoclasts are differentiated monocytes with multiple nuclei that can damage bone health by overtly destroying bone, leading to fractures [54]. The formation of osteoclasts is based on the presence of environmental factors such as receptor activator of NF-κB ligand (RANKL), which binds to RANK on precursor monocyte cells and macrophage-colony stimulating factor (M-CSF) [55]. The attraction of CD14+ monocytes in the blood circulation into the synovium of RA patients is a consequence of the presence of M-CSF on synovial endothelial cells [56]. M-CSF is a secreted cytokine that induces macrophages' survival, proliferation, and differentiation from hematopoietic stem cells. It also participates in osteoclast migration [57]. High expression levels of RANKL in the synovium of patients with RA can increase the formation of osteoclasts. It is found that RAFLSs induce osteoclastogenesis in the presence of 1,25-OH vitaminD3 which is of great importance in increasing the expression of RANKL, suppressing the expression of osteoprotegerin (OPG), and hence the formation of osteoclasts [58].
Osteomacs are a resident type of macrophages present in bone tissue and are distinguished by their F4/80, CD68, and Mac3 expression. They support bone formation, osteoblast maturation, and mineralization [59,60]. These cells help in the removal of bone destruction residues in the process of bone remodeling. The exact role of these cells in bone health and diseases is not defined yet and they could serve as a target for improving bone health in inflammatory bone diseases [61,62].
In RA patients, macrophages can affect the osteoporosis process by secreting various cytokines, which can either influence the functions of osteoclasts directly or through stimulation of RANKL expression by osteoblasts and fibroblasts [54,63]. For instance, the binding of TNF to its receptors (TNFr1) can induce RANKL expression, the formation of new osteoclasts, and the improvement of osteoclasts' survival [64]. Other cytokines secreted from macrophages that either enhance the formation of new osteoclasts, the survival of osteoclasts, or stimulate their functions include IL-1, 6, and 15 [65,66]. In addition, as we mentioned before, macrophages can favor the formation of Th17 lymphocytes by producing specific cytokines, which express RANKL and secrete IL-17 [67]. IL-17 increases RANK on the surface of precursor cells, which increases RANK/RANKL signaling and the formation of new osteoclasts [68]. In addition to osteoclasts formed from blood monocyte precursor cells, macrophages in the synovium can also differentiate into osteoclasts [69]. It has been found that if osteoprotegerin-ligand (OPGL) and M-CSF are present in the environment, synovial macrophages can transform into bone-destroying osteoclasts [70].
Conclusion
In this review article, we discussed the different aspects of macrophages as innate immunity cells in the pathogenesis of RA. We mentioned the variety of pathways by which macrophages can influence the bone and joint structures of RA patients. Indeed, we described two different types of macrophages, their origin, and their specific roles in shaping the immune system. Because of the tissue-repairing effects of tissue-resident macrophages, the clinical use of these cells in enhancing the protection and repair of synovial joints may change the prognosis of patients with rheumatoid disorders. These cells can affect the gene expression of fibroblasts and induce the production of structural molecules such as collagen. On the other hand, monocyte-derived macrophages secrete a great variety of immune molecules and communicate with other cells of the body through signaling pathways. Their secreted cytokines can stimulate and attract other immune cells to the affected joints and further destroy the bone and joint structures. This could be used as a new approach for the development of new drugs, by disrupting the inflammatory signaling of macrophages with other cells. Besides, one of the main problems of rheumatic disorders is the deterioration of bone health and disruption of homeostasis in bone formation and resorption. Therefore, osteoclasts and osteomacs are the most important therapeutic targets in these disorders. The current knowledge about the specific roles of these cells is not sufficient. Further research on understanding the various roles of macrophages would help in the development of new treatment strategies for RA patients.
Declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Competing interests
None.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors' contributions
MJ, SK, and MA contributed to the acquisition and interpretation of data and drafting the article. AJ, MM*, and EF* contributed to the interpretation of data, conception and design of the study, and critical revision of the article for important intellectual content. All authors contributed to the revision of the manuscript, read, and gave final approval for the submitted version of the article.
