Abstract
Stroke is a debilitating neurologic condition characterized by an interruption or complete blockage of blood flow to certain areas of the brain. While the primary injury occurs at the time of the initial ischemic event or hemorrhage, secondary injury mechanisms contribute to neuroinflammation, disruption of the blood-brain barrier (BBB), excitotoxicity, and cerebral edema in the days and hours after stroke. Of these secondary mechanisms of injury, significant dysregulation of various immune populations within the body plays a crucial role in exacerbating brain damage after stroke. Pathological activity of glial cells, infiltrating leukocytes, and the adaptive immune system promote neuroinflammation, BBB damage, and neuronal death. Chronic immune activation can additionally encourage the development of neurologic deficits, immunosuppression, and dysregulation of the gut microbiome. As such, immunotherapy has emerged as a promising strategy for the clinical management of stroke in a highly patient-specific manner. These strategies include regulatory T cells (Tregs), cell adhesion molecules, cytokines, and monoclonal antibodies. However, the use of immunotherapy for stroke remains largely in the early stages, highlighting the need for continued research efforts before widespread clinical use.
Keywords
Ischemic stroke, Hemorrhagic stroke, Immunotherapy, Immunology, Immunocompromised, Chronic immune activation
Introduction
Stroke is characterized by an interruption or complete blockage of blood flow to certain areas of the brain and is a significant cause of morbidity and mortality worldwide. Stroke can broadly be classified as ischemic, where an infarction of the brain or spinal cord occurs, or hemorrhagic, where a ruptured blood vessel causes bleeding within the brain. For both ischemic and hemorrhagic stroke, early medical intervention is critical for minimizing brain injury, preserving neurologic functioning, and improving patient outcomes overall. Ischemic stroke makes up approximately 71% of all stroke incidences [1] and the current standard of care therapy involves reperfusion via intravenous thrombolysis with tissue plasminogen activators. Likewise, the goal of acute hemorrhagic stroke management involves the prevention of additional intracranial bleeding, and medical interventions may include surgical hematoma evacuation, placement of external ventricular drains, and blood pressure control. Following the initial acute injury, additional secondary signaling cascades induce neuroinflammation, disruption of the BBB, apoptosis, excitotoxicity, and cerebral edema. Each of these factors has the potential to induce and exacerbate neurologic deficits, highlighting the critical importance of their careful management by a diverse team of medical providers.
Traditionally, stroke research and clinical management have focused on vascular and neuronal factors contributing to brain damage and functional impairment. However, recent investigations have unveiled the pivotal role of the immune system in stroke pathogenesis and recovery. Neuronal death in the acute phase of stroke triggers robust innate and adaptive immune responses in the brain. Increased neuroinflammation secondary to immune infiltration and cytokine release has the ability to worsen patient outcomes following stroke. Chronic activation of the immune system in response to stroke can lead to immunosuppression, leave patients susceptible to life-threatening infections, promote dysbiosis of the gut microbiome, and encourage neurologic deficits. Immunocompromised patients are especially at an increased risk for poor outcomes following a stroke due to increased susceptibility to infections.
Given the significant role of the immune system in stroke pathology and clinical management, novel immunotherapeutic treatment strategies aim to promote recovery in a highly patient-specific manner. Examples of such strategies include Tregs, cell adhesion molecules, cytokines, and monoclonal antibodies. Although immunotherapy holds great promise for stroke patients, many trials remain largely in the preclinical phase highlighting the need for further research before integration into clinical practice. Here, we provide an overview of stroke pathophysiology and delve into local and systemic immune responses to stroke. We then explore the consequences of chronic immune activation following stroke, highlight several current and novel immunotherapeutic treatment strategies, and provide future research directions.
Overview of Stroke Pathophysiology
Strokes, otherwise known as cerebrovascular accidents, are clinically defined as vascular injuries in the central nervous system (CNS) that result in severe neurological deficits lasting longer than 24 hours [2,3]. These acute injuries are the second leading cause of death globally [2,3]. There are two major categories of stroke, ischemic and hemorrhagic. Ischemic strokes account for approximately 87% of all strokes in the United States and are characterized by a focal blockage within the cerebrovascular network that leads to a limited blood supply to the brain [1,4]. Hemorrhagic stroke involves the rupture of a weakened blood vessel resulting in the accumulation of blood and subsequent compression of nearby brain tissue [5]. This category can further be divided into intracerebral hemorrhages (ICH) and subarachnoid hemorrhages, which respectively account for 10% and 3% of all strokes in the United States [4,5].
Stroke etiology
While there are many causes of ischemic stroke, they all fit within three major categories: cerebral small vessel disease (CSVD), large artery disease, and cardioembolic stroke, altogether accounting for approximately 75% of ischemic strokes [2]. In both CSVD and large artery disease, arteriosclerotic changes can lead to the narrowing of the vessel lumen, resulting in cerebral hypoperfusion. This, in turn, can be exacerbated by the rupture of arteriosclerotic plaques and subsequent platelet activation [2,6]. Lastly, cardioembolic strokes encompass any condition that satisfies the criteria of Virchow’s Triad of hypercoagulability, stasis, or endothelial injury. This includes conditions such as atrial fibrillation, patent foramen ovale, valvular heart disease, myocardial infarction, and ventricular and septal aneurysms [2]. The remaining 20-30% of patients suffer from cryptogenic ischemic stroke which is an ischemic stroke with no known cause [2].
Similarly, the etiology of hemorrhagic strokes is vast and encompasses a variety of underlying conditions. One of the main treatable causes of hemorrhagic strokes is arterial hypertension [7]. Untreated hypertension is thought to cause the remodeling of small vessels supplying the deep parts of the brain. Eventually, this can lead to the rupture of these vessels causing extraversion of blood into the brain parenchyma [7]. Comparably, cerebral amyloid angiopathy involves the buildup of amyloid within the arterial walls of small vessels, which also can weaken the vessel walls and cause rupture within the brain parenchyma [7]. Lastly, aneurysms, regardless of a spontaneous or genetic origin, can rupture into the brain tissue as well, thus causing hemorrhagic stroke in patients [7].
Stroke pathophysiology
With ischemic stroke, there is a deficient amount of blood reaching the brain tissue, thus causing a substantial decrease in oxygen delivery. Reduced oxygen delivery cannot sustain the metabolic demands of the brain tissue, causing necrotic cell death and irreversible damage [8,9]. In patients with hemorrhagic stroke, the resultant hematoma causes compression of the surrounding brain tissue, leading to neuronal and glial disruption [5]. Furthermore, the compression from the hematoma increases intracranial pressure [5]. This disruption leads to poor blood delivery to brain tissue, inappropriate neurotransmitter release, and inflammation via microglial activation [5].
Patients with stroke commonly present with neurologic deterioration due to its localization in the CNS. Among the chief deficits experienced by patients is neuromuscular dysfunction. This includes speech apraxia, incontinence, and spasticity of the extremities [10-12]. These factors significantly impact the quality of life for patients and are frequently indications of a poor outcome and permanent disability [10-12]. Additionally, patients often experience serious cognitive impairment symptoms, including memory loss, speech deficits, and lapses in reasoning and problem-solving skills [13]. Typically, elderly patients experience memory-related symptoms more often, but patients of all ages can experience these symptoms on a varied spectrum [13].
Additional risks for immunocompromised patients
Despite the known severity of strokes, there are additional risks and considerations to be made for immunocompromised patients. In general, certain immunocomprosed patients have an increased risk of developing strokes [14]. Of particular interest are patients with HIV. For example, the progression of atherosclerosis, a major cause of ischemic stroke, is accelerated by HIV infection. This is likely due to chronic inflammation or HIV treatment-associated dyslipidemia [14]. HIV infection may additionally lead to repeated endothelial damage and vasculopathy, possibly narrowing the vasculature lumen and weakening the vessel wall [14]. HIV infection also decreases CD4 count, which has been demonstrated to increase the risk of developing stroke due to decreased viral suppression [15-17]. These physiologic changes in patients with HIV all contribute to the multi-faceted predisposition of developing stroke. Furthermore, stroke results in activation of the autonomic nervous system, a phenomenon known as stroke-induced immunosuppression (SIIS) suppresses the activity of Th1 cells and the secretion of proinflammatory cytokines, such as TNF-α and interferon-gamma (IFN-γ). At the same time, SIIS increases the activation of anti-inflammatory Th2 cells and IL-10 [18]. Additionally, it is thought that peripheral innate immune cells have reduced function following stroke, including impaired neutrophil oxidative burst and NETosis [18]. Altogether, this increases a patient’s susceptibility to bacterial infections leading to deadly complications such as stroke-associated pneumonia [18]. Overall, this common phenomenon can disproportionately affect immunocompromised patients who already are more susceptible to infection and have difficulty mounting an appropriate immune response to infection.
Local Immune Responses to Stroke
The systemic and chronic effects of stroke extend far beyond the nervous system, disrupting the delicate balance between the CNS and other body systems [19,20]. The immune system in particular plays a crucial role in post-stroke care as stroke patients may become more susceptible to infections (Figure 1) [19,20]. Infectious complications such as pneumonia or urinary tract infections are commonly observed in stroke patients, with an incidence rate of approximately 30% [21-23]. Furthermore, infection is the leading cause of death in approximately 20% of stroke survivors [24]. To provide better care for stroke survivors, it is essential to comprehend the underlying mechanisms responsible for this immunosuppressed state.
