Keywords
IBD, IL-10, Anti-TNF, Macrophages, Colitis
Macrophages, IL-10 and Anti-TNF Therapy in IBD
Inflammatory bowel disease (IBD) is a multifactorial disease in which both genetic and environmental factors play an important role, although the precise cause remains obscure. It is clear that both the innate and adaptive immunity are involved in acquiring mucosal immune homeostasis in the intestine, which is dysregulated in IBD.
Interleukin-10 & IBD
Several studies have pinpointed Interleukin(IL)-10 signaling as a key player in the maintenance of intestinal mucosal immune homeostasis. IL-10 is a potent antiinflammatory cytokine that is produced by a wide variety of different cell types, like T-cells, macrophages, dendritic cells and epithelial cells [1]. Most hematopoietic cells can respond to IL-10 via expression of the IL-10 receptor (IL- 10 R), which is a heterodimer composed of IL-10 Rα and IL-10 Rβ [2]. Macrophages are among the cells that express the highest level of IL-10 R. Deleterious mutations in IL-10 or IL-10 R cause severe early onset IBD [3]. This situation is recapitulated in mice where deficiencies in either IL-10 or IL-10 R cause spontaneous colitis [4,5]. Also in genomewide association studies, single-nucleotide polymorphisms (SNPs) in the IL-10 pathway have been linked to the risk of developing IBD [6].
While initially it was assumed that IL-10 suppressed inflammatory responses by directly affecting T-cells [7], further experimental studies pointed out a pivotal role for IL-10 signaling within the myeloid compartment as the absence of IL-10 Rα in intestinal macrophages resulted in spontaneous colitis [8,9]. Therefore, IL-10 signaling within macrophages plays a central role in the regulation of intestinal mucosal homeostasis.
Recombinant IL-10 as a therapeutic strategy has been tested in various forms. Thus far, efforts to treat IBD patients with recombinant IL-10 systemically have not shown convincing evidence of therapeutic efficacy [10-13]. However, against the background of recent evidence that the primary issue in IBD seems to be a defect in IL-10 sensing rather than availability, administration of IL-10 is less likely to be effective, unless strategies to increase responsiveness will be developed.
Macrophages & IBD
Macrophages are part of the myeloid compartment and can orchestrate all stages of the immune response: 1) as a first line of defense against bacteria, 2) in the resolution of inflammation, and finally 3) in wound healing and tissue repair [14]. Macrophages are plastic cells that can adapt their phenotype to environmental stimuli. Macrophages have classically been subdivided into either a pro-inflammatory (M1) or a regulatory (M2) subtype, but it is now generally accepted that macrophages can adopt a spectrum of phenotypes in vivo as many different stimuli can influence their behavior [15]. In contrast to other organs, where tissue-resident macrophages are derived from the yolk-sac during embryogenesis, most macrophages in the intestine are constantly replenished by an influx of bone marrow derived monocytes [16]. In the intestine, macrophages are continuously exposed to microbial components that can potentially evoke an immune response. To prevent constant inflammation, they possess the capacity to clear pathogens and scavenge cell debris without further alerting other players of the immune cascade. This tolerogenic state is marked by a diminished production of inflammatory factors like nitric oxide, reactive oxygen species and proinflammatory cytokines, despite the presence of microbial stimuli [17,18]. This anergic state is thought to be actively maintained via inhibitory molecules like IL-10 [19]. Thus, while retaining their phagocytic and bactericidal capacity, intestinal macrophages play a crucial role in preventing an excessive immune response against the microbiome. In active IBD the balance between these seemingly opposing tasks is disrupted, leading to an excessive inflammatory cascade. Single-cell analysis confirmed the abundance of an inflammatory macrophage subset in active ileal Crohn’s disease (CD) compared to uninflamed lesions [20]. This subset was characterized by an increased expression of nuclear factor κB (NF-κB) and inflammatory molecules like IL-1, IL-6, IL-23, TNFα and Oncostatin M (OSM). A similar study for colonic CD showed that inflammatory CD14+CD64hi CD163-/dim IL-23 producing macrophages were enriched in inflamed lesions compared to uninflamed lesions [21]. Thus, during active disease the larger proportion of CD macrophages displays an inflammatory phenotype while during disease remission regulatory subsets prevail. Therefore, the interest in reprogramming macrophages to resolve inflammation in a therapeutic setting in IBD has grown over the last years.
