Abstract
Immune thrombocytopenia is an autoimmune disease predominantly caused by autoantibody mediated platelet and megakaryocyte destruction and or dysfunction, which leads to low platelet counts and risk of bleeding. Currently prognostic biomarkers are underdeveloped and there lacks a gold-standard for therapeutics, which leaves an inexplicable refractory subset of patients which are clinically challenging. Autoantibodies in ITP predominantly target the two most abundantly expressed platelet surface antigens integrin GPIIbIIIa and GPIb-IX. We and others have reported antibodies against GPIbα tend to exhibit more refractoriness to common ITP therapies such as steroids and intravenous IgG (IVIG). Here we discuss the mechanisms which could contribute to the increased resistance, including our recent finding of anti-GPIbα-, and some anti-GPIIbIIIa- mediated platelet activation and desialylation, leading to Fc-independent platelet clearance. Furthermore, we discuss the emerging clinical investigations of utilizing desialylation as a biomarker/prognostic tool in identifying the refractory subset, as well as the therapeutic benefits of targeting desialylation with sialidase inhibitors.
Keywords
Immune thrombocytopenia, Autoantibodies, Platelets, GPIbα, Integrin
Introduction
Platelets are small anuclear cells shed from the megakaryocyte, at a rate of ~1011/day [1], maintaining a blood concentration of 100-450 x 109/L in healthy adults. As the second most abundant circulating cells, they are becoming increasingly recognized for their versatility and cross-talks in cancer, development, immunology among others [2,3]. Classically, they are essential for hemostasis; as the first cellular responders to vascular injury, they sense and aggregate at sites of exposed subendothelial matrix proteins forming the initial cellular plug and scaffold for the coagulation cascade, leading to stabilized hemostatic plug formation and ultimately arrest of bleeding [4-6]. Thus, immune disorders such as autoimmune thrombocytopenia (ITP) that target one’s own platelets or megakaryocytes can lead to devasting bleeding and even death. ITP has an incidence of 3.3/100,000 and a prevalence of 9.5/100,000 worldwide and often occurs with insidious onset in adults [7,8]. Characterized by chronic thrombocytopenia, these patients experience bleeding symptoms and are at constant risk for fatal hemorrhage [ 9]. They frequently suffer co-morbidities such as fatigue, increased risk of infection, hematological malignances and an overall decrease in health-related quality of life [10,11]. Diagnosis is based on exclusion, and prognostic markers are underdeveloped [12]. Current therapies, mostly immunosuppressive or immunomodulatory in nature are aimed at only management of the disease, are based on empirical guidelines, have limited consensus and inexplicable refractoriness [13]. However, as research continues to unravel the complex pathophysiology of ITP it paves the way for novel, more effective therapeutic/ diagnostic regimens in particular targeting the difficult refractory patient subset [14,15]. Here we discuss recent discoveries into mechanisms of antibody mediated ITP, whereby antibody specificity may affect disease and response to treatment.
The Dichotomy of Anti-GPIb and Anti- GPIIbIIIa Mediated ITP and Challenges to First-line Therapies
GPIbα and GPIIbIIIa are distinct antigenic targets
Since Harrington’s seminal experiment in the 1950’s, it is now widely accepted that autoantibodies (immunoglobulin (IgG)) is the predominant mechanism mediating platelet/megakaryocyte destruction in ITP [16]. Although, cytotoxic T-cells and complement may also play a role [17-19]. Autoantibodies are generated from aberrant self-antigen presentation, which results in T-cell dependent B-cell isotype class switch and IgG production (predominantly IgG1) in the spleen [20]. The primary antibody targets are the two most abundantly expressed platelet surface receptors GPIb-IX complex and integrin GPIIbIIIa (αIIbβ3) (~25,000 copies and ~80,000 copies/platelet, respectively) [21]. While both these receptors are essential in hemostasis, they are distinct in function and structure.
GPIbα is the largest subunit of the GPIb-IX complex and possesses all the known ligand binding sites. It is comprised of a N-terminus leucine rich repeat followed by a long flexible “stalk” macroglycopeptide region (which constitutes an impressive ~60% of total platelet sialicacid content) and a juxtamembrane mechanosensory domain [22,23]. Possessing mechanosensory properties, it is one of the first receptors engaged in platelet activation, where stable binding to exposed A1 domain of immobilized von Willabrand (VWF) factor under arterial high shear stress initiates inside-out signaling and stable platelet adhesion [24,25]. This leads to platelet activation including platelet granule release and the critical GPIIbIIIa integrin activation [26]. In addition to binding VWF, other well-known ligands of GPIbα include thrombin, kininogen, P-selectin, thrombospodin and others [24]. GPIb-IX complex is exclusively expressed on platelet and megakaryocytes, and has been functionally implicated in a variety of processes beyond hemostasis and thrombosis including immune regulation (e.g. sepsis, infections), cancer, stroke, angiogenesis and megakaryopoesis [24]. And recently we have reported the requirement of GPIbα for platelet mediated hepatic TPO generation [27]. It is estimated that of those ITP patients with detectable autoantibodies, ~20-40% are positive for anti-GPIbα [21]. Given the cross functional roles of GPIbα, we are only beginning to elucidate the effects GPIbα antibody targeting.