Acknowledgments
None.
References
2. Chávez-Galán L, Olleros ML, Vesin D, Garcia I. Much More than M1 and M2 Macrophages, There are also CD169+ and TCR+ Macrophages. Frontiers in Immunology. 2015;6:263.
3. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8(12):958-69.
4. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090.
5. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317-43.
6. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. The Lancet. 2016;388(10055):2023-38.
7. Bullock J, Rizvi SAA, Saleh AM, Ahmed SS, Do DP, Ansari RA, et al. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med Princ Pract. 2018;27(6):501-7.
8. Grassi W, De Angelis R, Lamanna G, Cervini C. The clinical features of rheumatoid arthritis. Eur J Radiol. 1998;27 Suppl 1:S18-24.
9. Tamer TM. Hyaluronan and synovial joint: function, distribution and healing. Interdiscip Toxicol. 2013;6(3):111-25.
10. Kurowska-Stolarska M, Alivernini S. Synovial tissue macrophages: friend or foe? RMD Open. 2017;3(2):e000527.
11. Tu J, Wang X, Gong X, Hong W, Han D, Fang Y, et al. Synovial Macrophages in Rheumatoid Arthritis: The Past, Present, and Future. Mediators of Inflammation. 2020;2020:1583647.
12. Boutet MA, Courties G, Nerviani A, Le Goff B, Apparailly F, Pitzalis C, et al. Novel insights into macrophage diversity in rheumatoid arthritis synovium. Autoimmun Rev. 2021;20(3):102758.
13. Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. 2010;1209:23-9.
14. Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal. 2018;11(549):eaar3721.
15. Alivernini S, MacDonald L, Elmesmari A, Finlay S, Tolusso B, Gigante MR, et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nature Medicine. 2020;26(8):1295-306.
16. Sun W, Ma J, Zhao H, Xiao C, Zhong H, Ling H, et al. Resolvin D1 suppresses pannus formation via decreasing connective tissue growth factor caused by upregulation of miRNA-146a-5p in rheumatoid arthritis. Arthritis Research & Therapy. 2020;22(1):61.
17. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6:15-15.
18. Sakaguchi S, Tanaka S, Tanaka A, Ito Y, Maeda S, Sakaguchi N, et al. Thymus, innate immunity and autoimmune arthritis: Interplay of gene and environment. FEBS Letters. 2011;585(23):3633-39.
19. van Delft MAM, Huizinga TWJ. An overview of autoantibodies in rheumatoid arthritis. Journal of Autoimmunity. 2020;110:102392.
20. Daha NA, Banda NK, Roos A, Beurskens FJ, Bakker JM, Daha MR, et al. Complement activation by (auto-) antibodies. Mol Immunol. 2011;48(14):1656-65.
21. Trouw LA, Haisma EM, Levarht EW, van der Woude D, Ioan-Facsinay A, Daha MR, et al. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009;60(7):1923-31.
22. Firestein GS, McInnes IB. Immunopathogenesis of Rheumatoid Arthritis. Immunity. 2017;46(2):183-96.
23. Iwamoto T, Okamoto H, Toyama Y, Momohara S. Molecular aspects of rheumatoid arthritis: chemokines in the joints of patients. The FEBS Journal. 2008;275(18):4448-55.
24. Tu J, Huang W, Zhang W, Mei J, Zhu C. A Tale of Two Immune Cells in Rheumatoid Arthritis: The Crosstalk Between Macrophages and T Cells in the Synovium. Front Immunol. 2021;12:655477.
25. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nature Reviews Immunology. 2007;7(6):429-42.
26. Fox DA, Gizinski A, Morgan R, Lundy SK. Cell-cell interactions in rheumatoid arthritis synovium. Rheum Dis Clin North Am. 2010;36(2):311-23.
27. Kondo Y, Yokosawa M, Kaneko S, Furuyama K, Segawa S, Tsuboi H, et al. Review: Transcriptional Regulation of CD4+ T Cell Differentiation in Experimentally Induced Arthritis and Rheumatoid Arthritis. Arthritis Rheumatol. 2018;70(5):653-61.
