Abstract
Sleep is an evolutionarily conserved phenomenon which has survived tremendous evolutionary pressures. Its disruption has deleterious implications for human health. The importance of sleep is illustrated by the fact that sleep deprivation in many animals leads to death. While sleep is tightly regulated by a combination of intrinsic and extrinsic factors it becomes progressively disrupted in old age and in neurodegenerative diseases including Alzheimer’s disease (AD), frontotemporal dementia (FTD), Parkinson’s disease (PD) and Huntington’s disease (HD). One of the key effects of sleep disruption is increased levels of reactive oxygen/nitrogen species (ROS/RNS) and accumulation of protein aggregates, such as Amyloid β and alpha-Synuclein. A possible mechanism of protein plaque clearance is its autophagic degradation through endo-lysosomal pathways. In this review, we will discuss how sleep disruption is intimately linked with neurodegenerative diseases. We will also discuss the evidence that cellular autophagy and antioxidant defense are regulated by sleep, making it a target for future intervention strategies to tackle neurodegenerative diseases.
Keywords
ROS, RNS, REM, NREM, Autophagy, Glia
Introduction
Sleep is a state of behavioral quiescence when the animal shows reduced responsiveness to all kinds of sensory stimulation. The effects of sleep on our body are understood from studies on sleep deprivation in human and animal models. Lack of sleep leads to several detrimental effects on our health including increased risk to diabetes, forgetfulness, high blood pressure, weakening of immune system, cardiac problems and hallucinations, paranoia [1-7]. Recently, sleep has been identified as a risk factor for several neurodegenerative disease including AD, PD, and FTD [8-10].
In 1894, Marie de Manacéine, a Russian physician and scientist, reported that sleep deprived puppies kept in constant activity die after a few days [11]. In 1898, a similar outcome was reported by Italian physiologists Lamberto Daddi and Giulio Tarozzi in dogs. When they subjected dogs to constant sleep deprivation, the dogs died within 10 days [12]. Marie de Manacéine further showed that the need for sleep was greater than for food since dogs could be rescued after 20-25 days of starvation, but they were ‘irreparably lost’ after 4-5 days of sleep deprivation. Daddi and Tarozzi also reported degeneration of spinal ganglia, Purkinje cells of the cerebellum, and neurons of the frontal cortex. He would base these changes on ‘autointoxication’ due to sleep deprivation. While the toxicity of sleep deprivation and disorder have long been known to scientists and lay-people alike, research into the mechanisms of this damage has formed a perplexing web of biochemical processes which extend into the pathways of many prevalent diseases today. Furthermore, anatomical evidence has shown that sleep fragmentation is directly linked with neuron loss in the brain [13] demonstrating that sleep loss and loss of neurons have a direct relationship.
To understand the current evidence linking sleep to cellular metabolism and neurodegeneration, it is important to consider the role and origins of sleep. Sleep is a state of behavioral quiescence and reduced responsiveness to sensory stimulation, when most but vital motor movements cease and all active experiences, such as learning, stop [14]. Sleep has been observed in animals across different orders from jellyfish to humans emphasizing its evolutionary importance [15-18] . While sleep prevents most animals from beneficial activities such as foraging and gathering knowledge and experience, it must have a more important role to play in their survival that has allowed sleep to overcome such selective pressures. The characteristics of sleep including cessation of movement and behavioral quiescence are highly conserved between organisms such as C. elegans, Drosophila and humans indicating that similar molecular mechanisms govern the need and physiological effects of sleep [19,16]. This has allowed investigation of mechanisms of sleep, in simple model animals such as Drosophila where the complexity of biochemical and behavioral pathways is comparatively reduced. Sleep is yet to be found in organisms lacking a nervous system [20] suggesting a possibly fundamental role in the maintenance of neural tissue in particular.
Sleep deprivation has been shown to have profound cognitive and physiological detrimental effects in many organisms. In mice, chronic sleep deprivation can lead to several homeostatic abnormalities eventually leading to death [21,22]. In Drosophila and C. elegans sleep deprivation has been shown to shorten lifespan [19,23]. However, some other reports showed little effect of sleep disruption on Drosophila and C. elegans [24,25]. In both examples, however, the authors acknowledge that residual sleep may fulfil the necessity of sleep sufficient for survival in the absence of consolidated sleep. An important consideration that these studies may thus highlight is that while sleep is highly conserved, behavioral measures of sleep may not be sufficient to conclusively highlight the effects of sleep deprivation. For example, while sleep bouts are not essential for C. elegans survival, DAF-16/FOXO activation, a critical stress resistance pathway, is still essential to compensate for sleep loss [24]. This suggests that behavioral features of sleep may be of secondary importance to physiological functions.
Mammalian sleep has long been distinguished from invertebrate sleep in that in vertebrates sleep can be distinguished into different phases: rapid eye movement (REM) sleep and the more conserved non-REM (NREM) sleep [20]. NREM can be further subdivided into 4 stages - the deepest of which, stage 3 and 4, are also called slow-wave sleep (SWS) and seen outside the animal kingdom. REM sleep involves a more “active” stage of sleep, sometimes referred to as paradoxical sleep, due to wake-like neural function and rapid eye movements. REM sleep is differentiated from NREM sleep in mammals through delta wave frequency oscillations between 0.5 and 3.5 Hz [26] and many of the regenerative functions of sleep are associated with REM sleep in particular [27]. Others functions of sleep such as the Hippocampus dependent memory benefit more from the SWS and not from late REM sleep.
Determining sleep in Drosophila relies largely on inference and behavioral observations. More than 5 minutes of continuous inactivity is considered as sleep in Drosophila [28-30]. Recently, developments in the study of Drosophila sleep have provided some new perspectives on the distinction between mammalian and invertebrate sleep. REM-like sleep has been shown to exist in Drosophila where activation of the dorsal fan-shaped body (dFB) induces a state of wake-like neuronal function but no response to mechanical stimulation. This paradoxical-like sleep state cycles between the more and less active stages, reminiscent of mammalian sleep [31]. Similarly, a form of deep sleep is shown to exit in Drosophila characterized by proboscis extension (PE). Inhibition of PE doubled the rate of mortality of full-body injured flies compared to controls highlighting an important role in recovery [32]. Of particular interest however is the finding in this report that PEs facilitated waste clearance in Drosophila: using a luciferin feed protocol they demonstrated that metabolites were excreted more slowly in PE-immobilized flies than controls and that sleep deprivation yielded a similar result. Stress-induced sleep peptides flp-13 and nlp-8 in C. elegans are also known to be important in inhibiting defecation during sleep [18] but whether these or other peptides play a role in promoting metabolism has yet to be seen. Furthermore, some preliminary evidence suggests that flp family genes may be orthologous to NPF/NPY in Drosophila and humans [33,34]. However, the relationship between flp-13, nlp-8 and possible orthologs in other species warrant further investigation. These data implicate deep sleep as an evolutionarily conserved promoter of waste clearance and the mechanisms through which this process operates are a subject of interest in determining the therapeutic significance of sleep, in neurodegenerative disease. Additionally, emerging evidence of more complex sleep in Drosophila warrants further investigation to determine whether new, more applicable sleep models can be designed for studying human sleep.
Loss of sleep or sleep deprivation has many physiological and behavioural consequences such as neuronal oxidative stress [35,36,37], endoplasmic reticulum stress, mitochondrial dysfunction [38] and amyloid beta and tau accumulation [39-42,38], chromatolysis and vacuolization [43], abnormal spine homeostasis [44], disrupted synaptic AMPA channel [45] function neuronal loss [46] and impaired learning [44,47]. Discussion of all of these is beyond the scope of this review. The theme of autophagy is embedded throughout this article without dedicating a large section to describing autophagy on its own.
Autophagy and Sleep in Neurodegenerative Disease
While we do not intend to give an in-depth review of autophagy (for this, see Boland et al., 2018), a comprehensive knowledge of autophagy and its role in cellular homeostasis is important in understanding the scope of this review. Autophagy is a multistage process that involves the identification of undesirable substrates in the cell and their degradation by lysosomal enzymes in the multistage process [48]. Each of these stages depends on activation of several already identified genes. Mutations in these genes have been found in neurodegenerative disease patients [49]. The common hallmarks of autophagy disruption include oxidative stress, accumulation of amyloid beta, alpha-synuclein and mitochondrial stress [48,50]. While the contribution of autophagy defects to neurodegenerative disease has been long known, recent studies have suggested the role of sleep both as a cause and effect of autophagy disruption [51,52]. It is likely that autophagy sits at an interface between sleep and neurodegeneration where sleep and autophagy could be connected through a feedback loop (Figure 1). The idea of a tripartite model of sleep, neurodegeneration and autophagy has not been well explored and given the importance of this model in understanding the mechanisms of neurodegeneration it warrants further investigation.