GPIIbIIIa is a classical integrin, expressed as a heterodimer comprised of non-covalently associated αIIb and β3 subunits [28]. As with other integrins, GPIIbIIIa must undergo a divalent cation dependent conformation change from low-affinity (bent) to a high-affinity active state (extended) for stable adhesion to its ligand(s) [4,29]. Activation of GPIIbIIIa directly follows synergistic insideout signaling mediated by other platelet receptors such as GPIbα, GPVI, αIIβ1 etc. Receptor occupancy of GPIIbIIIa with fibrinogen or other ligands leads to platelet crosslinking and aggregation as well as GPIIbIIIa outside-in signaling and a second wave of platelet activation [30-33]. The canonical ligand of GPIIbIIIa usually consists of a simple RGD peptide sequence and binds at the interface between the β-propeller subunit of αIIb and the A like-domain of β3. However, GPIIbIIIa does not exclusively bind RGD-containing ligands, as in the case of fibrinogen (C-terminus KQAGDV sequence) or our recently identified novel anti-thrombotic Apolipoprotein A-IV [34,35]. While the principle ligands such as fibrinogen or VWF are not indispensable for platelet aggregation, GPIIbIIIa is essential, and thus critically important for hemostasis [31,36]. Interestingly, there exists quantitatively, a hemostatic threshold for platelet GPIIbIIIa, where ~50% of normal GPIIbIIIa expression is sufficient to maintain hemostasis (i.e. heterozygous Bernard-Soulier patients do not exhibit significant bleeding) [37]. Most autoantibodies in ITP are directed against GPIIbIIIa (~60%) and are predominantly targeted against αIIb subunit. However, it was found binding of these autoantibodies is usually divalent cation dependent and requires β3 subunit as an intact αIIbβ3 structure [38]. Putatively, autoantibodies that block the ligand binding site, or ‘lock’ the conformational state of GPIIbIIIa could inhibit platelet function in ITP [14] (Figure 1). Conversely, antibodies may also reveal cryptic ligand induced binding sites, causing abnormal GPIIbIIIa activation [39,40]. ITP sera that are activating or inhibitory have been reported, furthermore, there is evidence that platelet function in ITP patients may be a better predictor of bleeding tendency than platelet number [41].
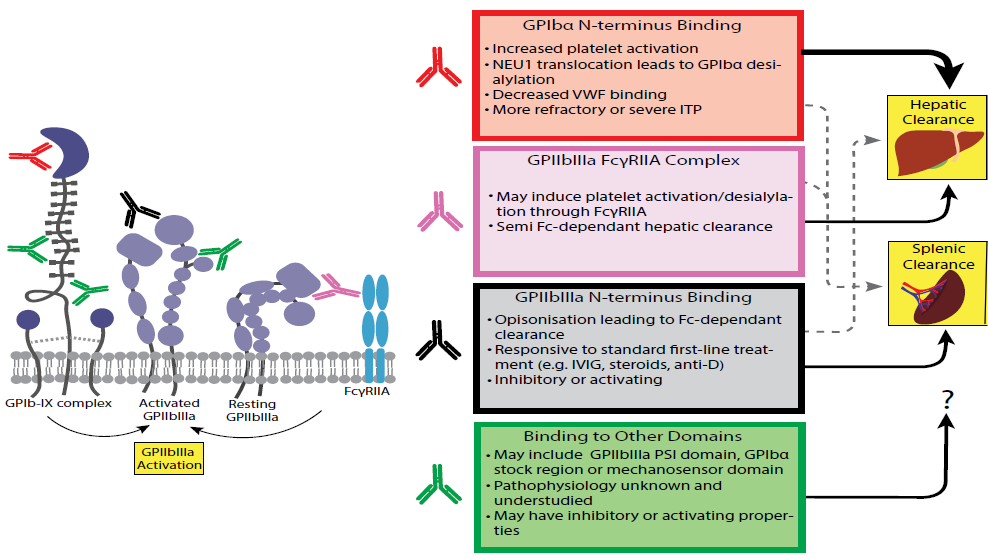
Given the unique roles of these respective platelet receptors, it is conceivable that antibody binding could manifest discreet outcomes in both bleeding tendency and response to treatment. We and others have demonstrated that in contrast to anti-GPIIbIIIa, anti-GPIbα mediated ITP can be Fc-independent whereby neither the Fc-region of antibody nor the corresponding FcγR are required for antibody mediated clearance [42,43]. This also led to IVIG resistant ITP [42,44]. Our initial observations in mouse models have been recapitulated in subsequent human clinical studies; it has since been reported that presence of anti-GPIbα antibodies in ITP is sufficient to render the patients more refractory to common first-line therapies including steroids, IVIG, and others including Rituximab [44-48].