28. Sokolove J, Zhao X, Chandra PE, Robinson WH. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcγ receptor. Arthritis & Rheumatism. 2011;63(1):53-62.
29. Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJM, Kraan MC, Baeten D, et al. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64(6):834-8.
30. Kinne RW, Stuhlmüller B, Burmester GR. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther. 2007;9(6):224.
31. Liu M, Sun H, Wang X, Koike T, Mishima H, Ikeda K, et al. Association of increased expression of macrophage elastase (matrix metalloproteinase 12) with rheumatoid arthritis. Arthritis Rheum. 2004;50(10):3112-7.
32. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529-43.
33. Yeo L, Adlard N, Biehl M, Juarez M, Smallie T, Snow M, et al. Expression of chemokines CXCL4 and CXCL7 by synovial macrophages defines an early stage of rheumatoid arthritis. Ann Rheum Dis. 2016;75(4):763.
34. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205-19.
35. Cascão R, Rosário HS, Souto-Carneiro MM, Fonseca JE. Neutrophils in rheumatoid arthritis: More than simple final effectors. Autoimmun Rev. 2010;9(8):531-35.
36. Chu CQ, Field M, Feldmann M, Maini RN. Localization of Tumor Necrosis Factor α in Synovial Tissues and at the Cartilage–Pannus Junction in Patients With Rheumatoid Arthritis. Arthritis & Rheumatism. 1991;34(9):1125-32.
37. Cauli A, Yanni G, Panayi GS. Interleukin-1, interleukin-1 receptor antagonist and macrophage populations in rheumatoid arthritis synovial membrane. Rheumatology. 1997;36(9):935-40.
38. Sidiropoulos PI, Goulielmos G, Voloudakis GK, Petraki E, Boumpas DT. Inflammasomes and rheumatic diseases: evolving concepts. Ann Rheum Dis. 2008;67(10):1382-89.
39. Fang Q, Zhou C, Nandakumar KS. Molecular and Cellular Pathways Contributing to Joint Damage in Rheumatoid Arthritis. Mediators of Inflammation. 2020;2020:3830212.
40. Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. The international journal of biochemistry & cell biology. 2004;36(3):372-78.
41. Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunological Reviews. 2010;233(1):233-55.
42. Champagne CM, Takebe J, Offenbacher S, Cooper LF. Macrophage cell lines produce osteoinductive signals that include bone morphogenetic protein-2. Bone. 2002;30(1):26-31.
43. Guihard P, Boutet MA, Brounais-Le Royer B, Gamblin AL, Amiaud J, Renaud A, et al. Oncostatin m, an inflammatory cytokine produced by macrophages, supports intramembranous bone healing in a mouse model of tibia injury. Am J Pathol. 2015;185(3):765-75.
44. Gu Q, Yang H, Shi Q. Macrophages and bone inflammation. Journal of orthopaedic translation. 2017;10:86-93.
45. Matsushima K, Yang D, Oppenheim JJ. Interleukin-8: An evolving chemokine. Cytokine. 2022;153:155828.
46. Hanlon MM, Rakovich T, Cunningham CC, Ansboro S, Veale DJ, Fearon U, et al. STAT3 Mediates the Differential Effects of Oncostatin M and TNFα on RA Synovial Fibroblast and Endothelial Cell Function. Frontiers in immunology. 2019;10:2056.
47. Han L, Yan J, Li T, Lin W, Huang Y, Shen P, et al. Multifaceted oncostatin M: novel roles and therapeutic potential of the oncostatin M signaling in rheumatoid arthritis. Frontiers in immunology. 2023;14.
48. Zhou P, Zheng T, Zhao B. Cytokine-mediated immunomodulation of osteoclastogenesis. Bone. 2022;164:116540.
49. Spector TD, Hall GM, McCloskey EV, Kanis JA. Risk of vertebral fracture in women with rheumatoid arthritis. Bmj. 1993;306(6877):558.
50. Okano T, Inui K, Tada M, Sugioka Y, Mamoto K, Wakitani S, et al. High frequency of vertebral fracture and low bone quality in patients with rheumatoid arthritis-Results from TOMORROW study. Mod Rheumatol. 2017;27(3):398-404.