Sleep and Oxidative Stress
Recent evidence has supported the idea that ROS accumulation may be a fundamental mechanism of sleep deprivation-related death. In both Drosophila melanogaster and mice, sleep loss has been shown to increase ROS levels in the gut which contribute to premature death [53]. However, both exogenous and transgenic antioxidants can rescue animals from sleep deprivation and allow them to live normal lifespans with little to no sleep [54]. While the rescue of ROS-damaged tissue in the gut may not be transferable to the CNS due to the gut’s high regenerative capacity compared to neural tissue [54-57] this does indicate a possible candidate for the physiological purpose of sleep. Moreover, it implicates sleep as a fundamental process in oxidative resistance. This notion of sleep as a promoter of oxidative resistance is supported by the contribution sleep makes to two key strategies cells employ to combat oxidative stress: antioxidant production and autophagy.
The central nervous system is at particular risk of oxidative stress due to the lower expression of antioxidants and their heavy dependence on polyunsaturated fatty acids (e.g. arachidonic acid) and catecholamines whose degradation release abundant free radicals (Figure 1) [58]. Furthermore, the structural and high energy demand leads to relatively higher levels of lipid demand in the brain which leads to many other fatty acids finding their way into neural tissue. The degradation of these molecules has been shown to increase the production of ROS through a cyclic activation of NMDA receptors driving calcium influx into the neurons followed by activation of PKCs leading to proliferative peroxide production even after the reactant has degraded [59,60]. Other processes that trigger NMDA-PKC chain activation (e.g. traumatic brain injury) show a similar increased production of ROS which are thought to contribute to secondary neuronal damage following injury [36]. Different neuronal populations also appear to show different selective neuronal vulnerability to oxidative stress depending on several factors such as high intrinsic ROS production [61,62], differential requirements for ROS in cellular signaling [63] and astrocyte inflammatory response [64]. These differences may underlie localization of pathological processes in neurodegenerative diseases.
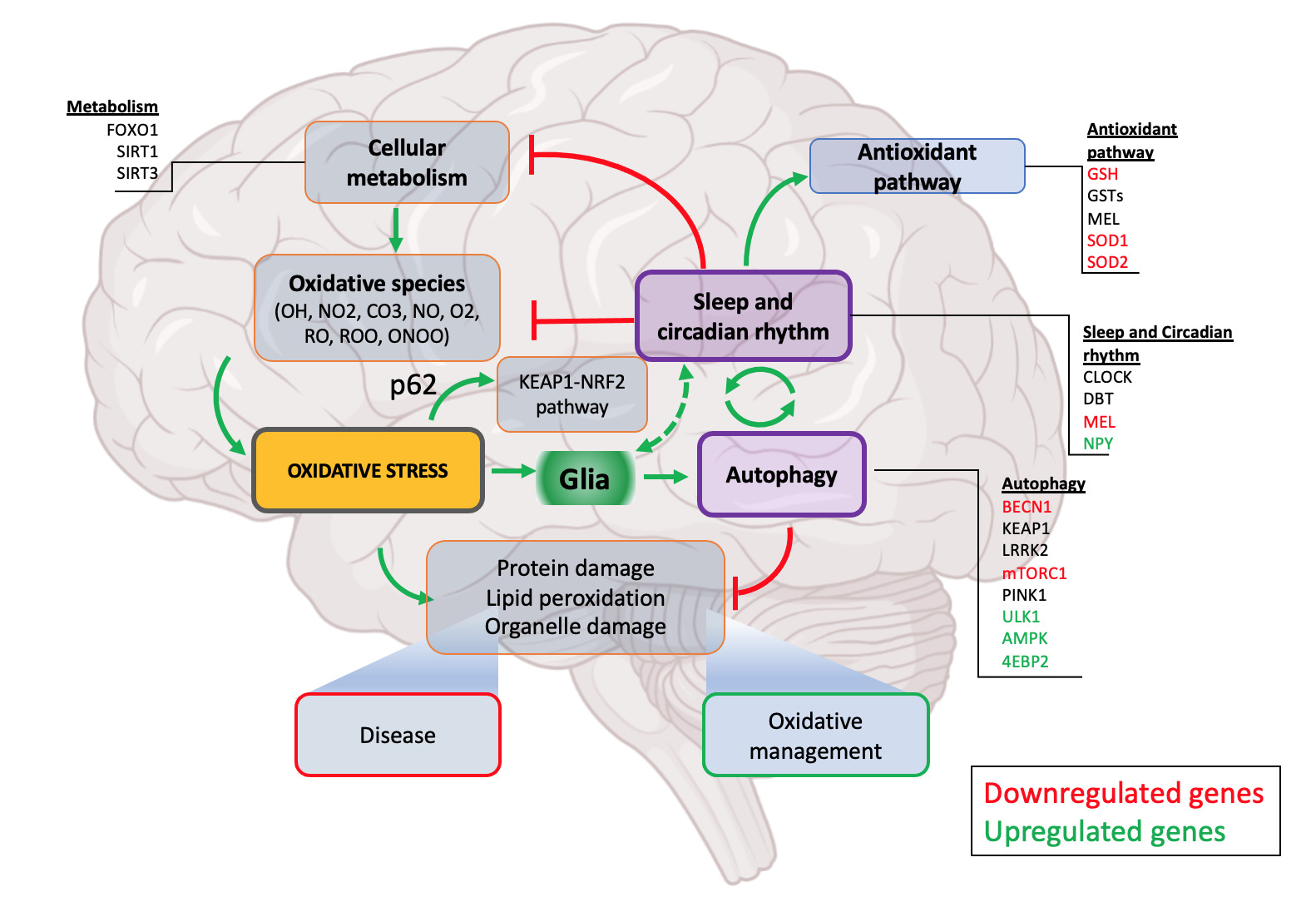
Figure 1. Sleep regulated autophagy. Sleep intervenes to alleviate oxidative stress and its effects at multiple levels. Firstly, in addition to sleep reducing overall metabolic rate, potential interactions exist between key sleep and metabolism genes. Secondly, several sleep-promoting genes are also important in the function of key antioxidant pathway (SODs, GSTs, MEL). Sleep is also associated with autophagy highlighting one of the mechanisms through which sleep regulates oxidative stress. Through both sleep’s inhibitory actions on the oxidative pathway and promotion of the antioxidative pathway, it plays an important role in the maintenance of healthy cellular metabolism and response to both oxidative processes and the products of oxidative damage. Glia act as a moderator of neuronal activity by providing additional support for the removal of waste materials and debri produced by stressed neurons. Glial activation is known to be tightly associated with sleep both microglia and astrocyte regulating sleep pressure and intensity. Sleep promotes autophagy by yet poorly understood mechanisms preventing accumulation of proteinaceous and lipid waste thus. Disruption of autophagy or sleep deprivation both lead to defective oxidative stress management, accumulation of cellular waste eventually leading to disease.
Sleep and Cellular Antioxidants
Due to their high metabolic demand, neurons produce a variety of oxidants which, if not metabolized, can lead to intracellular damage. Neurons thus produce a variety of oxidant scavengers (Figure 1) to counter metabolize the oxidative species it produces. Melatonin (MT) is an important hormone produced by the pineal gland that helps regulate circadian rhythm and is widely prescribed to patients suffering from insomnia [65,66]. In mice, melatonin has been shown to have a specific role depending on which receptor is bound: MT1 receptor knockout leads to disrupted REM sleep while MT2 receptor knockout leads to NREM sleep disruption [67] suggesting a differential role of MT which perhaps can adapt to need for sleep. Melatonin has also been shown to promote sleep and reduce sleep latency in both humans and C. elegans through activation of the BK channel and reducing neurotransmitter release [68] showing that its function in sleep is highly conserved. Beyond being a sleep promoting factor, melatonin is a low molecular weight antioxidant that has been shown to be not just linked with sleep, but also autophagy. Cyclosporine-induced oxidative stress can raise levels of autophagy, but subsequent co-administration of melatonin with Cyclosporin suppressed autophagy through direct suppression of LC3-II expression and increased expression of catalase [69]. This evidence suggests melatonin acts as a “peacekeeping” molecule, reducing ROS and inhibiting autophagy at the same time during asleep. In sleep deprived rats, decreased levels of superoxide dismutase in the liver were accompanied by decreased expression of autophagy receptor p62 and increased expression of LC3-II and Beclin-1, the latter two signaling an upregulation of autophagy [37], suggesting that increased autophagy may compensate for reduced antioxidant expression. A decrease in the expression of melatonin receptor 1 (MT1) in old age and AD brains when oxidative stress is higher and autophagy levels are lower further suggests a link between sleep and autophagy through MT receptor pathway [70-73]. Also, in preclinical cases of dementia, a decrease in melatonin release at night time has been reported with accompanying daytime sleepiness [71,74,75]. Administration of melatonin provides neuroprotection in genetic models of HD, suggesting perhaps a central position of melatonin in neurodegenerative disease [76]. A recent study has found that melatonin directly inhibits mTOR, which is accompanied by increased levels of phosphorylated AMPK (p-AMPK) and ULK1, promoting autophagy [77]. Sleep has similar effects on the levels of p-AMPK which then suppresses TORC1 complex and phosphorylation of 4EBP2 consequently influencing transcription of the 4EBP2 target genes [78]. This dual action of melatonin to inhibit autophagy in some contexts but promote it in others is curious as typically oxidant production signals an increase in p62-mediated autophagy through positive feedback from the KEAP1-NRF2 system to remove faulty organelles [79].
