Abstract
Macrophages play a crucial role in host innate immune defense against infection and tissue injury. Although macrophage activation and polarization has been well studied, we know less regarding the role of macrophage activation/polarization in inflammationassociated necrotic cell death. By using bone marrow-derived macrophages, we have recently demonstrated that M1 macrophages are much more susceptible than M0 and M2 subtypes of macrophages to necrotic cell death. Moreover, we showed that the enhanced necroptosis in M1 macrophages is dependent on the kinase activity of receptor-interacting protein kinase-3 (RIPK3) and may involve the upregulation of key necroptosis signaling molecules including RIPK3, mixed lineage kinase domain-like protein, and Z-DNA/ RNA binding protein 1. Our findings provide novel insights into the mechanisms of M1 macrophage engagement in inflammation and tissue injury.
Keywords
Macrophage polarization, M1/M2 phenotypes, Necroptosis, Inflammation, RIPK3, MLKL, ZBP1
Macrophage Phenotypes
Macrophages are important cells of the innate immune system and play a crucial role in host immune defense against infection and injury [1-3]. Macrophages form the first line of defense against airborne particles and microbes through multiple functions including phagocytosis, production of cytokines and chemokines, and antigen presentation. Macrophages are highly plastic cells and their phenotypes and functions can be regulated by the local microenvironment. Depending on the context, macrophages can be activated and polarized into different subsets. Macrophage polarization is a process whereby macrophages mount a specific phenotype and a functional response to the surrounding stimuli. Two major macrophage sub-populations with distinct functions have been characterized, and they are the M1 (also termed classically activated or inflammatory) and M2 (alternatively activated or anti-inflammatory) macrophages [2-4]. M1 macrophages are typically induced by pathogen-associated molecular patterns, such as doublestranded RNA (dsRNA) and lipopolysaccharide (LPS), and by Th1 cytokines including interferon γ (IFNγ) and tumor necrosis factor α (TNFα). The activated macrophages acquire transcriptional changes to produce higher levels of pro-inflammatory cytokines and chemokines as well as reactive oxygen species, through which they contribute to host defense against pathogens and tissue damage [3,4]. On the contrary, M2 macrophages can be induced by Th2 cytokines such as interlleukin-4 (IL-4) and IL-13 as well as anti-inflammatory cytokines IL-10 and transforming growth factor β (TGFβ). M2 macrophages are antiinflammatory and implicated in tissue repair, remodeling and vasculogenesis. M2 macrophages can be further divided into four different subsets that consist of M2a, M2b, M2c and M2d, depending on the stimuli received [5,6].
Necroptosis: A Critical Regulator of Inflammation
Programmed and regulated lytic cell death such as necroptosis is increasingly recognized as a driving factor in the pathogenesis of various forms of tissue injury and inflammation resulting from viral and bacterial infections, sepsis, trauma, sterile inflammation, mechanical ventilation, ischemia-reperfusion, and blood transfusion [7-13]. Unlike apoptosis, necroptosis causes cell membrane rupture, which triggers and amplifies inflammation thorough the release of damage-associated molecular patterns, such as high-mobility group protein B1, IL-1 family cytokines, nucleic acids, as well as S100 proteins [11,14]. Necroptosis is initiated by receptor-interacting protein kinase-3 (RIPK3) and executed by the effector mixed lineage kinase domain-like protein (MLKL) [9,15]. Activation of RIPK3 induces MLKL phosphorylation and membrane translocation and subsequent disruption of the plasma membrane, leading to necrotic cell death [16-19]. Necroptosis is negatively regulated by caspase-8 together with a caspase-like molecule c-FLIPL [20] and by the ubiquitin ligases cIAP1 and cIAP2 [21]. Thus necroptosis is sensitized under caspase inhibition and cIAPs for degradation by Smac mimetics [19]. Caspase 8 inhibitors have been identified in