51. Sinigaglia L, Nervetti A, Mela Q, Bianchi G, Del Puente A, Di Munno O, et al. A multicenter cross sectional study on bone mineral density in rheumatoid arthritis. Italian Study Group on Bone Mass in Rheumatoid Arthritis. J Rheumatol. 2000;27(11):2582-2589.
52. O'Gradaigh D, Bord S, Ireland D, Compston JE. Osteoclastic bone resorption in rheumatoid arthritis and the acute-phase response. Rheumatology. 2003;42(11):1429-30.
53. Kylmaoja E, Nakamura M, Tuukkanen J. Osteoclasts and Remodeling Based Bone Formation. Curr Stem Cell Res Ther. 2016;11(8):626-33.
54. Braun T, Zwerina J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis research & therapy. 2011;13(4):235.
55. Nakashima T, Takayanagi H. Osteoimmunology: crosstalk between the immune and bone systems. J Clin Immunol. 2009;29(5):555-67.
56. Nakano K, Okada Y, Saito K, Tanikawa R, Sawamukai N, Sasaguri Y, et al. Rheumatoid synovial endothelial cells produce macrophage colony-stimulating factor leading to osteoclastogenesis in rheumatoid arthritis. Rheumatology. 2007;46(4):597-603.
57. Plotkin LI, Bivi N. Chapter 3 - Local Regulation of Bone Cell Function. In: Burr DB, Allen MR, editors. Basic and Applied Bone Biology. San Diego: Academic Press; 2014. p. 47-73.
58. Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, et al. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheum. 2000;43(2):259-69.
59. Jilka RL, Weinstein RS, Parfitt AM, Manolagas SC. Quantifying osteoblast and osteocyte apoptosis: challenges and rewards. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2007;22(10):1492-501.
60. Alexander KA, Chang MK, Maylin ER, Kohler T, Müller R, Wu AC, et al. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2011;26(7):1517-32.
61. Terkawi MA, Matsumae G, Shimizu T, Takahashi D, Kadoya K, Iwasaki N. Interplay between Inflammation and Pathological Bone Resorption: Insights into Recent Mechanisms and Pathways in Related Diseases for Future Perspectives. Int J Mol Sci. 2022;23(3):1786.
62. Weivoda MM, Bradley EW. Macrophages and Bone Remodeling. J Bone Miner Res. 2023;38(3):359-69.
63. Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, et al. Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-kappaB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun. 2000;275(3):768-75.
64. Lee SE, Chung WJ, Kwak HB, Chung CH, Kwack KB, Lee ZH, et al. Tumor necrosis factor-alpha supports the survival of osteoclasts through the activation of Akt and ERK. J Biol Chem. 2001;276(52):49343-9.
65. Kitaura H, Marahleh A, Ohori F, Noguchi T, Shen W-R, Qi J, et al. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int J Mol Sci. 2020;21(14):5169.
66. Schett G. Review: Immune cells and mediators of inflammatory arthritis. Autoimmunity. 2008;41(3):224-229.
67. Jung SM, Kim KW, Yang C-W, Park S-H, Ju JH. Cytokine-mediated bone destruction in rheumatoid arthritis. J Immunol Res. 2014;2014:263625.
68. Adamopoulos IE, Chao C-c, Geissler R, Laface D, Blumenschein W, Iwakura Y, et al. Interleukin-17A upregulates receptor activator of NF-κB on osteoclast precursors. Arthritis Research & Therapy. 2010;12(1):R29.
69. Fujikawa Y, Sabokbar A, Neale S, Athanasou NA. Human osteoclast formation and bone resorption by monocytes and synovial macrophages in rheumatoid arthritis. Ann Rheum Dis. 1996;55(11):816-22.
70. Itonaga I, Fujikawa Y, Sabokbar A, Murray DW, Athanasou NA. Rheumatoid arthritis synovial macrophage-osteoclast differentiation is osteoprotegerin ligand-dependent. J Pathol. 2000;192(1):97-104.