Superoxide dismutase (SOD) and glutathione S-transferases (GSTs) are fundamental in the regulation of ROS and antioxidative processes. Ubiquitous decrease in SOD1 expression has been shown to decrease lifespan and accelerate age-related locomotive impairment in Drosophila [80]. Furthermore, a bidirectional relationship between these antioxidant enzymes and sleep has been proposed: upregulation GSTs and SOD1 expression has been demonstrated to be both a result of oxidative stress as well as a mechanism for promoting sleep in Drosophila [35]. This relationship, which merits further study in mammals, highlights the intimate relationship between antioxidants and sleep.
Nicotinamide adenine dinucleotide (NAD+) is a critical molecule in cellular metabolism [81]. Its role as an important oxidizing agent and electron transporter in processes such as the citric acid cycle and electron transport chain are well understood, and it is therefore no surprise that NAD+ should be implicated in cellular pathology. NAD+ exists in its oxidized state NAD+ and its reduced state NADH, which it cycles between to deliver electrons from the TCA to mitochondrial membrane complexes I through IV to drive ATP synthesis. Decreased NAD+ levels as a result of age have been suggested as a critical underlying mechanism of pathological aging [82]. While there is extensive research into NAD+, due to its ubiquity in cells and in particular its involvement in mitochondrial function, it is especially difficult to identify NAD+ as being a molecule causative of pathology or whether its disruption is a result of other pathological processes acting within the cell. Despite this, there has been evidence that administration of nicotinamide mononucleotide (NMN), a precursor molecule to NAD+, is capable of producing beneficial effects on cells in vitro and in vivo: NMN injections in transgenic AD (tg-AD) mice has been found to reverse oxygen consumption rate deficiencies to levels of control and a significant decrease in full length amyloid beta. Interestingly, administration of NAD+ also reduced SIRT1 immunoreactivity in the brain of tg-AD mice [83] which, as we will discuss, is a candidate gene for linking autophagy and oxidative management. A possible mechanism for the beneficial impacts of NMN on cellular health in AD is that NMN increases mitochondrial NAD+ stores as well as decreases SIRT3-mediated protein deacetylation [46]. NMN has also been shown to improve spatial learning and contextual memory in tg-AD rats when compared to untreated controls through inhibition of c-Jun N-terminal kinase (JNK) proteins: inhibition of JNK, which is especially prevalent in the hippocampus and cerebral cortex, leads to increased cleavage of amyloid-β precursor protein (APP) by non-oligomeric α-secretase and decreased cleavage by oligomeric β-secretase, suggesting that JNK inhibition by NMN can promote cleavage of APP into non-pathological Aβ [46]. This evidence implicates the NMN-JNK interaction in preventing localized Aβ oligomers and the behavioral manifestations of AD, and thus the importance of NMN in AD.
While NAD+ has been studied extensively in the context of aging, links between NAD+ and sleep are less well covered in literature. There are still clues that point to further lines of research into the impact sleep may have on NAD+ activity. Firstly, intracellular nicotinamide phosphoribosyl transferase (iNAMPT) is an enzyme that converts nicotinamide into NMN through the NAD salvage pathway and is encoded by the Nampt gene, whose expression is mediated by the fundamental circadian transcription factors CLOCK and BMAL1 [84]. The inhibition of CLOCK/BMAL1 by cytokines in chronic inflammation is believed to have a role in age-related circadian disturbances [85] but this may demonstrate that circadian disturbances through the CLOCK/BMAL1 pathway end up disrupting the salvage of cellular NAD+ through inhibition of Nampt. Secondly, recent evidence has suggested that NMN and SIRT1 may together play a role in both sleep- and age-related NAD+ depletion. NMN administration differentially influences gene expression in the presence of SIRT1 with a large subset of genes not differentially regulated in SIRT1 knockout mice presumably due to loss of BMAL1 binding to chromatin indicating that NAD+ transcription operates through a SIRT1/BMAL1 pathway [86]. BMAL1 and CLOCK are inhibited by clock suppressing proteins cryptochrome (CRY) and period-2 (PER2), which are sequestered in the cytoplasm during day but at night are present in the nucleus, inhibiting CLOCK/BMAL1 expression. In Drosophila SIRT1 knockout led to increased PER2 in the nucleus during the day which was more stable in response to NAD+ depletion, as well as impacting PER2 phosphorylation and acetylation [87]. Conversely, pharmacological inhibition of SIRT1 deacetylase activity has been shown to increase the cytoplasmic localisation of PER2 thereby inhibiting the PER/BMAL1 promoter driven circadian oscillation [88]. This evidence together suggests that SIRT1 is important for maintenance of circadian rhythms and in modulating the impact of NMN administration by regulating expression of several genes. Furthermore, PER2 is known to be upregulated in elderly mice [89] and 6 months of NR supplementation has been shown to both dramatically reduce PER2 levels and to inhibit PER2 binding at BMAL1 [86] suggesting that PER2 levels may be both up- and down-regulated by NAD under the control of SIRT1. The impact of NAD and other reduced coenzymes on sleep (or vice versa) is still largely unexplored. Similarly, further studies are required to establish a link between NMN and SIRT1 in the regulation of mitochondrial health and oxidative stress linked with sleep deprivation.
SIRT1 is the focal member of the Sirtuin family of seven histone deacetylase proteins, responsible for the regulation of post-translation protein acetylation [90]. SIRT1 has been shown to have neuroprotective qualities which are reduced as its expression decreases with age, demonstrated through the reduction in neurotransmitter enzymes, dendrites, axons and increased accumulation of lipofuscin proteins [91]. However, the increase in protein aggregation in the absence of SIRT1 identifies it as a possibly important gene in oxidative management and autophagy. As previously discussed, SIRT1 is understood to be highly dependent on NAD+ in performing its functions. SIRT3 is also expressed in the mitochondria and plays an essential role in regulating ROS by deacetylating the enzymes SOD2, Cytochrome C, Complex I and II [92] with SIRT3 knockout mice shown to be immune to the calorie restriction benefits of antioxidative protection [93]. SIRT1 has been found to activate AMPK and inhibit mTOR [94] indicating it is a prime candidate for regulation of autophagy. Indeed, SIRT1 knockout leads to increased damage resulting from peroxide infusion through downregulation of Beclin-1 and LC3-I to LC3-II conversion [95]. SIRT1 upregulation through resveratrol was also shown to have a protective effect against fluoride-induced cell-stress through upregulation of ATGs [46,96].
Mitophagy is a particularly relevant form of autophagy in the context of oxidative stress, given the role of damaged and faulty mitochondria in ROS production [97]. The impact of SIRT1 on mitophagy however remains far less clear. SIRT1 knockout may promote mitophagy by promoting ROS production through increased SOD2 acetylation, resulting in increased Parkin2 recruitment to mitochondrial membranes in prostate cancer cells in mice [98]. Whether SIRT1 bears the same impact on mitophagy in the brain has yet to be shown.