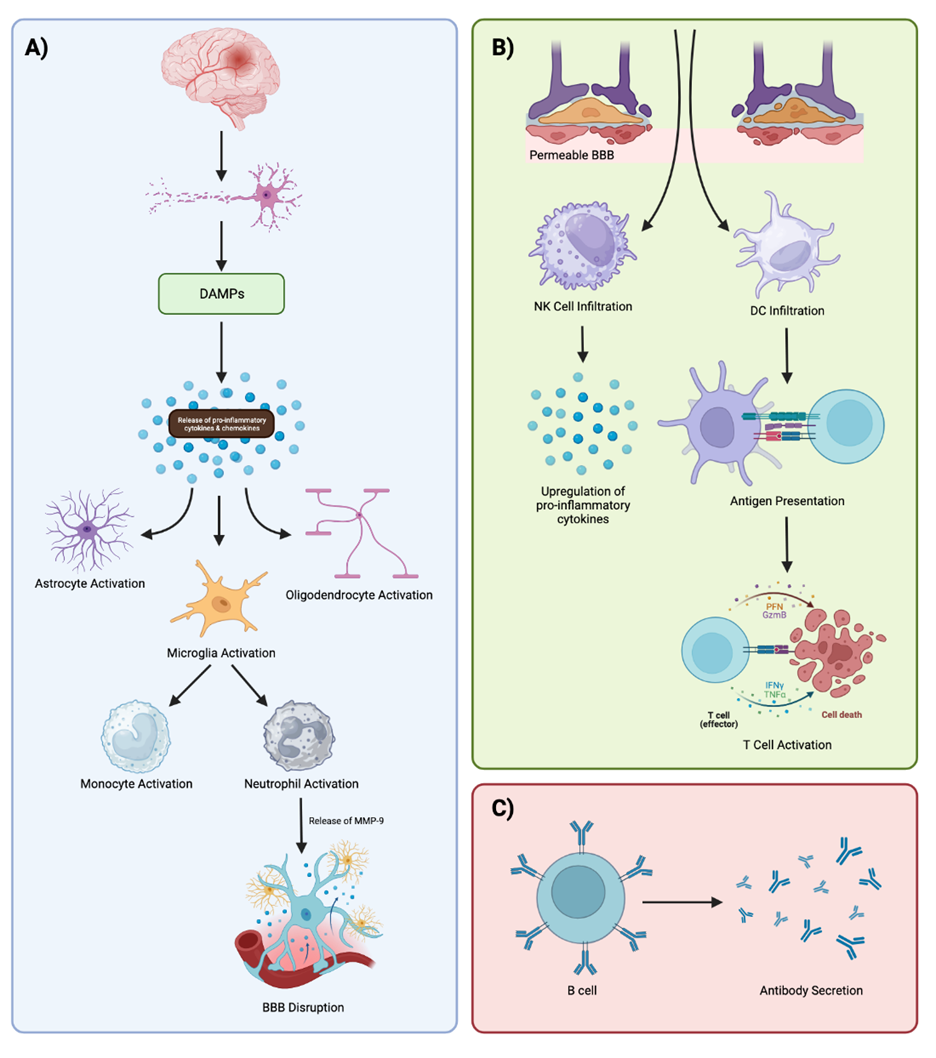
Figure 1. Visual Overview of the Immune Response to Ischemic Stroke. A) Ischemic stroke results in neuronal death. Dying neurons release DAMPs, which release pro-inflammatory cytokines that activate glial cells. Activated microglia promote monocyte and neutrophil proliferation and infiltration at the site of ischemia. MMP-9 produced by neutrophils disrupts the integrity of the BBB effectively exacerbating pathologic neuroinflammation. B) Disruption of the integrity of the BBB allows for the infiltration of additional immune cells into the damaged brain. NK cells at the ischemic lesion recruit additional neutrophils and secrete pro-inflammatory molecules. At the same time, DCs present antigens to T cells which launch specific immune responses at the lesion. C) B cells additionally work to produce antibodies and encourage immunologic memory following stroke. (Figure created with Biorender.com).
The CNS and the immune system are intricately interconnected, enabling the CNS to sense and modulate immune responses. Typically, this occurs through an anti-inflammatory stimulus triggered by a strong inflammatory signal detected by the autonomic nervous system. This response helps to contain both the infection and the associated inflammation [25]. However, brain or spinal cord injury can lead to the production of inflammatory mediators within the CNS or disruption of signaling in the neural-immune interaction control circuitry. These factors can result in systemic downregulation of both the innate and adaptive immune systems [25]. A closer examination of the effect of stroke on various components of the immune system, including the complement system, macrophages, neutrophils, lymphocytes, microglia, and T cells, will shed light on this immunosuppressed state.
As aforementioned, the obstruction of blood flow to specific regions of the brain during an ischemic stroke prevents the delivery of oxygen and nutrients to neurons ultimately resulting in cell death. This is because the depletion of ATP and subsequent glutamate release into neurons results in a pathological increase of calcium-ion influx into cells which activates proteolytic enzymes that trigger apoptosis [26]. Following ischemia, damage-associated molecular patterns (DAMPs) are released from dying cells and stimulate the production of inflammatory cytokines and chemokines including CCR2, CXCL8, CXCR4, CCR5, CCR6, and CXCR2 [27]. DAMPs ultimately function to activate innate immune receptors on glial cells in addition to acute and chronic post-stroke inflammatory responses through several key proteins and pattern recognition receptors such as toll-like receptors (TLRs), receptor for advanced glycation end products (RAGEs), High-mobility group box 1 (HMGB1), and heat shock proteins (Hsps) [26,28]. Specifically, HMGB1 released from necrotic cells binds and activates RAGEs, TLR-2, and TLR-4 which upregulate the expression of matrix metalloproteinase 9 (MMP-9), tumor necrosis factor-α (TNF-α), and interleukin (IL)-1β, all of which promote cellular death and neuroinflammation [26]. Similarly, Hsps such as Hsp70 bind to TLRs on macrophages and dendritic cells (DCs) resulting in the activation of pro-inflammatory nuclear factor kappa B (NF-κB) signaling [26].
Microglia, astrocytes, and oligodendrocytes are all glial cells activated by DAMP signaling. Microglia are brain cells largely responsible for the activation of innate immune responses in the brain and function to eliminate dead cells, microbes, protein aggregates, redundant synapses, and other antigens that endanger the CNS. In the acute phase, microglia secrete inflammatory cytokines including TNF-α, IL-6, and IL-1β which induce neuronal tissue injury [29]. However, microglia serve to aid in neurogenesis and tissue repair as the acute inflammatory response decreases [30]. Astrocytes additionally play a key role in the maintenance of CNS homeostasis through neurotransmitter regulation, BBB development and function, cerebral blood flow regulation, and synapse development. In contrast to microglia, astrocytes function to reduce neuroinflammation, support BBB regeneration, and promote CNS homeostasis following stroke [31]. However, glial scarring results from chronic astrocyte activation at the ischemic lesion which prevents axon regeneration and functional recovery [30]. Oligodendrocytes are the major myelinating cells of the CNS that function to promote neural regeneration following stroke [30]. Because oligodendrocytes do not have the ability to regenerate following injury, a large number of these cells die rapidly (within 3 hours) of ischemic stroke as a result of toxic levels of ATP and glutamate release [32].
Cytokine and chemokine secretion by activated microglia results in increased neutrophil proliferation and infiltration at the ischemic site [28,30]. Neutrophils are granulocytes that produce reactive oxygen species, MMP-9, perforins, and cytokines that directly damage the integrity of the BBB and exacerbate the ischemic lesion [28,33]. MMP-9 secreted by neutrophils specifically directly breaks down the BBB allowing for peripheral immune cell infiltration across the permeable BBB, activation of adhesion molecules in the brain endothelium, and increased cytokine production in the brain parenchyma [30]. This process results in increased neuroinflammation, tissue damage, and upregulation of TNF-α and IL-1β acutely and chronically following stroke [34]. Several animal models have demonstrated the detrimental effects neutrophil-induced neuroinflammation has on neurologic outcomes [26]. This effect is additionally observed in humans, where a high neutrophil count relative to a low lymphocyte count is considered to be a predictor of poor patient outcomes following stroke [35]. Interestingly, reactive microglia have been shown to engulf neutrophils at the periphery of the ischemic lesion, whereas microglial loss is associated with neutrophil accumulation in the brain parenchyma [36]. Furthermore, monocytes are additionally recruited to the site of ischemia around the same time as neutrophils in response to cytokine and chemokine secretion by activated microglia [27]. CCR2 is specifically responsible for monocyte recruitment to the brain parenchyma where they eventually mature and differentiate into macrophages [27].
Natural killer (NK) cells are lymphocytes that play a crucial role in innate immunity by recognizing and engulfing pathogens, producing cytokines, regulating immune responses, and assisting with antibody-dependent cell-mediated cytotoxicity. As such, NK cells contribute to the immune response following ischemia in the brain. NK cells have neurotoxic effects following ischemia as they express CX3CR1, a receptor that recruits and activates neutrophils [28]. Additionally, NK cells promote the upregulation of pro-inflammatory cytokines such as TNF-α and IFN-γ which contributes to neuroinflammation and can exacerbate tissue damage [30]. Both NK and DCs increase at the ischemic lesion until day 14 after stroke where they remain constant and continue to exert pro-inflammatory effects on the brain [27]. DCs are professional antigen-presenting cells that bridge the innate and adaptive immune systems. Specifically, DCs engulf and proteolyze pathogens by means of endocytosis or phagocytosis and express antigens on their surface through major histocompatibility complex (MHC) class I and II molecules. Once expressed on the surface of an MHC-I or -II receptor, T cells can then bind and recognize the specific antigen to launch a specific immune response. Within the ischemic environment, DCs engage in antigen presentation to T cells which secrete IL-12, IL-23, IL-6, and IL-1β, all of which promote neuroinflammation following stroke [28].
Following the innate immune response, T cells and B cells are activated to generate a highly specific immunologic defense and response to ischemia. After antigen presentation, CD8+ T cells exert cytotoxic effects on pathogens by introducing a large payload of molecules such as granzymes and perforin, both of which induce apoptosis via cell membrane lysis. T cells migrate to the ischemic region in an antigen-independent fashion within days following acute ischemia and continue to increase until up to 28 days after stroke [27,28,37]. At both the acute and chronic phases following stroke in the ischemic brain, CD8+ T cells produce significantly more IFN-γ, IL-17, and IL-10 [38]. Significant reductions in IL-4 production are additionally observed [38]. Both of these actions together can work to exacerbate symptoms of neuroinflammation following stroke. Another class of T cells known as Tregs function to regulate T cell activity and homeostasis. Tregs are present in the brain after stroke, though they exist and accumulate primarily in the chronic phase [38]. Tregs are believed to limit astrogliosis, suppress astrocyte toxicity, and promote functional recovery after an ischemic event through the secretion of IL-10, IL-2, and IL-33 [33,38].