Anti-GPIbα antibodies mediate platelet activation desialyation in a positive feed-back loop
In 2015, we were the first to elucidate one mechanism by which anti-GPIbα antibodies mediates Fc-independent platelet destruction. We found anti-GPIbα antibodies targeting the N-terminus of GPIbα cause receptor clustering, downstream signaling, leading to platelet activation, granule neuraminidase surface translocation and enzymatic removal of terminal surface sialic acids (desialylation) (Figure 1). This ultimately leads to platelet clearance via non-Fc receptors, of which the Ashwell-Morell Receptor (AMR) is a contributor [49]. We also demonstrated that antibody mediated platelet activation and desialylation exist in a positive feed-back loop whereby increased platelet desialylation potentiates further platelet activation, through enhanced facilitation of GPIbα signaling [49]. We also observed anti-GPIIbIIIa antibodies could induce human platelet activation, predominantly through immune complex binding and activation of human platelet FcγRIIa (Figure. 1). However, anti-GPIbα antibodies were more prone to mediate platelet activation/desialylation and Fc-independent clearance as it likely through N-terminus F(ab’)2 binding. An FcγRIIa-GPIbα complex mediated activation via N-terminus binding antibodies may be less feasible than GPIIbIIIa, due to the long length of GPIbα (~40 nm compared with ~20 nm, Figure 1) [50]. As bivalency of antibody is required (anti-GPIbα Fab does not induce Fc-independent platelet clearance), it is postulated that antibody crosslinking of GPIbα may mimic the signaling triggered by receptor binding of VWF multimers [49,51]. However, antibody binding site/epitope appears to be critical in anti-GPIbα antibody mediated platelet activation, as antibodies targeting non-N-terminus regions such as the mechanosensory region, which also causes receptor clustering, nevertheless fails to induce platelet activation and Fc-independent platelet clearance [ 52]. Recently it was suggested, shear is an important factor in this process, thus high affinity antibodies that can exert a pulling force under shear causing an unfolding of the mechanosensor domain, are more adept at inducing GPIbα activation [23]. However, the exact binding epitopes that confer activating properties to anti-GPIbα antibodies remains to be further studied.
The presence of platelet activating antibodies such as anti-GPIbα may predispose a more severe disease. Low antibody titers causing consumptive platelet microaggregate formation leads to an amplified and disproportionate thrombocytopenia. In addition, binding of anti-GPIbα at the N-terminus may block endogenous ligand binding, rendering the remaining platelets hemostatically impotent and exacerbating bleeding tendency (Figure 1). This culminates in a particularly severe and likely refractory disease, as we observed in an unfortunate fatal case of anti-GPIbα mediated ITP [53]. We also recently uncovered the requirement for GPIbα N-terminus for platelet mediated hepatic TPO generation, which could be blocked in the presence of anti-GPIbα antibodies [27]. This in combination with previous reports indicating the essential role of GPIbα for normal late stage megakaryopoesis (thrombopoiesis) and proplatelet formation, suggests platelet GPIbα N-terminus is a significant contributor in platelet production which may be significantly impacted in presence of anti-GPIbα antibodies [54,55].
Anti-GPIbα antibodies mediated platelet desialyation leads to non-classical Fcindependent clearance in the liver
A critical downstream consequence of anti-GPIbα antibody mediated platelet activation is platelet desialyation [23,49]. Removal of terminal sialic acids, particularly on GPIbα itself, primes platelets for rapid removal via non-FcγR pathways in the liver, rather than expected splenic Fc-FcγR mediated clearance. Attenuation of neuraminidase enzyme activity with pharmacological inhibitors such as Oseltamivir (Tamiflu©) has been demonstrated to decrease platelet desialylation and increase platelet counts in our murine models and human ITP patients [48,56-58]. Reciprocally, we and others have identified the AMR as one of the non-Fc receptors that contributes to desialylated platelet clearance [49,59]. However, contributions of other lectin receptors such as Mac-1 (αMβ2) on Kupffer cells are not well investigated. Kupffer cells are strategically positioned within the liver vessel lumen (lining the sinusoids) which facilitates interaction with circulating platelets [60]. Kupffer cellplatelet interactions have been reported to contribute to a variety of biological processes including liver injury, infection, and liver regeneration [61]. It is likely that they also significantly contribute to desialylated/apoptotic platelet clearance. There are emerging reports identifying αMβ2 cognate ligands on platelet surface including O-linked deglycosylated residues, but it’s role in ITP is not known [62]. Furthermore, the immunological consequences of platelet clearance within immunological disparate organs (spleen versus liver) has not been adequately studied, although our preliminary data show an immunosuppressive response associated with liver platelet clearance.