Neuropeptide Y (NPY) is one of the most abundant peptides in the mammalian brain [99]. It has several important roles in maintaining homeostasis in both the CNS and PNS, however in the CNS it is especially important for regulating food intake [100] and fat storage, blood pressure [101] and circadian rhythm [102,103] as well as being a molecule of interest in epilepsy [104,105]. NPY is also highly conserved, with the C. elegans homolog NPR1 being essential in maintaining homeostasis in response to sleep deprivation. NPR1 seems to be specifically associated with minor sleep disturbances (such as light exposure or vibrations) whereby it extends the duration of individual sleep bouts and this role seems to be separate from the DAF-16/FOXO pathway (which compensates for major sleep disturbances (such as vigorous agitation) by extending the overall duration of lethargus) [106]. In Drosophila, NPY homolog NPF has been shown to impact sleep quality but not duration and to mitigate some of the consequences of sleep deprivation [107]. While such mechanisms in mammals have yet to be shown, there is some evidence that NPY may be involved in regulating the structure of sleep without necessarily affecting the overall duration. Lateral hypothalamus and intracerebroventricular infusions of NPY in mice were shown to suppress REM and NREM sleep with higher doses eliminating REM sleep and suppressing the number but not the duration of NREM episodes [108]. Together these studies show a divergence in the role of NPY: in some mammalian studies such as in rodents [109] and humans [110] it promotes sleep, but it has equally been shown to promote waking [108,111]. Indeed, it has been shown that NPY overexpression and injection promotes sleep in zebrafish by inhibiting noradrenergic wake-promoting pathways, suggesting that NPY’s sleep promoting function is localised to specific neuronal populations [112]. How exactly this data could map onto mammalian models has yet to be seen, but NPY’s impact in reducing the arousal threshold during sleep could represent a conserved mechanism of NPY linking its role in minor sleep disturbance in C. elegans to its sleep promoting actions.
NPY has been shown to have neuroprotective effects in both PD [113] and AD [114]. In PD, one possible mechanism is NPY’s ability to inhibit microglia-mediated inflammatory markers which are responsible for the majority of dopaminergic neuron loss in PD [115]. Neprilysin (NEP) is a metalloprotease whose levels in the brain inversely correlate with deposition of Aβ plaques AD [116,117]. As well as its role in degradation of Aβ, NEP is involved in cleaving NPY producing NPY C-terminal fragments which show a protective function against the neurotoxic effects of Aβ in a transgenic mouse model [114]. Interestingly, NPY is also an autophagy promoting factor in hypothalamic neurons in rodents through activation of NPY Y1 and Y5 receptors associated with activation of PI3K, MEK/ERK and PKA pathways. As such NPY could provide a useful target in determining the efficacy of autophagy-promoting sleep treatments and strategies [118].
The Glial System
The glial system is a support system which neurons heavily rely upon for protection against toxins and for providing the necessary neurotrophic support for maintenance. There are three main cell types in the glial system: Astrocytes, microglia, and oligodendrocytes [119]. Activation of astrocytes and microglia is an indication of neuronal stress and is reported to increase with age and in various neurological diseases [120]. Glia in particular plays an important role during development, synapse formation, pruning and maintenance [121,122]. Similarly, dysfunction of astrocytes and microglia has also been associated with various neurodegenerative diseases [123]. Recently, progress has been made in the identification of glial genes which have been associated with neurodegenerative disease, which is highlighting molecular pathways required for microglial function [124-126].
Glia plays a crucial role in maintaining the ionic balance and clearance of waste materials from the brain through the glymphatic system [127]. Astrocytic function is necessary for maintaining the fluid volume during sleep-wake cycles which are essential for metabolic waste clearance from the brain dependent on aquaporin pump (AQP4) [128,129]. Interestingly sleep deprivation has been suggested to regulate the clearance of waste materials including neurotoxic proteins Tau and Aβ (Figure 2) [130,41]. Interestingly, AQP4 knockout mice show a compromised clearance of Aβ [129] suggesting that it is indeed through this mechanism that Aβ is cleared. In humans and rodents, sleep loss has been shown to increase inflammation markers [131]. It is now known that sleep deprivation leads to the activation of astrocytes and glia in mouse models, a phenomenon shared with aging and neurodegenerative disease [132]. The relevance of astrocytes in disease is demonstrated by the fact that they actively take up misfolded alpha-synuclein secreted by neurons in the brain of PD patients acting like a ‘sponge’ protecting from secreted alpha-synuclein [133]. In normal homeostatic state they are a partner of the tripartite structure including pre- and post- synaptic terminals where they take up some of the glutamate secreting into the synaptic cleft which they change into reusable Glutamine [134]. It is suggested that microglia and astrocytes activation shift between two different stages. In the case of microglia, these stages are termed ‘M1’ and ‘M2’ and in M2 state microglia release a host of anti-inflammatory cytokines including BDNF, TGFβ and IGF-1 which help neurons deal with stress factors. The effect of sleep deprivation on this shift is yet unknown but is thought to be a potential treatment for neurodegenerative diseases [135-137]. In fact, mice lacking the M2 macrophages show an impairment in restorative slow wave sleep (SWS) after sleep deprivation [138]. On the other hand, proinflammatory signals such as bacterial infections shift macrophages towards the proinflammatory state M1 releasing proinflammatory cytokines such as IL-1β and TNFα and Nitric oxide (NO). These cytokines stimulate non-restorative NREM sleep indicating a relationship between sleep and proinflammatory response together [139,140]. Recently, sleep deprivation in rats was demonstrated to directly cause neuroinflammation [141]. Interestingly, this study also showed an increase in AQP4 in sleep deprived rats. Together these data suggest that normal Glial function is regulated by sleep and its function is important for neuronal homeostasis and removal of waste materials from the brain.
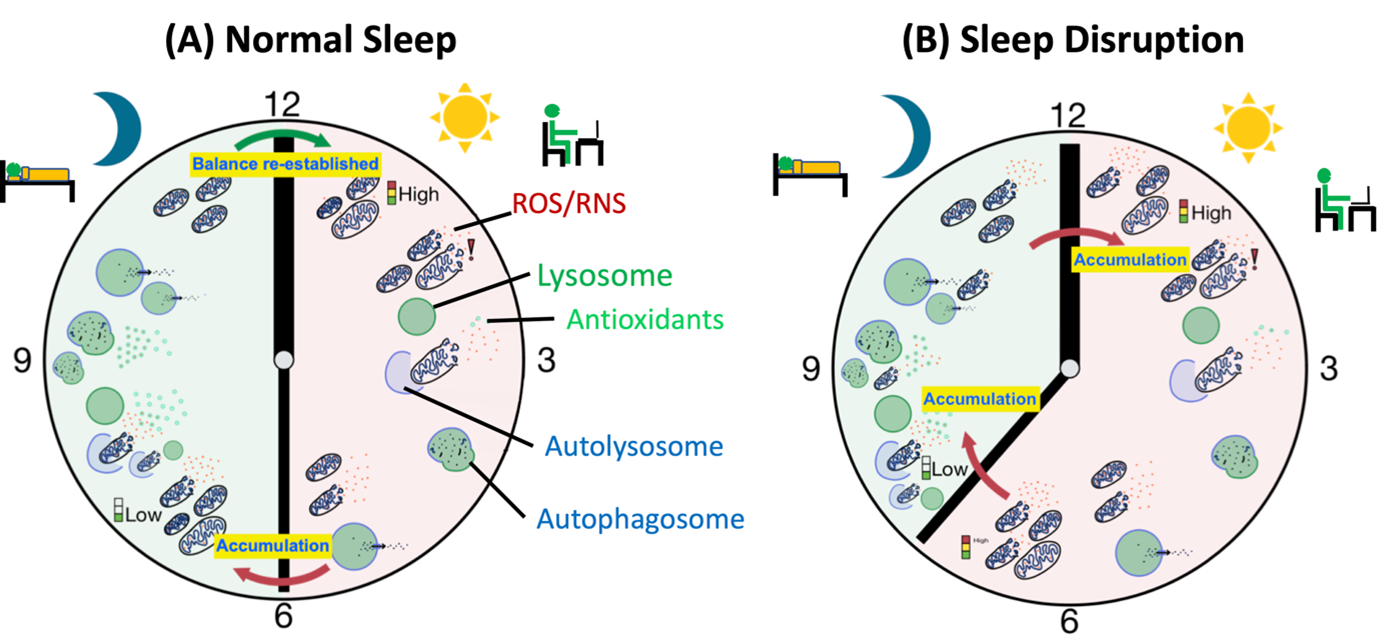
Figure 2. Sleep Disruption leads to metabolic waste build-up. (A) Metabolic activity during the day leads to accumulation of metabolic waste and the balance, between waste production and waste clearance, is tipped towards waste production leading to a build-up of metabolic waste. During the night when autophagy is higher waste clearance is high leading to cleanup of the cellular debris. (B) Sleep deprivation or loss of sleep tips the balance towards waste production and build-up which overpowers the autophagic waste clearance. Continuous imbalance in waste clearance leads to further accumulation of metabolic waste to toxic levels eventually leading to cell death.