murine cytomegalovirus and herpes family viruses [22], while loss of cIAPs can happen during cytokine stimulation [23]. Necroptosis can be induced by TNF superfamily death ligands, Tolllike receptor-3 (TLR3) and TLR4 ligands when caspases are inhibited and/or cIAPs are degraded [15-19,24]. TNFinduced necroptosis requires the complex formation of RIPK1-RIPK3-MLKL [19,25]. Direct phosphorylation of RIPK3 by RIPK1 has not been demonstrated; hence oligomerization of RIPK3 driven by the RHIM domains of RIPK1 and RIPK3 leads to RIPK3 autoactivation [26]. Consistent with that, RIPK3 can be activated by associating with other two RHIM-containing adaptor proteins such as TRIF and Z-DNA/RNA binding protein 1 (ZBP1; also known as DAI/DLM-1) to mediate necroptosis by TLR3/4 activation and virus infection, respectively [24,27,28]. RIPK3 activation is thus regulated by the competitive interactions with other three RHIM-containing proteins including RIPK1, TRIF and ZBP1 [9,15,24,29]. It has been shown that serum levels of RIPK3 are elevated in critically ill patients and are associated with the development of acute respiratory distress syndrome in sepsis, trauma and COVID-19 infection [30-32]. Recent experimental studies indicate that inhibition of necroptosis confers protection against acute lung injury and inflammation induced by LPS [33-35], hyperoxia [36], mechanical ventilation [37], sepsis/systemic inflammatory response syndrome [38-40], respiratory syncytial virus Infection [41], bacterial pneumonia [42-44], trauma [45], and blood transfusion [46]. Hence, the targeting of necroptosis holds significant promise for the treatment of acute lung injury and inflammation.
M1 Macrophages Are More Susceptible to Necroptosis
Although macrophage activation and polarization has been well studied [2-4], we know less regarding the role of macrophage activation/polarization in inflammationassociated necrotic cell death. The macrophage subtypes that are susceptible to necroptosis are not clear and the underlying mechanisms are likewise poorly understood.
Most necroptosis studies are performed in resting cells [15-19,24], which is a commonly used approach to define the necroptosis signaling pathway. As inflammation and tissue injury is often associated with release of cytokines among other mediators, innate immune cells such as macrophages and tissue structural cells are expected to encounter these pro-inflammatory effectors and be activated. Based on the idea, in our recent study, we pretreated bone marrow-derived macrophages with M1 (LPS, dsRNA and IFNγ) or M2 (IL-4, IL-10 and TGFβ) macrophage subtype inducers and then investigated the subtype-dependent responses to different necroptosis inducers. We found that macrophage necrotic cell death and the releases of lactate dehydrogenase and dead cell proteases were greatly augmented in M1 but not M2 macrophages, and the enhanced effects were blocked by two structurally distinct specific RIPK3 inhibitors GSK872 or GSK843 [47]. Our findings clearly demonstrate that M1 but not M2 subtypes of macrophages are much more susceptible to inflammation-related necrotic cell death in a RIPK3 kinase activity-dependent manner. The lytic cell death of M1 macrophages can result in release of not only damage-associated molecular patterns that are normally seen in resting cells [11,14], but also of high levels of newly synthesized pro-inflammatory cytokines and chemokines [3]. We thus posit that the burst of such immunestimulatory intracellular components could trigger and amplify inflammation, form a pro-inflammatory cycle and ultimately contribute to the pathogenesis of acute tissue injury and inflammation. Recent evidence has shown that necroptosis of alveolar macrophage plays an important role in the pathogenesis of acute lung injury and inflammation [41,48]. Our edifying findings also suggest that, like M1 macrophages, LPS-, dsRNA- or IFNγ-activated/primed tissue structural epithelial and endothelial cells could be susceptible to necroptosis, which merits further investigation. In contrast, the M1 macrophage inducers did not enhance macrophage susceptibility to apoptosis inducers [47].