B cells are an important component of the humoral immune system, responsible for producing antibodies and participating in immune memory. B cells originate from stem cells in the bone marrow and become activated when encountered by a pathogen in addition to CD8+ T cell stimulation. Activated B cells then differentiate into plasma cells which produce and secrete antibodies into the bloodstream. These antibodies can then identify and neutralize pathogens in the body with assistance from other cytotoxic cells. Memory B cells survive in the bloodstream longer than plasma cells and have the ability to rapidly respond to a previously encountered antigen upon re-exposure, effectively forming the basis of immunological memory. Although being relatively understudied, B cells have been shown to exhibit both detrimental and protective effects in the ischemic brain. For example, B cells have been shown to undergo isotype switching and secrete immunoglobulins to impair long-term functional recovery after stroke [8]. However, B cells have been reported to provide neuroprotection in an IL-10-dependent manner similar to Tregs [8]. Ultimately, the role of B cells in the post-stroke brain remains elusive and complex, highlighting the need for additional research to better understand the function of B cells.
Effects on the Immune System Following Stroke
Cells of the immune system
When examining peripheral blood as a whole, studies have reported a decrease in lymphocyte counts and impaired T- and NK-cell activity in patients following a stroke [39]. Specifically focusing on T cells, multiple studies have demonstrated reduced mitogen-induced cytokine production [40-42]. Cytokines play a critical role in immune system function as they facilitate communication between immune cells and non-immune cells during an immune response. Impairment of these molecules hinders the body's ability to fight infections and leaves patients more susceptible to serious consequences of infection.
Beyond their role in immune function, neutrophils also contribute to the pathogenesis of stroke. Neutrophils are among the first cells in the blood to respond after an ischemic stroke, contributing to the disruption of the BBB, cerebral edema, and brain injury [43]. Neutrophils are also involved in the major processes that cause ischemic stroke, including thrombosis, atherosclerosis, and clot formation [44-46]. Due to their significance in stroke pathogenesis, neutrophils have become a subject of great interest in modern research. In contrast to other immune cells, the number of neutrophils increases following the onset of a stroke [47]. This leads to an elevation in the neutrophil-to-lymphocyte ratio, and the extent of this increase has been correlated with both the size and severity of the stroke [48]. The rise in neutrophils results from enhanced production, increased release from the bone marrow and spleen, and possibly a reduction in neutrophil apoptosis [49]. Overall, neutrophil function appears to be enhanced following a stroke. However, this enhancement primarily occurs in the CNS, where the injury took place, as an attempt to clear necrotic debris. Neutrophils seem to play a role in the healing process, but the extent of their overactivity is still an area of intense study. Studies that have targeted neutrophil-recruiting chemokines have shown mixed results. For instance, in a mouse ischemic stroke model, inhibition of CXC chemokines with Evasin-3 impaired neutrophil activation but had no effect on stroke outcomes [50]. Conversely, inhibition of CXCR1 and CXCR2 with Reparixin in a rat stroke model reduced ischemic brain injury, improved motor outcomes, and decreased brain levels of myeloperoxidase MPO and IL-1β. However, when only the CXCR2 receptor was blocked with SB225002, no improvement in stroke outcomes occurred despite a reduction in neutrophil activation and infiltration [51,52]. Thus, therapies targeting neutrophil activation and recruitment are not completely understood, and further research is needed.
The function of macrophages must also be considered when examining the functional status of the immune system in stroke patients. The phagocytic ability of macrophages is impaired, leaving the host particularly susceptible to bacterial infections [53,54]. This is of particular concern as stroke patients spend a significant amount of time in hospitals receiving treatment, where the risk of bacterial infections is high [55]. In addition to decreased phagocytic ability, circulating monocytes from patients with acute brain injury show decreased expression of MHC II [53]. This reduced MHC II expression leaves stroke survivors susceptible to both initial infection and reinfection, as MHC II is necessary for the generation of memory B cells [56]. Overall, impaired monocyte function results in insufficient antigen presentation, which is a crucial function of macrophages as it links the innate with the adaptive immune system. Impaired macrophage function may, therefore, contribute to reduced lymphocyte responses and an impaired immune response.
In general, changes in cellular immune responses correlate with the severity of brain injury. These changes occur rapidly within hours after the injurious insult and can last for up to several weeks [25,40]. Furthermore, the extent and duration of impaired cell-mediated immune responses in patients with CNS injuries are usually correlated with an increased risk of infection and poor outcomes [57]. A study using a mouse model highlighted this phenomenon, demonstrating that stroke-induced a rapid and extensive apoptotic loss of lymphocytes in lymphoid organs and peripheral blood. This dysfunction was characterized by decreased production of IFN-γ and TNF-α, two key molecules involved in initiating and propagating immune responses [58]. The study further showed that these dysfunctions persisted for up to 6 weeks after the induction of stroke. Interestingly, immune reconstitution through the adoptive transfer of IFN-γ-producing lymphocytes from healthy littermates or treatment with recombinant IFN-γ greatly diminished bacterial burden. This suggests a causative link between the suppression of cell-mediated immune responses and increased susceptibility to bacterial infections following stroke.
The complement system
In addition to the role of immune cells, the complement system also plays a critical role in the immune response. Research has shown that complement components are strongly implicated in the progression of stroke in survivors [59]. C1q, a complement component, plays a crucial role in the CNS following ischemic injury. While C1q is normally present in the blood, it can also be synthesized in the brain after injury, such as viral infection, kainic acid treatment, and ischemic stroke [60,61]. Studies in mouse models have demonstrated that depletion of C1q results in impaired elimination of redundant synapses, highlighting the role of C1q following stroke [62]. This finding has been confirmed by other studies showing that ischemic neurons mostly express C1q, suggesting its involvement in the clearance of damaged neurons and cellular debris [63]. Further studies on C1q have shown that selective depletion of C1q in mice exposed to a hypoxia-ischemia state reduced neutrophil infiltration, oxidative injury, and brain infarct volume after 24 and 72 hours of cerebral ischemia [63]. This suggests that inhibiting C1q expression during the acute phase of cerebral ischemia could be beneficial in attenuating ischemic brain injury. However, the role of C1q is not as straightforward as this study suggests. Another study showed that C1q knockout in adult mice subjected to MCAO surgery did not affect brain infarct volume 24 hours after ischemia [64].
C3 is of particular importance in the complement system because of its role in creating both C3 convertase and C5 convertase, which are essential for the formation of the membrane attack complex. Clinical studies have found that plasma levels of C3 were higher in ischemic stroke patients compared to healthy controls, peaking 3 days after brain ischemia [65,66]. Longitudinal studies have shown that elevated C3 levels were associated with worse neurological outcomes at 3-month and 2-year time points after ischemic onset [67]. However, like the role of C1q, the role of C3 is complex. Studies have demonstrated that C3 levels increase in the brain after ischemic stroke, and inhibiting C3 activity could attenuate ischemic brain injury [68]. However, in another study using mice, C3 deficiency reduced brain infarct volume and neutrophil influx at 1 day of ischemic stroke but impaired subsequent neurogenesis at 7 to 28 days of brain ischemia [69]. Thus, it can be concluded that C3 has a dual role in the pathogenesis of ischemic stroke, both attenuating ischemic damage and enhancing neurogenesis during the healing process. This suggests that levels of C3 need to be modulated, and further studies should analyze the effect of modulating C3 levels following ischemic stroke.
Ischemic cascade
The ischemic cascade encompasses a series of events that ultimately lead to neuronal death in the context of ischemia. Due to the lack of oxygen, aerobic respiration fails to function, resulting in a greater reliance on anaerobic respiration. This energy imbalance leads to ionic imbalance and neurotransmitter release, particularly glutamate, along with inhibition of reuptake [70]. The increased extracellular glutamate levels cause a significant influx of calcium into neurons. Calcium then triggers numerous proteases and phospholipases that degrade proteins and membranes [70]. Additionally, the influx of glutamate leads to increased entry of sodium and water, resulting in swelling and shrinkage of the extracellular space [70]. Elevated levels of calcium, sodium, and adenosine diphosphate in ischemic cells stimulate excessive production of mitochondrial oxygen radicals, causing damage to lipids, proteins, nucleic acids, and carbohydrates within the neurons [71]. This is particularly detrimental to neurons because protective mechanisms against these reactive oxygen species (such as superoxide dismutase, catalase, glutathione, etc.) are too slow to counteract the sudden surge [72]. Ultimately, all of these processes culminate in cell death.
Experimental models have attempted to block glutamate receptors. Studies have shown that this approach reduces infarction volume by decreasing calcium influx and the activation of related proteases. It also attenuates cortical spreading depressions and triggers the subfamily of metabotropic mGluRs towards prosurvival or prodeath signaling [73]. These initial findings provided hope for a potential therapeutic target to improve stroke outcomes. However, subsequent trials have failed to demonstrate improved patient outcomes [74,75]. This area of research requires further investigation to better understand the underlying mechanisms by which blocking the glutamate receptor initially yielded positive outcomes. Such research may eventually lead to the blockade of GluRs being a viable therapeutic target.
Systemic Effects of Chronic Immune Activation Following Stroke
Cognitive and psychiatrics effects of chronic immune activation
Increasing evidence points towards various downstream effects of chronic immune activation following stroke in the CNS (Figure 2). Among the most extensively studied chronic sequelae in post-stroke patients are depression, fatigue, and dementia. The underlying mechanism is hypothesized to involve antigen-presenting cells in the brain, which pick up new antigens released from stroke-induced tissue damage. These antigen-presenting cells then reach lymphoid organs through the circulation and engage naïve lymphocytes, thereby initiating an adaptive immune response targeted at the brain [28]. The link between chronic inflammation and cognitive decline finds support in research that highlights strong correlations between higher erythrocyte sedimentation rates and poorer performances on memory tests in post-stroke patients [76].