Discussion
Sleep is one of those things that we all know we need to function at our prime, and yet we aren’t entirely sure why. It is often taken for granted in modern society where, through light/noise pollution, demanding work expectations or through general distractions, it can be tempting to push sleep aside despite an abundance of evidence citing the physiological, cognitive and behavioral effects of sleep deprivation as well as emerging evidence of an intimate relationship between sleep and neurodegenerative disease. Yet the biological mechanisms underlying these relationships are poorly understood. Here, we propose the idea that sleep plays a fundamental role as an evolutionarily conserved self-preservation mechanism of highly metabolically active neural tissue, inducing a pro-antioxidant and -autophagy environment to counteract the accumulation of cellular damage throughout waking. The abundance of mitochondria and charged molecules in neural tissue creates a hostile environment, vulnerable to oxidative damage, and disruptions to cellular maintenance processes (in part mediated by sleep) may underlie the profound effects of sleep disruption over time in humans and other organisms. Recent findings have shown that autophagosome production is decreased in sleep and that sustained upregulated autophagosome levels increase sleep duration [52]. While this hints at a regulatory role of autophagy in sleep it is uncertain whether this reduction of autophagosomes in sleep is, rather than being paradoxical, through the procession of autophagy to clear cellular debris. This highlights the need for further research in the genetic relationship between autophagy and sleep. Of particular interest are the SIRT family of genes. SIRTs are some of several exciting genes in aging research, with current evidence implicating them in mediating the longevity-promoting effects of calorie restrictive diets [142,143] though the exact contributions of SIRTs are still unknown. The relationship between SIRTs and sleep has been discussed above, however, the links between SIRTs and autophagy are not clear. For instance, the impact of SIRT1 on autophagy-mediated clearance of cellular debris is yet to be established. NPY, yet another protein implicated in longevity-promoting effects of calorie restriction [144] requires further investigation to determine its role in autophagy.
Sleep plays an important role in regulating various homeostatic functions of the brain which deteriorate with age and in neurodegenerative disease partly due to decreases in the expression of Melatonin. The role of sleep in regulating autophagy is of particular interest to neurodegenerative disease because of its potential to be used as an intervention strategy to treat neurodegenerative disease. Because of a direct relationship of sleep with autophagy it is important to consider sleep together with autophagy in this picture. Although melatonin is already in clinical use to treat various sleep disruption conditions, its impact on neurodegenerative disease has not yet been exploited. Furthermore, finding alternative targets and ways to boost autophagy as an intervention mechanism needs to be exploited.
Establishing a clearer picture of the links between metabolism, age-related genes (such as SIRTs) and sleep has strong implications for society. Sleep disruption is seen as part of natural aging [145]. Approximately 40-70% of older adults experience some degree of chronic sleep problems [146]. In the context of an aging population where neurodegenerative diseases are more common, such as in the Western world, a better understanding of mechanisms of sleep and finding possible intervention strategies can be immensely useful. Sleep disruption is not however an issue specific only to the elderly, with increasing numbers of younger individuals reporting chronic sleep disturbances especially relating to distractions such as smartphone use at night [147]. While younger people may feel immune to the effects of sleep deprivation, there is evidence that even in otherwise healthy young adults sleep deprivation leads to a significant increase in Aβ40 and reduction in Aβ42/40 ratio as a result of increased oxidative stress and reduced cellular clearance [42]. Whether this may impact long-term risk of AD or AD epidemiology in the future is yet to be seen, however some known impacts of sleep deprivation in young people (such as increased risk of depression) have been linked to significantly higher Aβ40 and lower Aβ42/40 ratios [148] suggesting that young people may not be immune from short-term pathological impacts of protein aggregation. It should be noted that this evidence does not imply a directional relationship and further research is needed into the implications of oxidized protein oligomerization in young people.
Recently, a direct relationship between sleep, autophagy and obesity was shown. Obesity is increasing in prevalence across both the developed and developing world and has been identified as a significant risk factor for neurodegenerative diseases such as AD and PD [149-151]. While obesity alters cellular metabolic health in a broad spectrum of ways, pathways of particular note are firstly the influence that a sustained positive energy balance induced by overnutrition has on promoting anabolic mTORC1 activity and inhibiting catabolism by autophagy [152]. Secondly, either independently or as a result of downregulation of autophagy, obesity results in increased ROS production which can then contribute to oxidative stress [153]. Sleep and obesity are directly linked, with obesity being associated with poorer sleep quality across age groups [154,155]. It thus appears that obesity and sleep may operate through similar mechanisms to increase neurodegenerative disease risk, providing possible biomolecular interaction between the two. It is therefore important to consider the possible interactions between both increasing chronic sleep disruption and obesity on neurodegenerative disease in the future.
Acknowledgements
The authors would like to thank Daniel Maddison, Saurabh Chaudhary for reviewing the manuscript. We would also like to thank Wellcome Trust for financial support to BM.
Author contribution
BM conceptualized the idea and both BM and SD wrote the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
References
2. Besedovsky L, Lange T, Born J. Sleep and immune function. Pflügers Archiv-European Journal of Physiology. 2012 Jan;463(1):121-37.
3. Broussard JL, Ehrmann DA, Van Cauter E, Tasali E, Brady MJ. Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Annals of Internal Medicine. 2012 Oct 16;157(8):549-57.
4. Cappuccio FP, Cooper D, D'Elia L, Strazzullo P, Miller MA. Sleep duration predicts cardiovascular outcomes: a systematic review and meta-analysis of prospective studies. European Heart Journal. 2011 Jun 1;32(12):1484-92.
5. Garbarino S, Lanteri P, Bragazzi NL, Magnavita N, Scoditti E. Role of sleep deprivation in immune-related disease risk and outcomes. Communications Biology. 2021 Nov 18;4(1):1-7.
6. Liu Y, Wheaton AG, Chapman DP, Croft JB. Sleep duration and chronic diseases among US adults age 45 years and older: evidence from the 2010 Behavioral Risk Factor Surveillance System. Sleep. 2013 Oct 1;36(10):1421-7.
7. Medic G, Wille M, Hemels ME. Short-and long-term health consequences of sleep disruption. Nature and Science of Sleep. 2017;9:151.
8. Lloret MA, Cervera-Ferri A, Nepomuceno M, Monllor P, Esteve D, Lloret A. Is sleep disruption a cause or consequence of Alzheimer's disease? Reviewing its possible role as a biomarker. International Journal of Molecular Sciences. 2020 Jan;21(3):1168.
9. McCarter SJ, St Louis EK, Boeve BF. Sleep disturbances in frontotemporal dementia. Current Neurology and Neuroscience Reports. 2016 Sep;16(9):1-8.
10. Rothman SM, Herdener N, Frankola KA, Mughal MR, Mattson MP. Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical A? and pTau in a mouse model of Alzheimer's disease. Brain Research. 2013 Sep 5;1529:200-8.
11. de Manacéïne M. Quelques observations experimentales sur l'influence de l'insomnie absolue. Arch Ital. Biol.. 1894;21:322-5.
12. Daddi L. Sulle alterazioni degli elementi del sistema nervoso centrale nell'insonnia sperimentale. Rivista di Patologia Nervosa e Mentale. 1898;3:1-2.
13. Lim AS, Ellison BA, Wang JL, Yu L, Schneider JA, Buchman AS, et al. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer's disease. Brain. 2014 Oct 1;137(10):2847-61.
14. Campbell SS, Tobler I. Animal sleep: a review of sleep duration across phylogeny. Neuroscience & Biobehavioral Reviews. 1984 Sep 1;8(3):269-300.
15. Cirelli C. The genetic and molecular regulation of sleep: from fruit flies to humans. Nature Reviews Neuroscience. 2009 Aug;10(8):549-60.
16. Cirelli C, Bushey D. Sleep and wakefulness in Drosophila melanogaster. Annals of the New York Academy of Sciences. 2008 May;1129(1):323-9.
17. Nath RD, Chow ES, Wang H, Schwarz EM, Sternberg PW. C. elegans stress-induced sleep emerges from the collective action of multiple neuropeptides. Current Biology. 2016 Sep 26;26(18):2446-5.
18. Nath RD, Bedbrook CN, Abrams MJ, Basinger T, Bois JS, Prober DA, et al. The jellyfish Cassiopea exhibits a sleep-like state. Current Biology. 2017 Oct 9;27(19):2984-90.
19. Bushey D, Hughes KA, Tononi G, Cirelli C. Sleep, aging, and lifespan in Drosophila. BMC Neuroscience. 2010 Dec;11(1):1-8.
20. Bringmann H. Sleep-active neurons: conserved motors of sleep. Genetics. 2018 Apr 1;208(4):1279-89.
21. Everson CA, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat: III. Total sleep deprivation. Sleep. 1989 Jan 1;12(1):13-21.