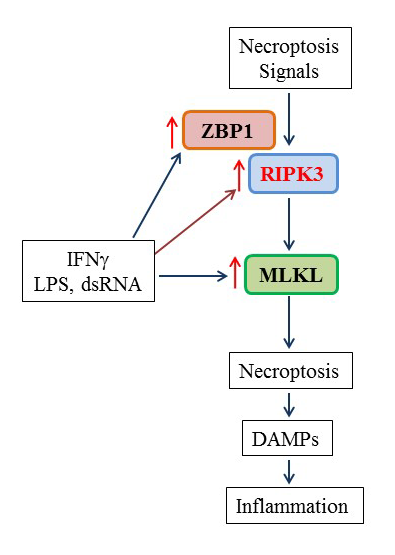
Delineation of the mechanisms participating in M1 macrophage necroptosis may offer a novel strategy to control aberrant host innate immune responses and tissue damage. Mechanistically, we found that the necroptosis effector MLKL and the key necroptosis signaling molecule ZBP1 were exclusively induced by M1 but not M2 macrophage subtype inducers [47] (Figure 1). We also found that the protein but not mRNA levels of RIPK3 were upregulated in M1 macrophages, which suggests that protein synthesis or posttranslational regulation (e.g. stability) may be involved in the upregulation of RIPK3 protein. Thus, enhanced necrotic cell death occurring in M1 macrophages may likely attribute to the upregulation of key necroptosis signaling molecules including RIPK3, MLKL and ZBP1 (Figure 1). In addition, we found that these three necroptosis signal molecules were readily upregulated in M1 macrophage inducer-primed dendritic cells [47]. These results suggest that like M1 macrophages, the activated dendritic cells could be susceptible to necroptosis, promoting antigen presentation to T lymphocytes as demonstrated previously [49]. More studies are needed to define the proposed mechanisms.
Conclusions and Perspectives
By using bone marrow-derived macrophages, we have recently reported that that M1 macrophages induced by LPS, IFNγ and dsRNA were much more sensitive than M0 and M2 subtypes of macrophages to various necrotic cell death inducers. The enhanced necroptosis in M1 macrophages is dependent on RIPK3 kinase activity and may involve the upregulation of key necroptosis signaling molecules including RIPK3, MLKL and ZBP1 (Figure 1). These findings provide novel insights into the mechanisms of M1 macrophage engagement in inflammation and tissue injury. Since we used bone marrow-derived macrophages for the study, it will be important to determine if M1 macrophages are more susceptible to necroptosis in vivo under disease conditions. Although inhibition of necroptosis confers protection against tissue injury and inflammation, the role of macrophages in the process is not clear. Generation of macrophage-specific knock out of RIPK3 will enable us to address this gap in current knowledge and to identify a specific therapeutic target that could be further developed to control various forms of inflammation and tissue injuries, in which macrophage death is a common feature.
Acknowledgments
This study was supported in part by a NIH grant AI135569 (to HT).
Author Contributions Statement
QH and HT wrote the manuscript. HT and SI critically reviewed and edited the manuscript. All authors approved the final version.
Conflict of Interest
The authors declare that they have no conflict of interest. Dr. Idell is a founder, board member and Chief Scientific Officer of Lung Therapeutics, Inc. and has an equity position in the company. He declares no conflict of interest as regards this work.
References
2. Byrne AJ, Maher TM, Lloyd CM. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol Med. 2016;22(4):303-16.
3. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425-40.
4. Patel U, Rajasingh S, Samanta S, Cao T, Dawn B, Rajasingh J. Macrophage polarization in response to epigenetic modifiers during infection and inflammation. Drug Discov Today. 2017;22(1):186-93.
5. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453-61.
6. Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol. 2015;184:436-45.
7. Zhao H, Jaffer T, Eguchi S, Wang Z, Linkermann A, Ma D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015;6:e1975.
8. Fujikura D, Miyazaki T. Programmed Cell Death in the Pathogenesis of Influenza. Int J Mol Sci. 2018;19(7).
9. He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol. 2018;19(9):912-22.
10. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151-64.
11. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311-20.
12. Faust H, Mangalmurti NS. Collateral damage: necroptosis in the development of lung injury. Am J Physiol Lung Cell Mol Physiol. 2020;318(2):L215-L25.
13. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019;4(15).
14. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209-23.
15. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352(6281):aaf2154.
16. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213-27.
17. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133-46.
18. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322-7.
19. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100- 11.
20. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase- 8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363-7.
21. Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43(3):432-48.
22. Guo H, Kaiser WJ, Mocarski ES. Manipulation of apoptosis and necroptosis signaling by herpesviruses. Med Microbiol Immunol. 2015;204(3):439-48.
23. Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P, et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol. 2008;182(1):171-84.
24. Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268-79.
25. Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112- 23.
26. Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150(2):339-50.
27. Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe. 2016;20(5):674-81.
28. Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/ DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290- 7.