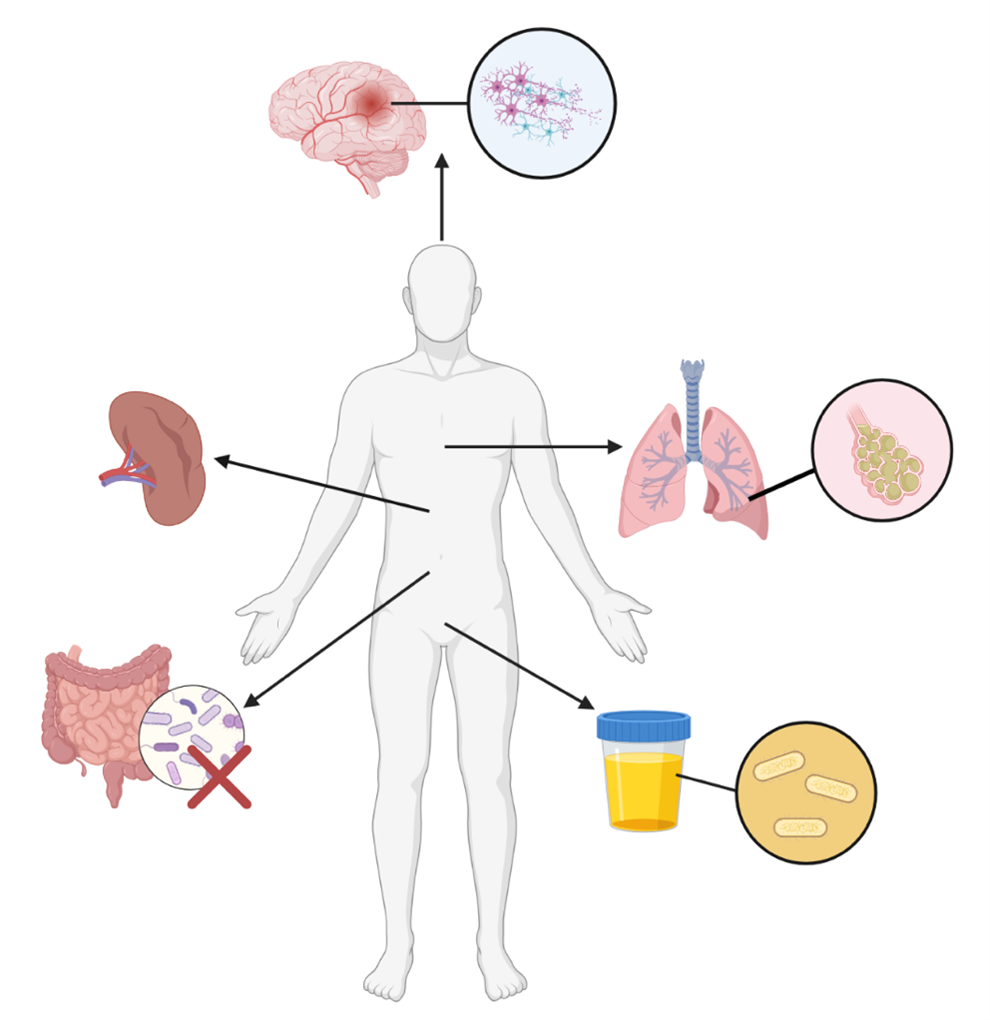
Figure 2. Systemic Effects of Chronic Immune Activation Following Ischemic Stroke. Chronic immune activation following stroke can result in a variety of pathologies. Significant neurologic and motor deficits may occur following ischemic stroke. Exhaustion of spleen-derived immune cells and consequent reduction in splenic volume results in immunosuppression. Significant immunosuppression following stroke can result in the development of pneumonia and UTIs, both of which can be fatal. Ischemic stroke can additionally result in dysbiosis of the natural gut microbiota, which may exacerbate symptoms. (Figure created with Biorender.com).
Gut microbiome dysbiosis secondary to chronic immune activation
Chronic immune activation following stroke has systemic effects beyond the CNS. For example, the gut-brain axis is a key signaling pathway that monitors and integrates gut functioning with the emotional and cognitive centers of the brain. In mice, acute middle cerebral artery occlusion has been linked with gut microbiome dysbiosis, most notably reductions in bacterial diversity and an overgrowth of Bacteroidetes [77]. These pathological changes to the gut microbiome ultimately resulted in worse outcomes [77]. Notably, when germ-free mice were recolonized with dysbiotic post-stroke microbiota, they exhibited increased stroke lesion volume, functional deficits, and inflammatory responses compared to controls. These findings highlight the significance of the gut-brain axis following stroke and the bidirectional effects that dysregulation can cause [77].
Immunosuppression following chronic immune activation
In humans, the spleen is the largest immune organ in the body, playing a significant role in both adaptive and innate immunity. Following stroke, there is a significant reduction in splenic volume which has severe consequences on the normal homeostatic functions of the immune system [78]. Elevated levels of IFN-γ, IL-6, IL-10, IL-12, and IL-13 have been associated with reduced splenic volume in humans [79]. The subsequent loss of splenic B, T, and NK cells results in activation of the hypothalamic-adrenal axis and autonomic nervous system [58]. This event results in pathological overexpression of norepinephrine, acetylcholine, and glucocorticoids which further exacerbate splenic atrophy, T cell apoptosis, and NK cell deficiency [80,81]. Spleen volume reduction is associated with decreased lymphocyte counts which can leave patients susceptible to life-threatening infections [82]. Lymphopenia refers to a disorder characterized by a significant reduction in white blood cell lymphocytes caused by infections, medications, and nutritional deficiencies [83]. Stroke-induced lymphopenia arises as a consequence of β3-adrenergic receptor suppression of IL-7, CXCL12, and angiopoietin-1 which encourages myeloid cell proliferation and the development of lymphopenia [28]. Common infections seen following stroke as a result of immunosuppression include urinary tract infections and pneumonia [84,85]. Overall, ongoing research on the effects of chronic inflammation following stroke holds important implications for stroke outcomes and therapeutic interventions.
Current and Emerging Treatment Strategies for Stroke
Current treatment strategies for stroke
Therapeutic intervention for the clinical management of stroke is heavily time-dependent and begins before reaching the hospital when emergency medical service personnel successfully identify the signs and symptoms of a stroke. The goal of treating ischemic stroke is to remove the embolus that causes blockage of blood flow to the ischemic area and tissue that receives blood from the affected blood vessels, known as the penumbra. Currently, the only FDA-approved medication for ischemic stroke is alteplase, a tissue plasminogen activator (tPA) [86]. When administered within 3 hours of the onset of symptoms, tPA provides optimal neurologic and functional improvement [87,88]. Certain patients may also receive treatment 4.5 hours after symptom onset; however, administering thrombolytics after the 3-4.5 hours post-stroke window can have deleterious effects [89-91]. These include an increased risk of symptomatic ICH, parenchymal hematoma, and hemorrhagic infarction [91,92].
Blood clots form when platelet cells aggregate and bind to fibrin which holds the blockage together. tPA, a serine protease, binds to plasminogen, activating it and forming plasmin which then breaks down the fibrin mesh of the embolus [93]. This allows recanalization and restores blood flow to the ischemic region. The effects of tPA are restricted to the site of the clot since plasmin breaks down rapidly with α2-antiplasmin [93]. This rapid removal of tPA from the body can also serve as a disadvantage, however, since its therapeutic effects are short-lasting. Moreover, tPA only exerts its effects on the surface of the clot, therefore, blood flow to regions blocked by larger clots is stopped for longer periods of time [93]. Moreover, recanalization after intravenous administration of tPA is only seen in 21% of cases and reocclusions are seen in 12% and although 30% of patients have demonstrated better functional outcomes, the drug was still associated with a greater risk of ICH [94-96].
Since the effectiveness of tPA in the treatment of proximal large vessel occlusion was moderate at best, a more invasive treatment, endovascular thrombectomy (EVT), was studied [97]. There are two approaches to mechanical thrombectomy; the thrombi can be approached either distally with retrievers or proximally with aspiration devices. Retrievers (devices such as MERCI, CATCH, Solitaire FR, TREVO, REVIVE) expand within the vessel and are similar to stents often coming equipped with baskets that retrieve thrombi [98]. Aspiration devices fragment the thrombus and subsequently aspirate them when connected to a suction device, and any residual fragments are removed using the thrombus removal ring [95]. Examples of aspiration devices include the Penumbra System (Penumbra Inc, California, USA) and QuickCat (DSM Inc, Philadelphia, USA). The window within which the EVT procedure must be conducted is longer than for IVT; however, greater success is seen if it is done within 3-4.5 hours after stroke symptoms with decreased success seen if 5 to 8 hours after stroke [98,99]. EVT procedure can be performed under general anesthesia or under conscious sedation [94]. Based on data from five recent randomized trials for EVT, the combined tPA and EVT treatment group demonstrated better outcomes than the tPA-only group including reduced severity of disability, higher rates of functional independence, and reduced mortality [100-104]. While some studies indicate the benefit of a combined therapy, other studies show the opposite [105]. Disadvantages of the treatment include the requirement for more resources, including a larger team of providers, and the need for a better-equipped facility. In addition, there is an increased risk of ICH [106]. However, it remains an option for patients who are ineligible for tPA therapy [107].
Secondary stroke prevention
Antithrombotic treatments play an important role in acute ischemic stroke treatment and prevention. Antiplatelet and anticoagulant therapy is recommended to prevent future ischemic events [108]. Dual antiplatelet treatment in the form of clopidogrel and aspirin prevents future ischemic events more effectively than aspirin alone but the dual treatment also increases the risk of hemorrhage [109]. Therefore, it is recommended to be used only short-term in a subset of patients with early arriving minor stroke and high-risk transient ischemic attack or severe symptomatic intercranial stenosis [108]. Patients are recommended to cease dual antiplatelet therapy after 10 to 21 days to balance the risks and benefits [108,109]. Ticagrelor may be used as an alternative to aspirin in patients with a contraindication to aspirin, however, it is not preferred [109,110]. In addition, several other preventative actions can be taken against secondary strokes including lifestyle changes to increase activity and improve diet [108]. Patients with extracranial carotid artery disease, a cause of stroke, must be treated soon after their ischemic stroke with either a carotid endarterectomy or carotid artery stenting [108].