Platelet Desialylation: A New Diagnostic Biomarker and Therapeutic Target in ITP
Current first line therapies in ITP include corticosteroids, IVIG, and/or anti-D. Although a good proportion of patients exhibit an initial response (up to 80%), durable response is difficult to achieve; long-term follow up frequently reveal significant drop in responders particularly following tapering or withdrawal of the drug (down to ~40%) [13]. In addition, cost, bioavailability (e.g. IVIG, anti-D) and drug related toxicities are of real concern. Paradoxically, risks associated with taking common treatments such as corticosteroids and IVIG sometimes outweigh the risk associated with ITP itself [11]. For those that fail first line therapies, second-line and third-line therapies including rituximab (B-cell depletion with an anti-CD20 monoclonal antibody), TPO receptor agonists (TPO-RA) (eltrombopag, avatrombopag, romiplostim), and less commonly fostamatinib (an FcγR Syk inhibitor) and other immunosuppressive drugs (mycophenolate, azathioprine, and cyclosporine) are either given alone or in combination. As there are no benchmark guidelines, these are administered on a ‘trial and error’ basis [63]. Splenectomy, a permanent surgical procedure with lifelong safety concerns, is now on the decline due to the conflicting data regarding its therapeutic superiority compared to pharmacological interventions such as rituximab or TPO-RA [13]. As with other ITP treatments prognostic markers are lacking, although it has been observed hepatic sequestration of radioisotope-labeled platelets is correlated with decreased response [64].
As most of the aforementioned ITP therapies target Fc-dependent platelet clearance mechanisms (e.g. IVIG, anti-D, rituximab, fostamatinib, splenectomy), it logically follows antibodies causing non-splenic Fc-independent platelet clearance will be less responsive. Since we have identified desialylation as a marker in both antibody mediated platelet activation and Fc-independent clearance, clinical studies have been initiated to assess prognostic potential of desialylation as a biomarker for refractory ITP and the therapeutic potential of neuraminidase inhibitor Oseltamivir (Tamiflu©) [48]. Recently, our clinical study of 61 ITP patients demonstrated higher platelet desialylation in patients was correlated with non-response to corticosteroids and IVIG first-line treatments [65]. The relatively simple protocol of detecting binding of Ricinus Agglutin I (RCA-1) to desialylated platelet residues via flow cytometry suggests a feasible prognostic assay for assessment of potentially refractory patients, although large scale clinical study is required to confirm this finding. Lessons learned from these emerging clinical studies suggest 1) increased refractoriness in ITP may be associated with platelet desialylation [48,65-67] and 2) platelet desialylation is not exclusively linked with anti-GPIbα antibodies, although presence of anti-GPIbα antibodies may increase propensity for platelet desialylation, other mechanisms such as CD8+ cytotoxic T-cells may also contribute [48,65,68]. Additionally, anti-GPIIbIIIa antibodies may also mediate platelet desialylation through human platelet FcγRIIa as we previously reported, which was also observed in human ITP patients [65,69]. Although in the latter study, there appeared to be a greater propensity for hepatic clearance in the presence of anti-GPIbα antibodies, despite not reaching statistical significance.
Tamiflu©, an FDA approved neuraminidase inhibitor typically used to treat influenza was previously shown to also effective on platelet sialidases and increased platelet sialylation [58]. Since we identified platelet desialylation as a Fc-independent clearance pathway in ITP, several case studies as well as ITP patient cohorts have demonstrated therapeutic benefit either alone or in combination with other ITP treatments, particularly in the refractory patient subset [48,56,57,70]. Although the data show platelet desialylation levels are decreased in ITP patients following successful response to sialidase inhibitors, other pathways which contributes to therapeutic effect including prolonging platelet lifespan through decreasing aged-related platelet desialylation or other off-target desialylation inhibition including that of pathogenic lymphocytes cannot be excluded.