22. Rechtschaffen A, Bergmann BM, Everson CA, Kushida CA, Carol AE. Sleep deprivation in the rat: I. Conceptual issues. Sleep. 1989 Jan 1;12(1):1-4.
23. Hill AJ, Mansfield R, Lopez JM, Raizen DM, Van Buskirk C. Cellular stress induces a protective sleep-like state in C. elegans. Current Biology. 2014 Oct 20;24(20):2399-405.
24. Bennett HL, Khoruzhik Y, Hayden D, Huang H, Sanders J, Walsh MB, et al. Normal sleep bouts are not essential for C. elegans survival and FoxO is important for compensatory changes in sleep. BMC Neuroscience. 2018 Dec;19(1):1-23.
25. Geissmann Q, Beckwith EJ, Gilestro GF. Most sleep does not serve a vital function: Evidence from Drosophila melanogaster. Science Advances. 2019 Feb 20;5(2):eaau9253.
26. Gronfier C, Simon C, Piquard F, Ehrhart J, Brandenberger G. Neuroendocrine processes underlying ultradian sleep regulation in man. The Journal of Clinical Endocrinology & Metabolism. 1999 Aug 1;84(8):2686-90.
27. Ciric J, Kapor S, Perovic M, Saponjic J. Alterations of sleep and sleep oscillations in the hemiparkinsonian rat. Frontiers in Neuroscience. 2019 Feb 25;13:148.
28. Andretic R, Shaw PJ. Essentials of sleep recordings in Drosophila: moving beyond sleep time. InMethods in Enzymology 2005 Jan 1 (Vol. 393, pp. 759-772). Academic Press.
29. Huber R, Hill SL, Holladay C, Biesiadecki M, Tononi G, Cirelli C. Sleep homeostasis in Drosophila melanogaster. Sleep. 2004 Jun 1;27(4):628-39.
30. Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. Correlates of sleep and waking in Drosophila melanogaster. Science. 2000 Mar 10;287(5459):1834-7.
31. Tainton-Heap LA, Kirszenblat LC, Notaras ET, Grabowska MJ, Jeans R, Feng K, et al. A paradoxical kind of sleep in Drosophila melanogaster. Current Biology. 2021 Feb 8;31(3):578-90.
32. Van Alphen B, Semenza ER, Yap M, Van Swinderen B, Allada R. A deep sleep stage in Drosophila with a functional role in waste clearance. Science Advances. 2021 Jan 20;7(4):eabc2999.
33. Fadda M, De Fruyt N, Borghgraef C, Watteyne J, Peymen K, Vandewyer E, et al. NPY/NPF-related neuropeptide FLP-34 signals from serotonergic neurons to modulate aversive olfactory learning in Caenorhabditis elegans. Journal of Neuroscience. 2020 Jul 29;40(31):6018-34.
34. Keating CD, Kriek N, Daniels M, Ashcroft NR, Hopper NA, Siney EJ, et al. Whole-genome analysis of 60 G protein-coupled receptors in Caenorhabditis elegans by gene knockout with RNAi. Current Biology. 2003 Sep 30;13(19):1715-20.
35. Hill VM, O’Connor RM, Sissoko GB, Irobunda IS, Leong S, Canman JC, et al. A bidirectional relationship between sleep and oxidative stress in Drosophila. PLoS Biology. 2018 Jul 12;16(7):e2005206.
36. Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK. Oxidative stress: major threat in traumatic brain injury. CNS & Neurological Disorders-Drug Targets (Formerly Current Drug Targets-CNS & Neurological Disorders). 2018 Nov 1;17(9):689-95.
37. Li Y, Zhang Y, Ji G, Shen Y, Zhao N, Liang Y, et al. Autophagy triggered by oxidative stress appears to be mediated by the AKT/mTOR signaling pathway in the liver of sleep-deprived rats. Oxidative Medicine and Cellular Longevity. 2020 Feb 13;2020.
38. Zhao H, Wu H, He J, Zhuang J, Liu Z, Yang Y, et al. Frontal cortical mitochondrial dysfunction and mitochondria-related ?-amyloid accumulation by chronic sleep restriction in mice. Neuroreport. 2016 Aug 17;27(12):916.
39. Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, et al. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. Journal of Neuroscience. 2013 Jan 23;33(4):1651-9.
40. Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, et al. Amyloid-? dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009 Nov 13;326(5955):1005-7.
41. Shokri-Kojori E, Wang GJ, Wiers CE, Demiral SB, Guo M, Kim SW, et al. ?-Amyloid accumulation in the human brain after one night of sleep deprivation. Proceedings of the National Academy of Sciences. 2018 Apr 24;115(17):4483-8.
42. Wei M, Zhao B, Huo K, Deng Y, Shang S, Liu J, et al. Sleep deprivation induced plasma amyloid-? transport disturbance in healthy young adults. Journal of Alzheimer's Disease. 2017 Jan 1;57(3):899-906.
43. Abushov BM. Morphofunctional analysis of the effects of total sleep deprivation on the CNS in rats. Neuroscience and Behavioral Physiology. 2010 May;40(4):403-9.
44. Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife. 2016 Aug 23;5:e13424.
45. Lanté F, Toledo-Salas JC, Ondrejcak T, Rowan MJ, Ulrich D. Removal of synaptic Ca2+-permeable AMPA receptors during sleep. Journal of Neuroscience. 2011 Mar 16;31(11):3953-61.
46. Yao Z, Yang W, Gao Z, Jia P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neuroscience Letters. 2017 Apr 24;647:133-40.
47. Yang G, Lai CS, Cichon J, Ma L, Li W, Gan WB. Sleep promotes branch-specific formation of dendritic spines after learning. Science. 2014 Jun 6;344(6188):1173-8.
48. Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nature reviews Drug discovery. 2018 Sep;17(9):660-88.
49. Malik BR, Maddison DC, Smith GA, Peters OM. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Molecular Brain. 2019 Dec;12(1):1-21.
50. Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochemical Journal. 2012 Jan 15;441(2):523-40.
51. Bedont JL, Toda H, Shi M, Park CH, Quake C, Stein C, et al. Short and long sleeping mutants reveal links between sleep and macroautophagy. Elife. 2021 Jun 4;10:e64140.
52. Kijak E, Pyza E. TOR signaling pathway and autophagy are involved in the regulation of circadian rhythms in behavior and plasticity of L2 interneurons in the brain of Drosophila melanogaster. PLoS One. 2017 Feb 14;12(2):e0171848.
53. Hindson J. Sleep loss lethality is caused by gut ROS in mice and flies. Nature Reviews Gastroenterology & Hepatology. 2020 Aug;17(8):452-.
54. Vaccaro A, Dor YK, Nambara K, Pollina EA, Lin C, Greenberg ME, et al. Sleep loss can cause death through accumulation of reactive oxygen species in the gut. Cell. 2020 Jun 11;181(6):1307-28.
55. Amcheslavsky A, Jiang J, Ip YT. Tissue damage-induced intestinal stem cell division in Drosophila. Cell Stem Cell. 2009 Jan 9;4(1):49-61.
56. Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell. 2009 Jun 26;137(7):1343-55.
57. Tsujioka H, Yamashita T. Neural circuit repair after central nervous system injury. International Immunology. 2021 Jun;33(6):301-9.
58. Singh E, Devasahayam G. Neurodegeneration by oxidative stress: a review on prospective use of small molecules for neuroprotection. Molecular Biology Reports. 2020 Apr;47(4):3133-40.
59. Alves JL, Figueira AS, Souto M, Lopes IL, Dionísio JC, Quinta-Ferreira RM, et al. Oleic acid enhances the production of reactive oxygen species in neuronal tissue. Energy Reports. 2020 Feb 1;6:885-90.
60. Murakami K, Routtenberg A. Direct activation of purified protein kinase C by unsaturated fatty acids (oleate and arachidonate) in the absence of phospholipids and Ca2+. FEBS Letters. 1985 Nov 18;192(2):189-93.
61. Mattiasson G, Friberg H, Hansson M, Elmér E, Wieloch T. Flow cytometric analysis of mitochondria from CA1 and CA3 regions of rat hippocampus reveals differences in permeability transition pore activation. Journal of Neurochemistry. 2003 Oct;87(2):532-44.
62. Wang X, Pal R, Chen XW, Limpeanchob N, Kumar KN, Michaelis EK. High intrinsic oxidative stress may underlie selective vulnerability of the hippocampal CA1 region. Molecular Brain Research. 2005 Oct 31;140(1-2):120-6.
63. Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, et al. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. Journal of Neuroscience. 2000 Oct 15;20(20):7631-9.
64. Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. Journal of Neuroscience. 2007 Apr 18;27(16):4253-60.