Immunotherapy for stroke
Although tPA has been a recommended treatment since 1996, the short window of administration in addition to the short half-life makes 25-70% of stroke patients ineligible for the treatment demonstrating a need for other therapies [111,112]. Stroke increases the permeability of the BBB, making the brain more susceptible to autoimmunity leading to the destruction of the ischemic tissue [113]. Thus, recent studies have aimed to target the immune response to improve stroke recovery (Table 1). Certain antibodies have been targeted for stroke management including myelin-associated proteins Nogo-A, myelin-associated glycoprotein (MAG), and Oligodendrocyte myelin glycoprotein (OMgp). Nogo-A (myelin-associated protein and antigen for monoclonal antibody IN-1) is produced by oligodendrocytes and inhibits axonal remodeling after injury [114]. Monoclonal antibody (IN-1), developed against Nogo-A, when administered improved neuroplasticity [113,115]. In a mouse model, genetic deletion or neutralization of Nogo-A led to functional improvement 3 weeks after stroke which may result from revascularization of the ischemic border zone [116]. Levels of MAG, an inhibitor of axon growth, are augmented after stroke. In a rodent model, an anti-MAG monoclonal antibody GSK249320, has been indicated as having restorative capacity after stroke [117]. Nasal administration of OMgp peptides reduced the ischemic infarct size and elevated anti-inflammatory cytokines TGF-β and IL-10 in a rodent model, suggesting that T cells and IL-10 have a role to play in decreasing stroke size via IL-10-dependent CD4+ T cells [118].
|
Immunotherapeutic Intervention |
Subject |
Outcome |
Source |
|
Myelin-Associated Proteins |
|||
|
Nogo-A – anti-Nogo-A antibody IN-1 |
Rodent
Rodent |
Improved neuroplasticity
Functional improvement 3 weeks after stroke resulting from recanalization of the ischemic border zone |
Seymour et al. [115]
Rust et al. [116] |
|
MAG – anti-MAG monoclonal antibody GSK249320 |
Rodent |
Improved functional outcomes |
Cash et al. [117] |
|
OMgp peptides |
Rodent |
Nasal administration reduced ischemic infarct size, and elevated anti-inflammatory cytokines TGF-beta and IL-10 |
Frenkel et al. [118] |
|
Treg therapy |
Rodent
Rodent |
Decreased infarct size, enhanced post-stroke recovery in a rodent model
Promotes oligodendrogenesis and tissue repair microglia after ischemic stroke . |
Becker et al. [123] Frenkel et al. [118] Gee et al. [124]
Shi et al. [125] |
|
Cell Adhesion Molecules |
|||
|
Enlimomab (ICAM-1 antibody) |
Rodent
Human |
Reduced infarct size and leukocyte adhesion
|
Yu et al. [113]
Enlimomab Acute Stroke Trial Investigators [129] |
|
β2 integrins inhibition |
Rodent |
Attenuated infarct volume, edema, and mortality |
Arumugam et al. [127] |
|
Anti-P-selectin |
Rodent
Rodent |
Reduced leukocyte infiltration
Decreased infarct size and brain edema |
Suzuki et al. [131]
Suzuki et al. [132] |
|
Anti-E-selectin |
Rodent
Human Phase II trials |
Increased blood flow, decreased leukocyte levels in the brain
Study was terminated. |
Yu et al. [113]
National Institute of Neurological Disorders and Stroke [133] |
|
Anti-L-selectin combined with tPA |
Rodent |
Moderately reduced brain damage after stroke |
Bednar et al. [136] |
|
Monoclonal Antibodies |
|||
|
Natalizumab |
Rodent
Rodent
Human |
Decreased leukocyte infiltration, reduced infarct volume, improved outcomes
No effect on stroke size or neurological impacts of stroke
No positive effect in stroke treatment |
Becker et al. [137]
Langhauser et al. [138]
ACTION and ACTION II clinical trials [139] |
|
Fingolimod |
Rodent
Human
|
Attenuated stroke size and improved neurological and functional outcomes
Decreased inflammation duration and attenuated secondary stroke injury |
Czech et al. [142] Kraft et al. [143] Dang et al. [144] Wei et al. [145] Brait et al. [52]
Fu et al. [140] |
Tregs: In response to ischemic stroke, T lymphocyte levels increase and persist in the brain up to 60 days after stroke is associated with an augmented risk of stroke recurrence and mortality [119,120]. While CD4+, CD8+, and γδ T cells increase tissue damage, Tregs have a protective effect and can be used as therapy for stroke [92,121,122]. Nasal administration of myelin basic protein (MBP) in a rodent model increased the frequency of Tregs, decreased infarct size, and enhanced post-stroke recovery while on the other hand, depletion of Treg cells in a mouse model after a stroke-induced sensorimotor issue many weeks after stroke indicating the role of these cells in recovery after stroke [118,123,124]. Administration of Treg cells also promoted oligodendrogenesis and tissue-repair microglia post-ischemic stroke [125]. This indicates that Treg may offer therapeutic advantages in stroke recovery.
Cell adhesion molecules and cytokines: After the occurrence of an ischemic stroke, there is growing evidence of post-reperfusion inflammation during which leukocytes are recruited with the help of adhesion molecules from the selectin family, integrins (β1 and β2), and intercellular adhesion molecule-1 (ICAM-1) [113,119]. Adhesion molecules are proteins found on the surface of the cell and play a role in cell-cell interaction or cell and extracellular matrix (ECM) interactions [126]. The infiltration of these leukocytes can cause additional damage as they cross the BBB and release reactive oxygen species, cytokines, and proteases, with groups indicating that increased circulating leukocytes and neutrophils are associated with increased stroke severity [119]. Thus, the adhesion molecules have been studied as targets of immunotherapies for stroke. β2 integrins may serve as a target to reduce inflammation with certain heterodimers reducing stroke severity and mortality in mice models [127,128].
ICAM-1 was considered a possible target to treat reperfusion-induced inflammation following stroke and Enlimomab an ICAM-1 antibody was developed and showed that it reduced infarct size and leukocyte adhesion in early studies [113]. However, when it was tested in humans, it worsened stroke outcome and was associated with ICH, cardiac arrest, and meningitis [113,129]. However, it has been shown to increase the therapeutic window when used in conjunction with tPA [130]. E- and P- selectin are upregulated after stroke, and in a rat model, anti-P-selectin antibody reduced leukocyte infiltration and decreased infarct size and brain edema [131,132]. Anti-E-selectin antibody had a neuroprotective effect as it increased blood flow and decreased leukocyte levels in the brain, however, Phase 2 studies administering it nasally to investigate whether it would be effective in the treatment for stroke, TIA, and secondary prevention of stroke were terminated before results could be gathered [113,133-135]. The anti-L-selectin antibody when used in conjunction with tPA moderately reduced brain damage after stroke [136].
Monoclonal antibodies: Currently, Natalizumab is used to treat multiple sclerosis and Crohn’s disease. It is a monoclonal antibody that targets the alpha chain of adhesion molecule integrin within very late antigen-4 and is associated with decreased leukocyte infiltration, reduced infarct volume, and improved outcomes in certain preclinical models [137,138]. However, the ACTION and ACTION II clinical studies indicated no positive effect in the treatment of stroke [139].
Fingolimod is a sphingosine I-phosphate receptor modulator that limits lymphocyte infiltration that has been approved for the treatment of multiple sclerosis [140]. So far, it has shown differing results in rodent models with most demonstrating its neuroprotective effects [141-146]. A recent early-stage clinical trial has so far demonstrated that the administration of fingolimod improves functional outcomes, reduces infarct growth, and increases cerebral blood flow (NCT02002390). Several stroke treatments have been researched over the past few decades, but the beneficial effects seen in animal models rarely translate over to clinical trials [147]. However, immunotherapy is an avenue that can be explored further to provide therapeutics for stroke recovery.
Conclusion and Future Research Directions
As our understanding of stroke pathophysiology continues to evolve, the role of the immune system has become of great importance. Dysregulation of critical signaling pathways and immune populations after stroke significantly contributes to pathologic neuroinflammation, excitotoxicity, and consequently, disease progression. Increasing evidence in the literature suggests chronic immune activation following a stroke can lead to the development of systemic pathologies that may worsen patient outcomes overall. Immunocompromised patients in particular are at an increased risk for significant morbidity and mortality following stroke as they are often more susceptible to life-threatening infections. As such, clinical management of stroke must be conducted in a highly patient-specific manner by a multidisciplinary team of providers to improve patient outcomes.
The significant role of the immune system in stroke pathogenesis has led to the investigation and development of novel treatment strategies that target the immune system. Immunotherapy for stroke has furthered our understanding of stroke pathogenesis at a biological level while simultaneously providing insight into novel treatment modalities that have the potential to revolutionize the clinical management of stroke. Despite the great promise of immunotherapy for stroke, significant research efforts must continue before clinical implementation. For example, significant disease heterogeneity exists amongst stroke patients as hemorrhagic and ischemic stroke have different disease etiologies and secondary injury can vary significantly between patients. Furthermore, individual differences persist amongst individual patients with the same stroke type. In addition, the BBB poses a significant challenge to the successful implementation of immunotherapy for stroke due to its highly selective nature. Many current immunotherapeutic strategies that have demonstrated safety and efficacy in other medical specialties are not suitable for stroke as they would not be able to permeate the BBB. The therapeutic administration route is another challenge experienced in immunotherapy. Intraoperative injection of immunotherapeutic agents can be expensive and pose a significant risk to patients, especially for those who are at a predisposed risk of infection, have significant neurologic or functional deficits, or who have already had multiple trips to the operating room. In-clinic injections or infusions can be safer and more convenient for patients due to the less invasive nature of the procedure. However, in-clinic injections or infusions may require multiple office visits which may complicate patient compliance for those who are unable to travel to the clinic as a result of work, school, personal obligations, lack of transportation, or other medical comorbidities. As such, future research directions should work to investigate these issues to determine optimal immunotherapeutic platforms including dosages, formulations, and administration routes. Individual patient differences including age, sex, and preexisting comorbidities should be accounted for.
The introduction of immunotherapeutic agents into the body can result in the release of pathological concentrations of proinflammatory cytokines into the bloodstream. This phenomenon, known as “cytokine storm”, can result in significant morbidity and mortality rates if not properly controlled and accounted for. Although the impact and role of cytokine storm is not well characterized following immunotherapy for stroke, it is reasonable to hypothesize cytokine storm could occur following the introduction of immunotherapeutic agents for the treatment of stroke. Thus, drug formulations need to be refined in a way that ensures patient safety while also promoting the efficacy of the platform. In addition, immunotherapeutics can take a significant amount of time to process and develop due to their patient-specific mechanism of action. This can ultimately impact treatment cost and access to care while also providing provide time for the lesion and surrounding ECM to further develop in a way that combats the efficacy of the immunotherapeutic platform, potentially rendering it ineffective. As aforementioned, future research efforts should be conducted to determine the optimal therapeutic formulation and processing method.
In sum, immunotherapy has the potential to revolutionize the clinical management of stroke. Future research endeavors must address key issues concerning immunotherapy formulation, administration, and access to care before clinical use.