Future Perspectives
It is becoming increasingly clear that ITP can no longer be considered a homogeneous disease. Indeed, patients often experience great variability in bleeding tendency independent of platelet count, as well as inconsistent response to therapeutics. Our recent findings highlighting differential pathways of antibody mediated effects on platelets and platelet clearance represent one facet of the heterogeneity that may exist within the disease pathology. While these findings were discussed in the context of ITP, they may also be applicable to other immune-mediated thrombocytopenic disorders in which anti-platelet antibodies are present, such as alloimmune thrombocytopenias (e.g. fetal neonatal alloimmune thrombocytopenia, post-transfusion purpura etc.) or secondary ITP. Although great strides have been made to elucidate pathobiological differences between refractory or severe ITP subset and responders, our understanding of its mechanisms are still in its infancy. Larger scale clinical trials are needed to evaluate the utility of reliable prognostic biomarkers (e.g. anti-GPIbα antibodies and desialyation) which will optimize personalized treatment, as well as alleviate healthcare cost burden.
Financial Support and Sponsorship
This work was supported in part by Canadian Institutes of Health Research (MOP 97918, MOP 119540, MOP 119551) and Canadian Blood Services- Canadian Institutes of Health Research partnership fund (CIHRBUC201403- HN-326400). J.L. is a recipient of a Ph.D. Graduate Student Fellowship from Canadian Blood Services. J.A.S. is a recipient of the Canadian Institutes of Health Research Canadian Graduate Student – Master Award and the Laboratory Medicine and Pathobiology Departmental Fellowship from the University of Toronto.
Conflicts of Interest
No conflicts of interest to disclose.
References
2. Xu XR, Zhang D, Oswald BE, Carrim N, Wang X, Hou Y, Zhang Q, Lavalle C, McKeown T, Marshall AH, Ni H. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Critical reviews in clinical laboratory sciences. 2016 Nov 1;53(6):409-30.
3. Xu XR, Yousef GM, Ni H. Cancer and platelet crosstalk: opportunities and challenges for aspirin and other antiplatelet agents. Blood. 2018 Apr 19;131(16):1777-89.
4. Ni H, Freedman J. Platelets in hemostasis and thrombosis: role of integrins and their ligands. Transfusion and Apheresis Science. 2003 Jun 1;28(3):257-64.
5. Wang Y, Gallant RC, Ni H. Extracellular matrix proteins in the regulation of thrombus formation. Current opinion in hematology. 2016 May 1;23(3):280-7.
6. Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circulation research. 2007 Jun 22;100(12):1673-85.
7. Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: a critical review of published reports. American journal of hematology. 2010 Mar;85(3):174-80.
8. Fogarty PF. Chronic immune thrombocytopenia in adults: epidemiology and clinical presentation. Hematology/Oncology Clinics. 2009 Dec 1;23(6):1213- 21.
9. Frederiksen H, Christiansen CF, Nørgaard M. Risk and prognosis of adult primary immune thrombocytopenia. Expert review of hematology. 2012 Jan 1;5(2):219-28.
10. Snyder CF, Mathias SD, Cella D, Isitt JJ, Wu AW, Young J. Health-related quality of life of immune thrombocytopenic purpura patients: results from a web-based survey. Current medical research and opinion. 2008 Oct 1;24(10):2767-76.
11. Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood. 2001 May 1;97(9):2549-54.
12. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, Bussel JB, Cines DB, Chong BH, Cooper N, Godeau B. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009 Mar 12;113(11):2386-93.
13. Provan D, Arnold DM, Bussel JB, Chong BH, Cooper N, Gernsheimer T, Ghanima W, Godeau B, González- López TJ, Grainger J, Hou M. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood advances. 2019 Nov 26;3(22):3780-817.
14. Li J, Sullivan JA, Ni H. Pathophysiology of immune thrombocytopenia. Current opinion in hematology. 2018 Sep 1;25(5):373-81.
15. Miltiadous O, Hou M, Bussel JB. Identifying and Treating Refractory ITP: Difficulty in Diagnosis and Role of Combination Treatment. Blood. 2019 Nov.
16. Harrington WJ, Minnich V, Hollingsworth JW, Moore CV. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. The Journal of laboratory and clinical medicine. 1951 Jul 1;38(1):1-0.
17. Peerschke EI, Andemariam B, Yin W, Bussel JB. Complement activation on platelets correlates with a decrease in circulating immature platelets in patients with immune thrombocytopenic purpura. British journal of haematology. 2010 Feb;148(4):638-45.
18. Chow L, Aslam R, Speck ER, Kim M, Cridland N, Webster ML, Chen P, Sahib K, Ni H, Lazarus AH, Garvey MB. A murine model of severe immune thrombocytopenia is induced by antibody-and CD8+ T cell–mediated responses that are differentially sensitive to therapy. Blood. 2010 Feb 11;115(6):1247-53.
19. Ma L, Simpson E, Li J, Xuan M, Xu M, Baker L, Shi Y, Yougbaré I, Wang X, Zhu G, Chen P. CD8+ T cells are predominantly protective and required for effective steroid therapy in murine models of immune thrombocytopenia. Blood. 2015 Jul 9;126(2):247-56.