65. Auld F, Maschauer EL, Morrison I, Skene DJ, Riha RL. Evidence for the efficacy of melatonin in the treatment of primary adult sleep disorders. Sleep Medicine Reviews. 2017 Aug 1;34:10-22.
66. Ferracioli-Oda E, Qawasmi A, Bloch MH. Meta-analysis: melatonin for the treatment of primary sleep disorders. PloS One. 2013 May 17;8(5):e63773.
67. Gobbi G, Comai S. Differential function of melatonin MT1 and MT2 receptors in REM and NREM sleep. Frontiers in Endocrinology. 2019:87.
68. Niu L, Li Y, Zong P, Liu P, Shui Y, Chen B, et al. Melatonin promotes sleep by activating the BK channel in C. elegans. Proceedings of the National Academy of Sciences. 2020 Oct 6;117(40):25128-37.
69. Yoo YM, Jeung EB. Melatonin suppresses cyclosporine A?induced autophagy in rat pituitary GH3 cells. Journal of Pineal Research. 2010 Apr;48(3):204-11.
70. Wu YH, Zhou JN, Van Heerikhuize J, Jockers R, Swaab DF. Decreased MT1 melatonin receptor expression in the suprachiasmatic nucleus in aging and Alzheimer's disease. Neurobiology of Aging. 2007 Aug 1;28(8):1239-47.
71. Wu YH, Feenstra MG, Zhou JN, Liu RY, Torano? JS, Van Kan HJ, et al. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. The Journal of Clinical Endocrinology & Metabolism. 2003 Dec 1;88(12):5898-906.
72. Ohashi Y, Okamoto N, Uchida K, Iyo M, Mori N, Morita Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer's type. Biological psychiatry. 1999 Jun 15;45(12):1646-52.
73. Uchida K, Okamoto N, Ohara K, Morita Y. Daily rhythm of serum melatonin in patients with dementia of the degenerate type. Brain Research. 1996 Apr 22;717(1-2):154-9.
74. Videnovic A, Noble C, Reid KJ, Peng J, Turek FW, Marconi A, et al. Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurology. 2014 Apr 1;71(4):463-9.
75. Waller KL, Mortensen EL, Avlund K, Fagerlund B, Lauritzen M, Gammeltoft S, et al. Melatonin and cortisol profiles in late midlife and their association with age-related changes in cognition. Nature and Science of Sleep. 2016;8:47.
76. Wang X, Sirianni A, Pei Z, Cormier K, Smith K, Jiang J, et al. The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. Journal of Neuroscience. 2011 Oct 12;31(41):14496-507.
77. Chen WR, Yang JQ, Liu F, Shen XQ, Zhou YJ. Melatonin attenuates vascular calcification by activating autophagy via an AMPK/mTOR/ULK1 signaling pathway. Experimental Cell Research. 2020 Apr 1;389(1):111883.
78. Tudor JC, Davis EJ, Peixoto L, Wimmer ME, van Tilborg E, Park AJ, et al. Sleep deprivation impairs memory by attenuating mTORC1-dependent protein synthesis. Science Signaling. 2016 Apr 26;9(425):ra41-.
79. Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. Journal of Biological Chemistry. 2010 Jul 16;285(29):22576-91.
80. Martin I, Jones MA, Grotewiel M. Manipulation of Sod1 expression ubiquitously, but not in the nervous system or muscle, impacts age-related parameters in Drosophila. FEBS Letters. 2009 Jul 7;583(13):2308-14.
81. Garten A, Schuster S, Penke M, Gorski T, De Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nature Reviews Endocrinology. 2015 Sep;11(9):535-46.
82. Zhang M, Ying W. NAD+ deficiency is a common central pathological factor of a number of diseases and aging: mechanisms and therapeutic implications. Antioxidants & Redox Signaling. 2019 Feb 1;30(6):890-905.
83. Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, Schuh RA. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer's disease-relevant murine model. BMC Neurology. 2015 Dec;15(1):1-4.
84. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014 Aug;24(8):464-71.
85. Hood S, Amir S. Neurodegeneration and the circadian clock. Front Aging Neurosci 9: 170.
86. Levine DC, Hong H, Weidemann BJ, Ramsey KM, Affinati AH, Schmidt MS, et al. NAD+ controls circadian reprogramming through PER2 nuclear translocation to counter aging. Molecular cell. 2020 Jun 4;78(5):835-49.
87. Ye R, Selby CP, Chiou YY, Ozkan-Dagliyan I, Gaddameedhi S, Sancar A. Dual modes of CLOCK: BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes & Development. 2014 Sep 15;28(18):1989-98.
88. Ashimori A, Nakahata Y, Sato T, Fukamizu Y, Matsui T, Yoshitane H, et al. Attenuated SIRT1 activity leads to PER2 cytoplasmic localization and dampens the amplitude of bmal1 promoter-driven circadian oscillation. Frontiers in Neuroscience. 2021;15.
89. Wang RH, Zhao T, Cui K, Hu G, Chen Q, Chen W, et al. Negative reciprocal regulation between Sirt1 and Per2 modulates the circadian clock and aging. Scientific Reports. 2016 Jun 27;6(1):1-5.
90. Verdin E, Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nature reviews Molecular Cell Biology. 2015 Apr;16(4):258-64.
91. Panossian L, Fenik P, Zhu Y, Zhan G, McBurney MW, Veasey S. SIRT1 regulation of wakefulness and senescence-like phenotype in wake neurons. Journal of Neuroscience. 2011 Mar 16;31(11):4025-36.
92. Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Molecular cell. 2011 Jun 10;42(5):561-8.
93. Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010 Nov 24;143(5):802-12.
94. Tang BL. Sirt1 and the mitochondria. Molecules and cells. 2016 Feb 29;39(2):87.
95. Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proceedings of the National Academy of Sciences. 2008 Mar 4;105(9):3374-9.
96. Yun JM, Chien A, Jialal I, Devaraj S. Resveratrol up-regulates SIRT1 and inhibits cellular oxidative stress in the diabetic milieu: mechanistic insights. The Journal of Nutritional Biochemistry. 2012 Jul 1;23(7):699-705.
97. Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009 Jan 1;417(1):1-3.
98. Di Sante G, Pestell TG, Casimiro MC, Bisetto S, Powell MJ, Lisanti MP, et al. Loss of Sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays PARK2 translocation to mitochondria. The American Journal of Pathology. 2015 Jan 1;185(1):266-79.
99. Heilig M, Widerlöv E. Neurobiology and clinical aspects of neuropeptide Y. Critical Reviews in Neurobiology. 1995 Jan 1;9(2-3):115-36.
100. Mercer RE, Chee MJ, Colmers WF. The role of NPY in hypothalamic mediated food intake. Frontiers in Neuroendocrinology. 2011 Oct 1;32(4):398-415.
101. Dahltf C, Dahltf P, Lundberg JM. Enhancement of blood pressure increase upon alpha-adrenoceptor activation and direct pressor effects in pithed rats. Eur. J. Pharmacol. 1985;109:289-92.
102. Soscia SJ, Harrington ME. Neuropeptide Y does not reset the circadian clock in NPY Y2?/? mice. Neuroscience letters. 2005 Jan 20;373(3):175-8.
103. Biello SM, Janik D, Mrosovsky N. Neuropeptide Y and behaviorally induced phase shifts. Neuroscience. 1994 Sep 1;62(1):273-9.
104. Cattaneo S, Verlengia G, Marino P, Simonato M, Bettegazzi B. NPY and gene therapy for epilepsy: how, when,... and Y. Frontiers in Molecular Neuroscience. 2021 Jan 22;13:261.
105. Colmers WF, Bahh BE. Neuropeptide Y and epilepsy. Epilepsy Currents. 2003 Mar;3(2):53-8.
106. Nagy S, Tramm N, Sanders J, Iwanir S, Shirley IA, Levine E, et al. Homeostasis in C. elegans sleep is characterized by two behaviorally and genetically distinct mechanisms. Elife. 2014 Dec 4;3:e04380.
107. He C, Yang Y, Zhang M, Price JL, Zhao Z. Regulation of sleep by neuropeptide Y-like system in Drosophila melanogaster. PLoS One. 2013 Sep 11;8(9):e74237.
108. Szentirmai E, Krueger JM. Central administration of neuropeptide Y induces wakefulness in rats. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2006 Aug;291(2):R473-80.
109. Akanmu MA, Ukponmwan OE, Katayama Y, Honda K. Neuropeptide-Y Y2-receptor agonist, PYY3–36 promotes non-rapid eye movement sleep in rat. Neuroscience Research. 2006 Mar 1;54(3):165-70.