29. Kuriakose T, Kanneganti TD. ZBP1: Innate Sensor Regulating Cell Death and Inflammation. Trends Immunol. 2018;39(2):123-34.
30. Shashaty MGS, Reilly JP, Faust HE, Forker CM, Ittner CAG, Zhang PX, et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: a cohort study. Crit Care. 2019;23(1):235.
31. Nakamura H, Kinjo T, Arakaki W, Miyagi K, Tateyama M, Fujita J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit Care. 2020;24(1):484.
32. Ma KC, Schenck EJ, Siempos, II, Cloonan SM, Finkelsztein EJ, Pabon MA, et al. Circulating RIPK3 levels are associated with mortality and organ failure during critical illness. JCI Insight. 2018;3(13).
33. Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie W, et al. Receptor Interacting Protein 3-Mediated Necroptosis Promotes Lipopolysaccharide-Induced Inflammation and Acute Respiratory Distress Syndrome in Mice. PLoS One. 2016;11(5):e0155723.
34. Chen J, Wang S, Fu R, Zhou M, Zhang T, Pan W, et al. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J Transl Med. 2018;16(1):233.
35. Lin B, Jin Z, Chen X, Zhao L, Weng C, Chen B, et al. Necrostatin1 protects mice from acute lung injury by suppressing necroptosis and reactive oxygen species. Mol Med Rep. 2020;21(5):2171-81.
36. Syed MA, Shah D, Das P, Andersson S, Pryhuber G, Bhandari V. TREM-1 Attenuates RIPK3-mediated Necroptosis in Hyperoxia-induced Lung Injury in Neonatal Mice. Am J Respir Cell Mol Biol. 2019;60(3):308-22.
37. Siempos, II, Ma KC, Imamura M, Baron RM, Fredenburgh LE, Huh JW, et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight. 2018;3(9):e97102.
38. Bolognese AC, Yang WL, Hansen LW, Denning NL, Nicastro JM, Coppa GF, et al. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery. 2018;164:110-116.
39. Hansen LW, Jacob A, Yang WL, Bolognese AC, Prince J, Nicastro JM, et al. Deficiency of receptor-interacting protein kinase 3 (RIPK3) attenuates inflammation and organ injury in neonatal sepsis. J Pediatr Surg. 2018;53(9):1699-705.
40. Sharma A, Matsuo S, Yang WL, Wang Z, Wang P. Receptor-interacting protein kinase 3 deficiency inhibits immune cell infiltration and attenuates organ injury in sepsis. Crit Care. 2014;18(4):R142.
41. Santos LD, Antunes KH, Muraro SP, de Souza GF, da Silva AG, de Souza Felipe J, et al. TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during Respiratory Syncytial Virus infection. Eur Respir J. 2020 Dec 10;2003764
42. Gonzalez-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, et al. Pore-forming toxinmediated ion dysregulation leads to death receptorindependent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017;24(5):917- 28.
43. Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, et al. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015;11(12):e1005337.
44. Gonzalez-Juarbe N, Riegler AN, Jureka AS, Gilley RP, Brand JD, Trombley JE, et al. Influenza-Induced Oxidative Stress Sensitizes Lung Cells to Bacterial-Toxin- Mediated Necroptosis. Cell Rep. 2020;32(8):108062.
45. Cui YL, Qiu LH, Zhou SY, Li LF, Qian ZZ, Liu XM, et al. Necroptosis as a potential therapeutic target in multiple organ dysfunction syndrome. Oncotarget. 2017;8(34):56980-90.
46. Qing DY, Conegliano D, Shashaty MG, Seo J, Reilly JP, Worthen GS, et al. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med. 2014;190(11):1243-54.
47. Hao Q, Kundu S, Kleam J, Zhao ZJ, Idell S, Tang H. Enhanced RIPK3 kinase activity-dependent lytic cell death in M1 but not M2 macrophages. Mol Immunol. 2021;129:86-93.
48. Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. 2018;19(1):50.
49. Hartmann BM, Albrecht RA, Zaslavsky E, Nudelman G, Pincas H, Marjanovic N, et al. Pandemic H1N1 influenza A viruses suppress immunogenic RIPK3-driven dendritic cell death. Nat Commun. 2017;8(1):1931.