20. Chan H, Moore JC, Finch CN, Warkentin TE, Kelton JG. The IgG subclasses of platelet‐associated autoantibodies directed against platelet glycoproteins IIb/IIIa in patients with idiopathic thrombocytopenic purpura. British journal of haematology. 2003 Sep;122(5):818-24.
21. McMillan R. Autoantibodies and autoantigens in chronic immune thrombocytopenic purpura. InSeminars in hematology 2000 Jul 1 (Vol. 37, No. 3, pp. 239-248). WB Saunders.
22. Li R, Emsley J. The organizing principle of the platelet glycoprotein Ib–IX–V complex. Journal of thrombosis and haemostasis. 2013 Apr;11(4):605-14.
23. Quach ME, Dragovich MA, Chen W, Syed AK, Cao W, Liang X, Deng W, De Meyer SF, Zhu G, Peng J, Ni H. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood. 2018 Feb 15;131(7):787-96.
24. Li, R. The Glycoprotein Ib-IX-V complex. in Platelets 4th Edition. 2019: 193-211.
25. Lei X, Reheman A, Hou Y, Zhou H, Wang Y, Marshall AH, Liang C, Dai X, Li BX, Vanhoorelbeke K, Ni H. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thrombosis and haemostasis. 2014 Aug;112(02):279-89.
26. Kroll MH, Harris TS, Moake JL, Handin RI, Schafer AI. von Willebrand factor binding to platelet GpIb initiates signals for platelet activation. The Journal of clinical investigation. 1991 Nov 1;88(5):1568-73.
27. Xu M, Li J, Neves MA, Zhu G, Carrim N, Yu R, Gupta S, Marshall J, Rotstein O, Peng J, Hou M. GPIbα is required for platelet-mediated hepatic thrombopoietin generation. Blood. 2018 Aug 9;132(6):622-34.
28. Fujimura K, Phillips DR. Calcium cation regulation of glycoprotein IIb-IIIa complex formation in platelet plasma membranes. Journal of Biological Chemistry. 1983 Sep 10;258(17):10247-52.
29. Shimaoka M, Springer TA. Therapeutic antagonists and conformational regulation of integrin function. Nature Reviews Drug Discovery. 2003 Sep;2(9):703.
30. Rocco M, Rosano C, Weisel JW, Horita DA, Hantgan RR. Integrin conformational regulation: uncoupling extension/tail separation from changes in the head region by a multiresolution approach. Structure. 2008 Jun 11;16(6):954-64.
31. Yang H, Reheman A, Chen P, Zhu G, Hynes RO, Freedman J, Wagner DD, Ni H. Fibrinogen and von Willebrand factor‐independent platelet aggregation in vitro and in vivo. Journal of Thrombosis and Haemostasis. 2006 Oct;4(10):2230-7.
32. Dunne E, Spring CM, Reheman A, Jin W, Berndt MC, Newman DK, Newman PJ, Ni H, Kenny D. Cadherin 6 has a functional role in platelet aggregation and thrombus formation. Arteriosclerosis, thrombosis, and vascular biology. 2012 Jul;32(7):1724-31.
33. Reheman A, Tasneem S, Ni H, Hayward CP. Mice with deleted multimerin 1 and α-synuclein genes have impaired platelet adhesion and impaired thrombus formation that is corrected by multimerin 1. Thrombosis research. 2010 May 1;125(5):e177-83.
34. Kloczewiak M, Timmons S, Hawiger J. Recognition site for the platelet receptor is present on the 15-residue carboxy-terminal fragment of the γ chain of human fibrinogen and is not involved in the fibrin polymerization reaction. Thrombosis research. 1983 Jan 15;29(2):249-54.
35. Xu XR, Wang Y, Adili R, Ju L, Spring CM, Jin JW, Yang H, Neves MA, Chen P, Yang Y, Lei X. Apolipoprotein A-IV binds αIIbβ3 integrin and inhibits thrombosis. Nature communications. 2018 Sep 6;9(1):3608.
36. Ni H, Denis CV, Subbarao S, Degen JL, Sato TN, Hynes RO, Wagner DD. Persistence of platelet thrombus formation in arterioles of mice lacking both von Willebrand factor and fibrinogen. The Journal of clinical investigation. 2000 Aug 1;106(3):385-92.
37. Coller BS, Seligsohn U, Zivelin A, Zwang E, Lusky A, Modan M. Immunologic and biochemical characterization of homozygous and heterozygous Glanzmann thrombasthenia in the Iraqi‐Jewish and Arab populations of Israel: comparison of techniques for carrier detection. British journal of haematology. 1986 Apr;62(4):723-35.