Abbreviations
BBB: Blood Brain Barrier; CNS: Central Nervous System; CSVD: Cerebral Small Vessel Disease; DAMP: Damage-Associated Molecular Pattern; DC - Dendritic Cell; ECM: Extracellular Matrix; EVT: Endovascular Thrombectomy; Hsp: Heat Shock Protein; ICAM-1: Intercellular Adhesion Molecule-1; ICH: Intracranial Hemorrhage; IFN-γ: Interferon gamma; IL – Interleukin; MAG: Myelin-Associated Glycoprotein; MBP: Myelin Basic Protein; MHC: Major Histocompatibility Complex; MMP-9: Matrix Metalloproteinase; NF-kB: Nuclear Factor kappa B; NK: Natural Killer; OMgp: Oligodendrocyte Myelin Glycoprotein; SIIS: Stroke-Induced Immunosuppression; TLR: Toll-Like Receptor; TNF-α: Tumor Necrosis Factor alpha; Tregs: Regulatory T cells; tPA: Tissue Plasminogen Activator
References
2. Murphy SJ, Werring DJ. Stroke: causes and clinical features. Medicine. 2020 Sep 1;48(9):561-6.
3. Tadi P, Lui F, Budd LA. Acute Stroke (Nursing). StatPearls. 2023
4. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation. 2022 Feb 22;145(8):e153-639.
5. Unnithan AK, Mehta P. Hemorrhagic Stroke. StatPearls. 2023.
6. Li Q, Yang Y, Reis C, Tao T, Li W, Li X, et al. Cerebral small vessel disease. Cell Transplantation. 2018 Dec;27(12):1711-22.
7. Magid-Bernstein J, Girard R, Polster S, Srinath A, Romanos S, Awad IA, et al. Cerebral hemorrhage: pathophysiology, treatment, and future directions. Circulation Research. 2022 Apr 15;130(8):1204-29.
8. Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009 May 1;40(5):e331-9.
9. Datta A, Sarmah D, Mounica L, Kaur H, Kesharwani R, Verma G, et al. Cell death pathways in ischemic stroke and targeted pharmacotherapy. Translational Stroke Research. 2020 Dec;11:1185-202.
10. Esmailzade Moghimi S, Mohammadi F, Yadegari F, Dehghan M, Hojjati SM, Saadat P, et al. Verbal and oral apraxia in patients with acute stroke: Frequency, relationship, and some risk factors. Applied Neuropsychology: Adult. 2022 Feb 4:1-12.
11. Bavikatte G, Subramanian G, Ashford S, Allison R, Hicklin D. Early identification, intervention and management of post-stroke spasticity: expert consensus recommendations. Journal of Central Nervous System Disease. 2021 Sep 23;13:11795735211036576.
12. Thomas L, French B, Watkins C, Leathley M, Sutton CJ, Cross S, et al. Treating urinary incontinence in post-stroke adults. Continence UK. 2008;2008(1):CD004462.
13. Al-Qazzaz NK, Ali SH, Ahmad SA, Islam S, Mohamad K. Cognitive impairment and memory dysfunction after a stroke diagnosis: a post-stroke memory assessment. Neuropsychiatric Disease and Treatment. 2014 Sep 9:1677-91.
14. Benjamin LA, Bryer A, Lucas S, Stanley A, Allain TJ, Joekes E, et al. Arterial ischemic stroke in HIV: defining and classifying etiology for research studies. Neurology-Neuroimmunology Neuroinflammation. 2016 Aug 1;3(4).
15. Chow FC, Bacchetti P, Kim AS, Price RW, Hsue PY. Effect of CD4 cell count and viral suppression on risk of ischemic stroke in HIV infection. AIDS (London, England). 2014 Nov 11;28(17):2573-7.
16. Chow FC, Wilson MR, Wu K, Ellis RJ, Bosch RJ, Linas BP. Stroke incidence is highest in women and non-Hispanic blacks living with HIV in the AIDS Clinical Trials Group Longitudinal Linked Randomized Trials cohort. Aids. 2018 Jun 1;32(9):1125-35.
17. Bogorodskaya M, Chow FC, Triant VA. Stroke in HIV. Canadian Journal of Cardiology. 2019 Mar 1;35(3):280-7.
18. Faura J, Bustamante A, Miró-Mur F, Montaner J. Stroke-induced immunosuppression: implications for the prevention and prediction of post-stroke infections. Journal of Neuroinflammation. 2021 Dec;18(1):127.
19. Liu Q, Sanai N, Jin WN, La Cava A, Van Kaer L, Shi FD. Neural stem cells sustain natural killer cells that dictate recovery from brain inflammation. Nature Neuroscience. 2016 Feb;19(2):243-52.
20. Shi K, Wood K, Shi FD, Wang X, Liu Q. Stroke-induced immunosuppression and poststroke infection. Stroke and Vascular Neurology. 2018 Mar 1;3(1):34-41.
21. Westendorp WF, Nederkoorn PJ, Vermeij JD, Dijkgraaf MG, de Beek DV. Post-stroke infection: a systematic review and meta-analysis. BMC Neurology. 2011 Dec;11(1):110.
22. Gong S, Zhou Z, Zhou M, Lei Z, Guo J, Chen N, et al. Validation of risk scoring models for predicting stroke-associated pneumonia in patients with ischaemic stroke. Stroke and Vascular Neurology. 2016 Sep 1;1(3):126.
23. Zheng H, Cao N, Yin Y, Feng W. Stroke recovery and rehabilitation in 2016: A year in review of basic science and clinical science. Stroke and Vascular Neurology. 2017 Dec 1;2(4):222-9.
24. Lord AS, Lewis A, Czeisler B, Ishida K, Torres J, Kamel H, et al. Majority of 30-day readmissions after intracerebral hemorrhage are related to infections. Stroke. 2016 Jul;47(7):1768-71.
25. Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nature Reviews Neuroscience. 2005 Oct 1;6(10):775-86.
26. Zera KA, Buckwalter MS. The local and peripheral immune responses to stroke: implications for therapeutic development. Neurotherapeutics. 2020 Apr;17(2):414-35.
27. DeLong JH, Ohashi SN, O’Connor KC, Sansing LH. Inflammatory responses after ischemic stroke. Seminars in Immunopathology. 2022 Sep;44(5):625-48.
28. Iadecola C, Buckwalter MS, Anrather J. Immune responses to stroke: mechanisms, modulation, and therapeutic potential. The Journal of Clinical Investigation. 2020 Jun 1;130(6):2777-88.
29. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. Journal of Cerebral Blood Flow & Metabolism. 2012 Sep;32(9):1677-98.
30. Xu S, Lu J, Shao A, Zhang JH, Zhang J. Glial cells: role of the immune response in ischemic stroke. Frontiers in Immunology. 2020 Feb 26;11:294.
31. Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014 Aug;62(8):1227-40.
32. Mifsud G, Zammit C, Muscat R, Di Giovanni G, Valentino M. Oligodendrocyte pathophysiology and treatment strategies in cerebral ischemia. CNS Neuroscience & Therapeutics. 2014 Jul;20(7):603-12.
33. Anrather J, Iadecola C. Inflammation and stroke: an overview. Neurotherapeutics. 2016 Oct;13:661-70.
34. Wanrooy BJ, Wen SW, Shim R, Wilson JL, Prame Kumar K, Wong CH. Brain-associated innate leukocytes display diverse inflammatory states following experimental stroke. Immunology and Cell Biology. 2022 Aug;100(7):482-96.
35. Petrone AB, Eisenman RD, Steele KN, Mosmiller LT, Urhie O, Zdilla MJ. Temporal dynamics of peripheral neutrophil and lymphocytes following acute ischemic stroke. Neurological Sciences. 2019 Sep 1;40:1877-85.
36. Otxoa-de-Amezaga A, Miró-Mur F, Pedragosa J, Gallizioli M, Justicia C, Gaja-Capdevila N, et al. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathologica. 2019 Feb 11;137:321-41.
37. Xiao W, Guo S, Chen L, Luo Y. The role of Interleukin-33 in the modulation of splenic T-cell immune responses after experimental ischemic stroke. Journal of Neuroimmunology. 2019 Aug 15;333:576970.
38. Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 2019 Jan 10;565(7738):246-50.
39. Woiciechowsky C, Schöning B, Cobanov J, Lanksch WR, Volk HD, Döcke WD. Early IL-6 plasma concentrations correlate with severity of brain injury and pneumonia in brain-injured patients. Journal of Trauma and Acute Care Surgery. 2002 Feb 1;52(2):339-45.
40. Cruse JM, Lewis RE, Bishop GR, Kliesch WF, Gaitan E. Neuroendocrine-immune interactions associated with loss and restoration of immune system function in spinal cord injury and stroke patients. Immunologic Research. 1992 Jun;11:104-16.
41. Członkowska A, Cyrta B, Korlak J. Immunological observations on patients with acute cerebral vascular disease. Journal of the Neurological Sciences. 1979 Nov 1;43(3):455-64.
42. Meert KL, Long M, Kaplan J, Sarnaik AP. Alterations in immune function following head injury in children. Critical Care Medicine. 1995 May 1;23(5):822-8.
43. Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. Journal of Cerebral Blood Flow & Metabolism. 2015 Jun;35(6):888-901.
44. Ruf W, Ruggeri ZM. Neutrophils release brakes of coagulation. Nature Medicine. 2010 Aug;16(8):851-2.
45. Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F. Pathophysiological role of neutrophils in acute myocardial infarction. Thrombosis and Haemostasis. 2013;110(09):501-14.
46. Mócsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. Journal of Experimental Medicine. 2013 Jul 1;210(7):1283-99.
47. Ross AM, Hurn P, Perrin N, Wood L, Carlini W, Potempa K. Evidence of the peripheral inflammatory response in patients with transient ischemic attack. Journal of Stroke and Cerebrovascular Diseases. 2007 Sep 1;16(5):203-7.
48. Gökhan S, Ozhasenekler A, Durgun HM, Akil E, Ustündag M, Orak M. Neutrophil lymphocyte ratios in stroke subtypes and transient ischemic attack. European Review for Medical and Pharmacological Sciences. 2013 Mar;17(5):653-7.
49. Tsai NW, Chang WN, Shaw CF, Jan CR, Lu CH. Leucocyte apoptosis in patients with acute ischaemic stroke. Clinical and Experimental Pharmacology and Physiology. 2010 Sep;37(9):884-8.
50. Copin JC, Da Silva RF, Fraga-Silva RA, Capettini L, Quintao S, Lenglet SB, et al. Treatment with Evasin-3 reduces atherosclerotic vulnerability for ischemic stroke, but not brain injury in mice. Journal of Cerebral Blood Flow & Metabolism. 2013 Apr;33(4):490-8.
51. Villa P, Triulzi S, Cavalieri B, Di Bitondo R, Bertini R, Barbera S, et al. The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Molecular Medicine. 2007 Mar;13(3):125-33.