110. Antonijevic IA, Murck H, Bohlhalter S, Frieboes RM, Holsboer F, Steiger A. Neuropeptide Y promotes sleep and inhibits ACTH and cortisol release in young men. Neuropharmacology. 2000 Jul 1;39(8):1474-81.
111. Ushimura A, Tsuji T, Tanaka S, Kogo M, Yamamoto T. Neuropeptide-Y modulates eating patterns and masticatory muscle activity in rats. Behavioural Brain Research. 2015 Feb 1;278:520-6.
112. Singh C, Rihel J, Prober DA. Neuropeptide Y regulates sleep by modulating noradrenergic signaling. Current Biology. 2017 Dec 18;27(24):3796-811.
113. Decressac M, Pain S, Chabeauti PY, Frangeul L, Thiriet N, Herzog H, et al. Neuroprotection by neuropeptide Y in cell and animal models of Parkinson's disease. Neurobiology of Aging. 2012 Sep 1;33(9):2125-37.
114. Rose JB, Crews L, Rockenstein E, Adame A, Mante M, Hersh LB, et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer's disease. Journal of Neuroscience. 2009 Jan 28;29(4):1115-25.
115. Pain S, Vergote J, Gulhan Z, Bodard S, Chalon S, Gaillard A. Inflammatory process in Parkinson disease: neuroprotection by neuropeptide Y. Fundamental & Clinical Pharmacology. 2019 Oct;33(5):544-8.
116. Madani R, Poirier R, Wolfer DP, Welzl H, Groscurth P, Lipp HP, et al. Lack of neprilysin suffices to generate murine amyloid?like deposits in the brain and behavioral deficit in vivo. Journal of Neuroscience Research. 2006 Dec;84(8):1871-8.
117. Wang DS, Iwata N, Hama E, Saido TC, Dickson DW. Oxidized neprilysin in aging and Alzheimer's disease brains. Biochemical and Biophysical Research Communications. 2003 Oct 10;310(1):236-41.
118. Aveleira CA, Botelho M, Cavadas C. NPY/neuropeptide Y enhances autophagy in the hypothalamus: a mechanism to delay aging?. Autophagy. 2015 Aug 3;11(8):1431-3.
119. Jäkel S, Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Frontiers in Cellular Neuroscience. 2017 Feb 13;11:24.
120. Thameem Dheen S, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Current medicinal Chemistry. 2007 May 1;14(11):1189-97.
121. Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001 Jan 26;291(5504):657-61.
122. Ullian EM, Harris BT, Wu A, Chan JR, Barres BA. Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Molecular and Cellular Neuroscience. 2004 Feb 1;25(2):241-51.
123. Stevenson R, Samokhina E, Rossetti I, Morley JW, Buskila Y. Neuromodulation of glial function during neurodegeneration. Frontiers in Cellular Neuroscience. 2020:278.
124. Bonham LW, Sirkis DW, Yokoyama JS. The transcriptional landscape of microglial genes in aging and neurodegenerative disease. Frontiers in Immunology. 2019 Jun 4;10:1170.
125. Sims R, Van Der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nature Genetics. 2017 Sep;49(9):1373-84.
126. Gleichman AJ, Carmichael ST. Glia in neurodegeneration: drivers of disease or along for the ride?. Neurobiology of Disease. 2020 Aug 1;142:104957.
127. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013 Oct 18;342(6156):373-7.
128. Ding F, O'donnell J, Xu Q, Kang N, Goldman N, Nedergaard M. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science. 2016 Apr 29;352(6285):550-5.
129. Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid ?. Science Translational Medicine. 2012 Aug 15;4(147):147ra111-.
130. Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019 Feb 22;363(6429):880-4.
131. Everson CA. Clinical assessment of blood leukocytes, serum cytokines, and serum immunoglobulins as responses to sleep deprivation in laboratory rats. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2005 Oct;289(4):R1054-63.
132. Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, Cirelli C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. Journal of Neuroscience. 2017 May 24;37(21):5263-73.
133. Song YJ, Halliday GM, Holton JL, Lashley T, O'Sullivan SS, McCann H, et al. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. Journal of Neuropathology & Experimental Neurology. 2009 Oct 1;68(10):1073-83.
134. Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends in Neurosciences. 1999 May 1;22(5):208-15.
135. Park HJ, Oh SH, Kim HN, Jung YJ, Lee PH. Mesenchymal stem cells enhance ?-synuclein clearance via M2 microglia polarization in experimental and human parkinsonian disorder. Acta Neuropathologica. 2016 Nov;132(5):685-701.
136. Zhang L, Zhang J, You Z. Switching of the microglial activation phenotype is a possible treatment for depression disorder. Frontiers in Cellular Neuroscience. 2018:306.
137. Zhou T, Huang Z, Sun X, Zhu X, Zhou L, Li M, et al. Microglia polarization with M1/M2 phenotype changes in rd1 mouse model of retinal degeneration. Frontiers in Neuroanatomy. 2017 Sep 5;11:77.
138. Massie A, Boland E, Kapás L, Szentirmai É. Mice lacking alternatively activated (M2) macrophages show impairments in restorative sleep after sleep loss and in cold environment. Scientific Reports. 2018 Jun 5;8(1):1-1.
139. Kapas L, Krueger JM. Nitric oxide donors SIN-1 and SNAP promote nonrapid-eye-movement sleep in rats. Brain Research Bulletin. 1996 Jan 1;41(5):293-8.
140. Krueger JM, Walter JA, Dinarello CA, Wolff SM, Chedid L. Sleep-promoting effects of endogenous pyrogen (interleukin-1). American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 1984 Jun 1;246(6):R994-9.
141. Xing C, Zhou Y, Xu H, Ding M, Zhang Y, Hu M, et al. Sleep Deprivation Induces Neuroinflammation and Depressive-like Behaviors by Impairing the Regulation of Circadian Clock Genes Expression in Rats.
142. Mercken EM, Hu J, Krzysik?Walker S, Wei M, Li Y, McBurney MW, et al. SIRT 1 but not its increased expression is essential for lifespan extension in caloric?restricted mice. Aging Cell. 2014 Feb;13(1):193-6.
143. Yu W, Qin J, Chen C, Fu Y, Wang W. Moderate calorie restriction attenuates age?associated alterations and improves cardiac function by increasing SIRT1 and SIRT3 expression. Molecular Medicine Reports. 2018 Oct 1;18(4):4087-94.
144. Komatsu T, Park S, Hayashi H, Mori R, Yamaza H, Shimokawa I. Mechanisms of calorie restriction: a review of genes required for the life-extending and tumor-inhibiting effects of calorie restriction. Nutrients. 2019 Dec;11(12):3068.
145. Mander BA, Winer JR, Walker MP. Sleep and human aging. Neuron. 2017 Apr 5;94(1):19-36.
146. Avidan A. Atlas of Clinical Sleep Medicine Chapter 4: Normal Sleep. Saunders Elsevier. 2010.
147. Lemola S, Perkinson-Gloor N, Brand S, Dewald-Kaufmann JF, Grob A. Adolescents' electronic media use at night, sleep disturbance, and depressive symptoms in the smartphone age. Journal of Youth and Adolescence. 2015 Feb;44(2):405-18.
148. Kita Y, Baba H, Maeshima H, Nakano Y, Suzuki T, Arai H. Serum amyloid ? protein in young and elderly depression: a pilot study. Psychogeriatrics. 2009 Dec;9(4):180-5.
149. Alamri Y, MacAskill M, Anderson T, Benamer H. Parkinson's disease in the Gulf countries: an updated review. European Neurology. 2015;74(3-4):222-5.
150. Procaccini C, Santopaolo M, Faicchia D, Colamatteo A, Formisano L, De Candia P, et al. Role of metabolism in neurodegenerative disorders. Metabolism. 2016 Sep 1;65(9):1376-90.
151. Uchoa MF, Moser VA, Pike CJ. Interactions between inflammation, sex steroids, and Alzheimer's disease risk factors. Frontiers in Neuroendocrinology. 2016 Oct 1;43:60-82.
152. Dann SG, Selvaraj A, Thomas G. mTOR Complex1–S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends in Molecular Medicine. 2007 Jun 1;13(6):252-9.
153. Pietrocola F, Pedro BS, Manuel J. Targeting autophagy to counteract obesity-associated oxidative stress. Antioxidants. 2021 Jan;10(1):102.
154. Morrissey B, Taveras E, Allender S, Strugnell C. Sleep and obesity among children: a systematic review of multiple sleep dimensions. Pediatric Obesity. 2020 Apr;15(4):e12619.
155. Ogilvie RP, Patel SR. The epidemiology of sleep and obesity. Sleep Health. 2017 Oct 1;3(5):383-8.