38. Fujisawa K, Tani P, McMillan R. Platelet-associated antibody to glycoprotein IIb/IIIa from chronic immune thrombocytopenic purpura patients often binds to divalent cation-dependent antigens. Blood. 1993 Mar 1;81(5):1284-9.
39. Armstrong PC, Peter K. GPIIb/IIIa inhibitors: from bench to bedside and back to bench again. Thrombosis and haemostasis. 2012;107(05):808-14.
40. Xu XR, Carrim N, Neves MA, McKeown T, Stratton TW, Coelho RM, Lei X, Chen P, Xu J, Dai X, Li BX. Platelets and platelet adhesion molecules: novel mechanisms of thrombosis and anti-thrombotic therapies. Thrombosis journal. 2016 Oct 1;14(1):29.
41. Frelinger III AL, Grace RF, Gerrits AJ, Carmichael SL, Forde EE, Michelson AD. Platelet function in ITP, independent of platelet count, is consistent over time and is associated with both current and subsequent bleeding severity. Thrombosis and haemostasis. 2018 Jan;118(01):143-51.
42. Webster ML, Sayeh E, Crow M, Chen P, Nieswandt B, Freedman J, Ni H. Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbα antibodies. Blood. 2006 Aug 1;108(3):943-6.
43. Nieswandt B, Bergmeier W, Rackebrandt K, Gessner JE, Zirngibl H. Identification of critical antigenspecific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood. 2000 Oct 1;96(7):2520-7.
44. Go RS, Johnston KL, Bruden KC. The association between platelet autoantibody specificity and response to intravenous immunoglobulin G in the treatment of patients with immune thrombocytopenia. Haematologica. 2007 Feb 1;92(2):283-4.
45. Arnold DM, Vrbensky JR, Karim N, Smith JW, Liu Y, Ivetic N, Kelton JG, Nazy I. The effect of rituximab on anti‐platelet autoantibody levels in patients with immune thrombocytopenia. British journal of haematology. 2017 Jul;178(2):302-7.
46. Peng J, Ma SH, Liu J, Hou Y, Liu XM, Niu T, Xu RR, Guo CS, Wang XM, Cheng YF, Ni H. Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: a multicenter cohort study. Journal of Thrombosis and Haemostasis. 2014 Apr;12(4):497- 504.
47. Zeng Q, Zhu L, Tao L, Bao J, Yang M, Simpson EK, Li C, van der Wal DE, Chen P, Spring CM, Wang M. Relative efficacy of steroid therapy in immune thrombocytopenia mediated by anti‐platelet GPIIbIIIa versus GPIbα antibodies. American journal of hematology. 2012 Feb;87(2):206-8.
48. Revilla N, Corral J, Miñano A, Mingot-Castellano ME, Campos RM, Velasco F, Gonzalez N, Galvez E, Berrueco R, Fuentes I, Gonzalez-Lopez TJ. Multirefractory primary immune thrombocytopenia; targeting the decreased sialic acid content. Platelets. 2019 Aug 18;30(6):743-51.
49. Li J, Van Der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, Vadasz B, Carrim N, Grozovsky R, Ruan M, Zhu L. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nature communications. 2015 Jul 17;6:7737.
50. Erlandsen SL, Bittermann AG, White J, Leith A, Marko M. High-resolution CryoFESEM of individual cell adhesion molecules (CAMs) in the glycocalyx of human platelets: detection of P-selectin (CD62P), GPI-IX complex (CD42a/CD42bα, bβ), and integrin GPIIbIIIa (CD41/CD61) by immunogold labeling and stereo imaging. Journal of Histochemistry & Cytochemistry. 2001 Jul;49(7):809-19.
51. Cauwenberghs N, Meiring M, Vauterin S, Van Wyk V, Lamprecht S, Roodt JP, Novák L, Harsfalvi J, Deckmyn H, Kotzé HF. Antithrombotic effect of platelet glycoprotein Ib–blocking monoclonal antibody Fab fragments in nonhuman primates. Arteriosclerosis, thrombosis, and vascular biology. 2000 May;20(5):1347-53.
52. Liang X, Syed AK, Russell SR, Ware J, Li R. Dimerization of glycoprotein Ibα is not sufficient to induce platelet clearance. Journal of Thrombosis and Haemostasis. 2016 Feb;14(2):381-6.
53. Li J, Callum JL, Lin Y, Zhou Y, Zhu G, Ni H. Severe platelet desialylation in a patient with glycoprotein Ib/ IX antibody-mediated immune thrombocytopenia and fatal pulmonary hemorrhage. Haematologica. 2014 Apr;99(4):e61.