52. Brait VH, Rivera J, Broughton BR, Lee S, Drummond GR, Sobey CG. Chemokine-related gene expression in the brain following ischemic stroke: no role for CXCR2 in outcome. Brain Research. 2011 Feb 4;1372:169-79.
53. Woiciechowsky C, Asadullah K, Nestler D, Eberhardt B, Platzer C, Schöning B, et al. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nature Medicine. 1998 Jul 1;4(7):808-13.
54. Campagnolo DI, Bartlett JA, Keller SE, Sanchez W, Oza R. Impaired phagocytosis of staphylococcus aureus in complete tetraplegics1. American Journal of Physical Medicine & Rehabilitation. 1997 Jul 1;76(4):276-80.
55. Cheng K, He M, Shu Q, Wu M, Chen C, Xue Y. Analysis of the risk factors for nosocomial bacterial infection in patients with COVID-19 in a tertiary hospital. Risk Management and Healthcare Policy. 2020 Nov 13:2593-9.
56. Zola H. T cell memory: A role for MHC class II molecules on T cells?. Immunology and Cell Biology. 1992 Oct;70(5):337-41.
57. Asadullah K, Woiciechowsky C, Docke WD, Liebenthal C, Wauer H, Kox W, et al. Immunodepression following neurosurgical procedures. Critical Care Medicine. 1995 Dec 1;23(12):1976-83.
58. Prass K, Meisel C, Höflich C, Braun J, Halle E, Wolf T, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1–like immunostimulation. The Journal of Experimental Medicine. 2003 Sep 1;198(5):725-36.
59. Arumugam TV, Woodruff TM, Lathia JD, Selvaraj PK, Mattson MP, Taylor SM. Neuroprotection in stroke by complement inhibition and immunoglobulin therapy. Neuroscience. 2009 Feb 6;158(3):1074-89.
60. Dietzschold B, Schwaeble W, Schäfer MK, Hooper DC, Zehng YM, Petry F, et al. Expression of C1q, a subcomponent of the rat complement system, is dramatically enhanced in brains of rats with either Borna disease or experimental allergic encephalomyelitis. Journal of the Neurological Sciences. 1995 May 1;130(1):11-6.
61. Goldsmith SK, Wals P, Rozovsky I, Morgan TE, Finch CE. Kainic acid and decorticating lesions stimulate the synthesis of C1q protein in adult rat brain. Journal of Neurochemistry. 1997 May;68(5):2046-52.
62. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS Synapse Elimination. Cell. 2007 Dec 14;131(6):1164-78.
63. Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. Journal of Neurochemistry. 2010 Feb;112(3):733-43.
64. De Simoni MG, Rossi E, Storini C, Pizzimenti S, Echart C, Bergamaschini L. The powerful neuroprotective action of C1-inhibitor on brain ischemia-reperfusion injury does not require C1q. The American Journal of Pathology. 2004 May 1;164(5):1857-63.
65. Tamam Y, Iltumur K, Apak I. Assessment of acute phase proteins in acute ischemic stroke. The Tohoku Journal of Experimental Medicine. 2005;206(2):91-8.
66. Cojocaru IM, Cojocaru M, Tanasescu R, Burcin CE, Atanasiu AN, Petrescu AM, et al. Changes in plasma levels of complement in patients with acute ischemic stroke. Rom J Intern Med. 2008 Jan 1;46(1):77-80.
67. Stokowska A, Olsson S, Holmegaard L, Jood K, Blomstrand C, Jern C, et al. Plasma C3 and C3a levels in cryptogenic and large-vessel disease stroke: associations with outcome. Cerebrovascular Diseases. 2011 Jun 28;32(2):114-22.
68. Ma Y, Liu Y, Zhang Z, Yang GY. Significance of complement system in ischemic stroke: a comprehensive review. Aging and Disease. 2019 Apr;10(2):429.
69. Alawieh A, Elvington A, Zhu H, Yu J, Kindy MS, Atkinson C, et al. Modulation of post-stroke degenerative and regenerative processes and subacute protection by site-targeted inhibition of the alternative pathway of complement. Journal of Neuroinflammation. 2015 Dec;12(1):247.
70. Lipton P. Ischemic cell death in brain neurons. Physiological Reviews. 1999 Jan 10;79(4):1431-568.
71. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nature Reviews Neuroscience. 2003 May;4(5):399-415.
72. Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. International Journal of Stroke. 2012 Jul;7(5):378-85.
73. Bruno V, Battaglia G, Copani A, D'Onofrio M, Di Iorio P, De Blasi A, et al. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. Journal of Cerebral Blood Flow & Metabolism. 2001 Sep;21(9):1013-33.
74. Ginsberg MD. Current status of neuroprotection for cerebral ischemia: synoptic overview. Stroke. 2009 Mar 1;40(3_suppl_1):S111-4.
75. O'Collins VE, Macleod MR, Donnan GA, Horky LL, Van Der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Annals of Neurology. 2006 Mar;59(3):467-77.
76. Kliper E, Bashat DB, Bornstein NM, Shenhar-Tsarfaty S, Hallevi H, Auriel E, et al. Cognitive decline after stroke: relation to inflammatory biomarkers and hippocampal volume. Stroke. 2013 May;44(5):1433-5.
77. Singh V, Roth S, Llovera G, Sadler R, Garzetti D, Stecher B, et al. Microbiota dysbiosis controls the neuroinflammatory response after stroke. Journal of Neuroscience. 2016 Jul 13;36(28):7428-40.
78. Yu H, Cai Y, Zhong A, Zhang Y, Zhang J, Xu S. The “Dialogue” between central and peripheral immunity after ischemic stroke: Focus on spleen. Frontiers in Immunology. 2021 Dec 16;12:792522.
79. Vahidy FS, Parsha KN, Rahbar MH, Lee M, Bui TT, Nguyen C, et al. Acute splenic responses in patients with ischemic stroke and intracerebral hemorrhage. Journal of Cerebral Blood Flow & Metabolism. 2016 Jun;36(6):1012-21.
80. Rosas-Ballina M, Tracey KJ. The neurology of the immune system: neural reflexes regulate immunity. Neuron. 2009 Oct 15;64(1):28-32.
81. Wong CH, Jenne CN, Lee WY, Léger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science. 2011 Oct 7;334(6052):101-5.
82. Nous A, Peeters I, Nieboer K, Vanbinst AM, De Keyser J, De Raedt S. Post-stroke infections associated with spleen volume reduction: a pilot study. PLoS One. 2020 May 11;15(5):e0232497.
83. Sheu TT, Chiang BL. Lymphopenia, lymphopenia-induced proliferation, and autoimmunity. International Journal of Molecular Sciences. 2021 Apr 16;22(8):4152.
84. Stott DJ, Falconer A, Miller H, Tilston JC, Langhorne P. Urinary tract infection after stroke. QJM: An International Journal of Medicine. 2009 Apr 1;102(4):243-9.
85. Hannawi Y, Hannawi B, Rao CP, Suarez JI, Bershad EM. Stroke-associated pneumonia: major advances and obstacles. Cerebrovascular Diseases. 2013 May 31;35(5):430-43.
86. Bansal S, Sangha KS, Khatri P. Drug treatment of acute ischemic stroke. American Journal of Cardiovascular Drugs. 2013 Feb;13:57-69.
87. Khaja AM, Grotta JC. Established treatments for acute ischaemic stroke. The Lancet. 2007 Jan 27;369(9558):319-30.
88. Albers GW. Expanding the window for thrombolytic therapy in acute stroke: the potential role of acute MRI for patient selection. Stroke. 1999 Oct;30(10):2230-7.
89. Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. New England Journal of Medicine. 2008 Sep 25;359(13):1317-29.
90. Lansberg MG, Bluhmki E, Thijs VN. Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours after acute ischemic stroke: a metaanalysis. Stroke. 2009 Jul 1;40(7):2438-41.
91. dela Peña I, Borlongan C, Shen G, Davis W. Strategies to extend thrombolytic time window for ischemic stroke treatment: an unmet clinical need. Journal of Stroke. 2017 Jan;19(1):50-60.
92. Li Y, McBride DW, Tang Y, Doycheva D, Zhang JH, Tang Z. Immunotherapy as a Treatment for Stroke: Utilizing Regulatory T Cells. Brain Hemorrhages. 2023 Feb 16.
93. Gravanis I, Tsirka SE. Tissue-type plasminogen activator as a therapeutic target in stroke. Expert Opinion on Therapeutic Targets. 2008 Feb 1;12(2):159-70.
94. National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. New England Journal of Medicine. 1995 Dec 14;333(24):1581-8.
95. Balasubramaian A, Mitchell P, Dowling R, Yan B. Evolution of endovascular therapy in acute stroke: implications of device development. Journal of Stroke. 2015 May;17(2):127.
96. IMS Study Investigators. Combined intravenous and intra-arterial recanalization for acute ischemic stroke: the Interventional Management of Stroke Study. Stroke. 2004 Apr;35(4):904-11.
97. Bhatia R, Hill MD, Shobha N, Menon B, Bal S, Kochar P, et al. Low rates of acute recanalization with intravenous recombinant tissue plasminogen activator in ischemic stroke: real-world experience and a call for action. Stroke. 2010 Oct 1;41(10):2254-8.
98. Papanagiotou P, Ntaios G. Endovascular thrombectomy in acute ischemic stroke. Circulation: Cardiovascular Interventions. 2018 Jan;11(1):e005362.
99. Nogueira RG, Liebeskind DS, Sung G, Duckwiler G, Smith WS. Predictors of good clinical outcomes, mortality, and successful revascularization in patients with acute ischemic stroke undergoing thrombectomy: pooled analysis of the Mechanical Embolus Removal in Cerebral Ischemia (MERCI) and Multi MERCI Trials. Stroke. 2009 Dec 1;40(12):3777-83.
100. Jovin TG, Chamorro A, Cobo E, de Miquel MA, Molina CA, Rovira A, et al. Thrombectomy within 8 hours after symptom onset in ischemic stroke. New England Journal of Medicine. 2015 Jun 11;372(24):2296-306.
101. Campbell BC, Mitchell PJ, Kleinig TJ, Dewey HM, Churilov L, Yassi N, et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. New England Journal of Medicine. 2015 Mar 12;372(11):1009-18.