54. Strassel C, Eckly A, Léon C, Petitjean C, Freund M, Cazenave JP, Gachet C, Lanza F. Intrinsic impaired proplatelet formation and microtubule coil assembly of megakaryocytes in a mouse model of Bernard-Soulier syndrome. Haematologica. 2009 Jun 1;94(6):800-10.
55. Iraqi M, Perdomo J, Yan F, Choi PY, Chong BH. Immune thrombocytopenia: antiplatelet autoantibodies inhibit proplatelet formation by megakaryocytes and impair platelet production in vitro. Haematologica. 2015 May 1;100(5):623-32.
56. Shao L, Wu Y, Zhou H, Qin P, Ni H, Peng J, Hou M. Successful treatment with oseltamivir phosphate in a patient with chronic immune thrombocytopenia positive for anti-GPIb/IX autoantibody. Platelets. 2015 Jul 4;26(5):495-7.
57. Jansen AJ, Peng J, Zhao HG, Hou M, Ni H. Sialidase inhibition to increase platelet counts: A new treatment option for thrombocytopenia. American journal of hematology. 2015 May;90(5):E94-5.
58. Alioglu B, Tasar A, Ozen C, Selver B, Dallar Y. An experience of oseltamivir phosphate (tamiflu™) in a pediatric patient with chronic idiopathic thrombocytopenic purpura: a case report. Pathophysiology of haemostasis and thrombosis. 2010;37(2-4):55-8.
59. Hoffmeister KM, Falet H. Platelet clearance by the hepatic Ashwell-Morrell receptor: mechanisms and biological significance. Thrombosis research. 2016 May 1;141:S68-72.
60. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Comprehensive Physiology. 2013 Jan;3(2):785-97.
61. Murata S, Maruyama T, Nowatari T, Takahashi K, Ohkohchi N. Signal transduction of platelet-induced liver regeneration and decrease of liver fibrosis. International journal of molecular sciences. 2014 Apr;15(4):5412-25.
62. Li Y, Fu J, Ling Y, Yago T, McDaniel JM, Song J, Bai X, Kondo Y, Qin Y, Hoover C, McGee S. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proceedings of the National Academy of Sciences. 2017 Aug 1;114(31):8360-5.
63. Peter K, Schwarz M, Ylänne J, Kohler B, Moser M, Nordt T, Salbach P, Kübler W, Bode C. Induction of Fibrinogen Binding and Platelet Aggregation as a Potential Intrinsic Property of Various Glycoprotein IIb/IIIa (IIbβ3) Inhibitors. Blood. 1998 Nov 1;92(9):3240-9.
64. Sarpatwari A, Provan D, Erqou S, Sobnack R, David Tai FW, Newland AC. Autologous 111In‐labelled platelet sequestration studies in patients with primary immune thrombocytopenia (ITP) prior to splenectomy: a report from the United Kingdom ITP Registry. British journal of haematology. 2010 Dec;151(5):477-87.
65. Tao L, Zeng Q, Li J, Xu M, Wang J, Pan Y, Wang H, Tao Q, Chen Y, Peng J, Hou M. Platelet desialylation correlates with efficacy of first-line therapies for immune thrombocytopenia. Journal of hematology & oncology. 2017 Dec;10(1):46.
66. Raskova Kafkova L, Brokesova D, Novak Z, Raska M, Pospisilova D, Volejnikova J. Platelet Desialylation As a Predictive Marker in Childhood Immune Thrombocytopenia (ITP).
67. Monzón Manzano E, Justo Sanz R, Hernández D, Álvarez Roman T, Fernandez-Bello I, Martín M, Valor L, Rivas Pollmar MI, Canales MA, Jimenez-Yuste V, Butta N. Platelet and Immune Characteristics of Patients with Immune Thrombocytopaenia Non Responders to Therapeutic Treatments.
68. Qiu J, Liu X, Li X, Zhang X, Han P, Zhou H, Shao L, Hou Y, Min Y, Kong Z, Wang Y. CD8+ T cells induce platelet clearance in the liver via platelet desialylation in immune thrombocytopenia. Scientific reports. 2016 Jun 20;6:27445.
69. Cantoni S, Carpenedo M, Nichelatti M, Sica L, Rossini S, Milella M, Popescu C, Cairoli R. Clinical relevance of antiplatelet antibodies and the hepatic clearance of platelets in patients with immune thrombocytopenia. Blood. 2016 Oct 27;128(17):2183-5.
70. Sun L, Liu X, Zhou H, Zhao H, Yuan C, Li D, Wang Z, Hou Y, Peng J, Hou M. High-Dose Dexamethasone (HD-DXM) Plus Oseltamivir Versus High-Dose Dexamethasone for Treatment of Adult Primary Immune Thrombocytopenia: Interim Analysis of a Prospective, Multicenter, Randomized, Controlled Trial.