102. Saver JL, Goyal M, Bonafe A, Diener HC, Levy EI, Pereira VM, et al. Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. New England Journal of Medicine. 2015 Jun 11;372(24):2285-95.
103. Goyal M, Menon BK, Van Zwam WH, Dippel DW, Mitchell PJ, Demchuk AM, et al. Endovascular thrombectomy after large-vessel ischaemic stroke: a meta-analysis of individual patient data from five randomised trials. The Lancet. 2016 Apr 23;387(10029):1723-31.
104. Bracard S, Ducrocq X, Mas JL, Soudant M, Oppenheim C, Moulin T, et al. Mechanical thrombectomy after intravenous alteplase versus alteplase alone after stroke (THRACE): a randomised controlled trial. The Lancet Neurology. 2016 Oct 1;15(11):1138-47.
105. Broderick JP, Palesch YY, Demchuk AM, Yeatts SD, Khatri P, Hill MD, et al. Endovascular therapy after intravenous t-PA versus t-PA alone for stroke. The New England Journal of Medicine. 2013 Mar 7;368(10):893-903.
106. Neuberger U, Kickingereder P, Schönenberger S, Schieber S, Ringleb PA, Bendszus M, et al. Risk factors of intracranial hemorrhage after mechanical thrombectomy of anterior circulation ischemic stroke. Neuroradiology. 2019 Apr 12;61:461-9.
107. Smith WS, Sung G, Starkman S, Saver JL, Kidwell CS, Gobin YP, et al. Safety and efficacy of mechanical embolectomy in acute ischemic stroke: results of the MERCI trial. Stroke. 2005 Jul 1;36(7):1432-8.
108. Kleindorfer DO, Towfighi A, Chaturvedi S, Cockroft KM, Gutierrez J, Lombardi-Hill D, et al. 2021 guideline for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline from the American Heart Association/American Stroke Association. Stroke. 2021 Jul;52(7):e364-467.
109. Qiu S, Xu Y. Guidelines for acute ischemic stroke treatment. Neuroscience Bulletin. 2020 Oct;36(10):1229-32.
110. Johnston SC, Amarenco P, Albers GW, Denison H, Easton JD, Evans SR, et al. Ticagrelor versus aspirin in acute stroke or transient ischemic attack. New England Journal of Medicine. 2016 Jul 7;375(1):35-43.
111. Barber PA, Zhang J, Demchuk AM, Hill MD, Buchan AM. Why are stroke patients excluded from TPA therapy?: An analysis of patient eligibility. Neurology. 2001 Apr 24;56(8):1015-20.
112. Messé SR, Khatri P, Reeves MJ, Smith EE, Saver JL, Bhatt DL, et al. Why are acute ischemic stroke patients not receiving IV tPA?: Results from a national registry. Neurology. 2016 Oct 11;87(15):1565-74.
113. Yu CY, Ng G, Liao P. Therapeutic antibodies in stroke. Translational Stroke Research. 2013 Oct;4:477-83.
114. Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, et al. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000 Jan 27;403(6768):434-9.
115. Seymour AB, Andrews EM, Tsai SY, Markus TM, Bollnow MR, Brenneman MM, et al. Delayed treatment with monoclonal antibody IN-1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. Journal of Cerebral Blood Flow & Metabolism. 2005 Oct;25(10):1366-75.
116. Rust R, Grönnert L, Gantner C, Enzler A, Mulders G, Weber RZ, et al. Nogo-A targeted therapy promotes vascular repair and functional recovery following stroke. Proceedings of the National Academy of Sciences. 2019 Jul 9;116(28):14270-9.
117. Cash D, Easton AC, Mesquita M, Beech J, Williams S, Lloyd A, et al. GSK249320, a monoclonal antibody against the axon outgrowth inhibition molecule myelin-associated glycoprotein, improves outcome of rodents with experimental stroke. Journal of Neurology and Experimental Neuroscience. 2016;2(2):28-33.
118. Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, et al. Nasal vaccination with myelin oligodendrocyte glycoprotein reduces stroke size by inducing IL-10-producing CD4+ T cells. The Journal of Immunology. 2003 Dec 15;171(12):6549-55.
119. Edwards DN, Bix GJ. The inflammatory response after ischemic stroke: targeting β2 and β1 integrins. Frontiers in Neuroscience. 2019 May 28;13:540.
120. Tarkowski E, Rosengren L, Blomstrand C, Wikkelsö C, Jensen C, Ekholm S, et al. Early intrathecal production of interleukin-6 predicts the size of brain lesion in stroke. Stroke. 1995 Aug;26(8):1393-8.
121. Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood, The Journal of the American Society of Hematology. 2012 Nov 1;120(18):3793-802.
122. Cai W, Shi L, Zhao J, Xu F, Dufort C, Ye Q, et al. Neuroprotection against ischemic stroke requires a specific class of early responder T cells in mice. Journal of Clinical Investigation. 2022 Aug 1;132(15):e157678.
123. Becker KJ. Sensitization and tolerization to brain antigens in stroke. Neuroscience. 2009 Feb 6;158(3):1090-7.
124. Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke. 2008 May 1;39(5):1575-82.
125. Shi L, Sun Z, Su W, Xu F, Xie D, Zhang Q, et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity. 2021 Jul 13;54(7):1527-42.
126. Ren G, Roberts AI, Shi Y. Adhesion molecules. Cell Adh Migr. 2011 2011;5(1):20-22.
127. Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. American Journal of Physiology-Heart and Circulatory Physiology. 2004 Dec;287(6):H2555-60.
128. Zhang A, Liu Y, Wang X, Xu H, Fang C, Yuan L, et al. Clinical Potential of Immunotherapies in Subarachnoid Hemorrhage Treatment: Mechanistic Dissection of Innate and Adaptive Immune Responses. Aging and Disease. 2023 Jan 29.
129. Enlimomab Acute Stroke Trial Investigators. Use of anti-ICAM-1 therapy in ischemic stroke: results of the Enlimomab Acute Stroke Trial. Neurology. 2001 Oct 23;57(8):1428-34.
130. Bowes MP, Rothlein R, Fagan SC, Zivin JA. Monoclonal antibodies preventing leukocyte activation reduce experimental neurologic injury and enhance efficacy of thrombolytic therapy. Neurology. 1995 Apr 1;45(4):815-9.
131. Suzuki H, Abe K, Tojo SJ, Kitagawa H, Kimura K, Mizugaki M, et al. Reduction of ischemic brain injury by anti-P-selectin monoclonal antibody after permanent middle cerebral artery occlusion in rat. Neurological Research. 1999 Apr 1;21(3):269-76.
132. Suzuki H, Hayashi T, Tojo SJ, Kitagawa H, Kimura K, Mizugaki M, Itoyama Y, Abe K. Anti-P-selectin antibody attenuates rat brain ischemic injury. Neuroscience letters. 1999 Apr 23;265(3):163-6.
133. National Institute of Neurological D, Stroke. Induction of Mucosal Tolerance to E-Selectin for the Secondary Prevention of Stroke. Clinical trial registration. 2010. NCT00012454. 2010/05/03/. Accessed 2023/07/21/. https://clinicaltrials.gov/study/NCT00012454
134. Chen Y, Ruetzler C, Pandipati S, Spatz M, McCarron RM, Becker K, et al. Mucosal tolerance to E-selectin provides cell-mediated protection against ischemic brain injury. Proceedings of the National Academy of Sciences. 2003 Dec 9;100(25):15107-12.
135. Takeda H, Spatz M, Ruetzler C, McCarron R, Becker K, Hallenbeck J. Induction of mucosal tolerance to E-selectin prevents ischemic and hemorrhagic stroke in spontaneously hypertensive genetically stroke-prone rats. Stroke. 2002 Sep 1;33(9):2156-64.
136. Bednar M, Gross C, Russell S, Fuller S, Ellenberger C, Schindler E, et al. Humanized anti-L-selectin monoclonal antibody DREC200 therapy in acute thromboembolic stroke. Neurological research. 1998 Jul 1;20(5):403-8.
137. Becker K, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the α4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001 Jan;32(1):206-11.
138. Langhauser F, Kraft P, Göb E, Leinweber J, Schuhmann MK, Lorenz K, et al. Blocking of α4 integrin does not protect from acute ischemic stroke in mice. Stroke. 2014 Jun;45(6):1799-806.
139. Elkind MS, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Natalizumab in acute ischemic stroke (ACTION II): a randomized, placebo-controlled trial. Neurology. 2020 Aug 25;95(8):e1091-104.
140. Fu Y, Zhang N, Ren LI, Yan Y, Sun N, Li YJ, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proceedings of the National Academy of Sciences. 2014 Dec 23;111(51):18315-20.
141. Mandeville ET, Li W, Quinto-Alemany D, Zhang F, Esposito E, Nakano T, et al. Fingolimod Does Not Reduce Infarction After Focal Cerebral Ischemia in Mice During Active or Inactive Circadian Phases. Stroke. 2022 Dec;53(12):3741-50.
142. Czech B, Pfeilschifter W, Mazaheri-Omrani N, Strobel MA, Kahles T, Neumann-Haefelin T, et al. The immunomodulatory sphingosine 1-phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochemical and biophysical research communications. 2009 Nov 13;389(2):251-6.
143. Kraft P, Göb E, Schuhmann MK, Göbel K, Deppermann C, Thielmann I, et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke. 2013 Nov;44(11):3202-10.
144. Dang C, Lu Y, Li Q, Wang C, Ma X. Efficacy of the sphingosine-1-phosphate receptor agonist fingolimod in animal models of stroke: an updated meta-analysis. International Journal of Neuroscience. 2021 Jan 2;131(1):85-94.
145. Wei Y, Yemisci M, Kim HH, Yung LM, Shin HK, Hwang SK, et al. Fingolimod provides long‐term protection in rodent models of cerebral ischemia. Annals of neurology. 2011 Jan;69(1):119-29.
146. Brait VH, Tarrasón G, Gavaldà A, Godessart N, Planas AM. Selective sphingosine 1-phosphate receptor 1 agonist is protective against ischemia/reperfusion in mice. Stroke. 2016 Dec;47(12):3053-6.
147. Schmidt‐Pogoda A, Bonberg N, Koecke MH, Strecker JK, Wellmann J, Bruckmann NM, et al. Why most acute stroke studies are positive in animals but not in patients: a systematic comparison of preclinical, early phase, and phase 3 clinical trials of neuroprotective agents. Annals of neurology. 2020 Jan;87(1):40-51.