Anti-TNF therapy in IBD
Anti-Tumor Necrosis Factor-(TNF)-α monoclonal antibodies (mAbs) are considered a breakthrough in the therapeutic treatment of IBD in clinical practice. Interestingly, a range of clinical observations has shown that only the full mAbs against TNF, adalumimab and infliximab, have a substantial therapeutic effect in IBD, in contrast to other anti-TNF agents like certolizumab (pegylated Fab’ fragment), onercept (soluble TNF receptor I) and etanercept (TNF receptor II-Fc fusion protein) [22]. While these TNF blocking strategies established their value in several immune mediated diseases like Rheumatoid Arthritis (RA), only the full mAbs demonstrated induction of complete endoscopic remission in IBD. This suggests that additional features besides TNF neutralization are necessary for therapeutic efficacy and for the induction of mucosal healing in IBD. The Fc region of an antibody can bind to Fcγ receptors (FcγR) which are highly expressed by myeloid cells. Over the last decade we have found strong evidence for FcγR dependent macrophage modulation as a core mechanism of action for the efficacy of anti-TNF mAbs in IBD. The hypothesis that the Fc region might play a role in the therapeutic effect of anti-TNFs originated from unsuccessful experiments to find direct effects of anti-TNFs on T-cells, in particular induction of apoptosis [23]. It wasn’t until we used a mixed lymphocyte reaction (MLR), created by mixing peripheral blood mononuclear cells (PBMCs) of two different donors, where monocytes of one donor activate the T-cells of the other donor, that we found an effect of anti-TNFs. In this set-up only the anti-TNF mAbs (infliximab, adalimumab and golimumab) inhibited T-cell proliferation, in contrast to other TNF blocking agents like etanercept or certolizumab. The effect was completely dependent on the Fc-mediated polarization of the monocytes in the MLR to an ‘M2- like’ regulatory macrophage phenotype that suppressed T-cell proliferation [23]. We were subsequently able to reproduce the dependence on the Fc region in preclinical experiments in vivo. First, we showed that an anti-TNF mAb with high Fc binding affinity was effective in reducing colitis in contrast to an anti-TNF mAb with a low Fc binding affinity in an experimental IBD mouse model, while both mAbs were equally effective in an experimental model of RA [24]. This showed that Fc interaction was necessary for therapeutic efficacy in the IBD model but not in the model for RA, where reducing arthritis seemed solely dependent on TNF blockade. We found that mice lacking activating FcγR were completely unresponsive to anti-TNF mAb therapy in the same IBD model, while an afucosylated anti-TNF mAb with increased affinity for FcγR showed increased efficacy [25]. In both mice and humans Fc engagement by anti-TNF promoted the formation of regulatory macrophages. Characterization of the intestinal myeloid compartment of mice with active colitis showed an increase in CD206+ regulatory macrophages after anti- TNF treatment compared to treatment with an isotype control antibody. The number of CD206+ macrophages was even higher with an afucosylated anti-TNF and, conversely, percentages remained low in anti-TNF treated mice that were deficient for activating FcγR [25]. Likewise, the number of CD206+ macrophages increased in the lamina propria of IBD patients responding to anti-TNF mAb therapy. In contrast, this was not the case in anti- TNF non-responders [26].
Further characterization of regulatory features showed that anti-TNF mAbs increased the ratio of IL- 10 versus IL-12/IL-23p40 production in inflammatory macrophages differentiated in vitro [27]. Similarly, anti- TNF mAbs suppressed IL-12/IL-23 p40 in lamina propria macrophages isolated from IBD patients with active disease.
Overall, these data strongly suggest that Fc engagement of activating Fc receptors polarizes intestinal macrophages to an ‘M2-like’ regulatory phenotype and that this is required for therapeutic efficacy.
Anti-TNF therapy effectiveness depends on IL-10 signaling in macrophages in IBD
In our recent paper regarding anti-TNF in IBD we show that IL-10 signaling in macrophages is necessary for CD206+ macrophage induction and for response to anti- TNF mAbs in experimental colitis. Several reports describe patients with germline mutations in the IL-10 pathway to be unresponsive to anti-TNF [3,28]. Interestingly, although IL-10 deficient mice were refractory to anti-TNF therapy as shown previously [29], they did respond to antibodies against IL-12/IL-23p40. Perhaps macrophages become refractory to Fc engagement when IL-10 signaling is disrupted, locking them in an IL-12/IL-23 secreting inflammatory state that can only be blocked further downstream.
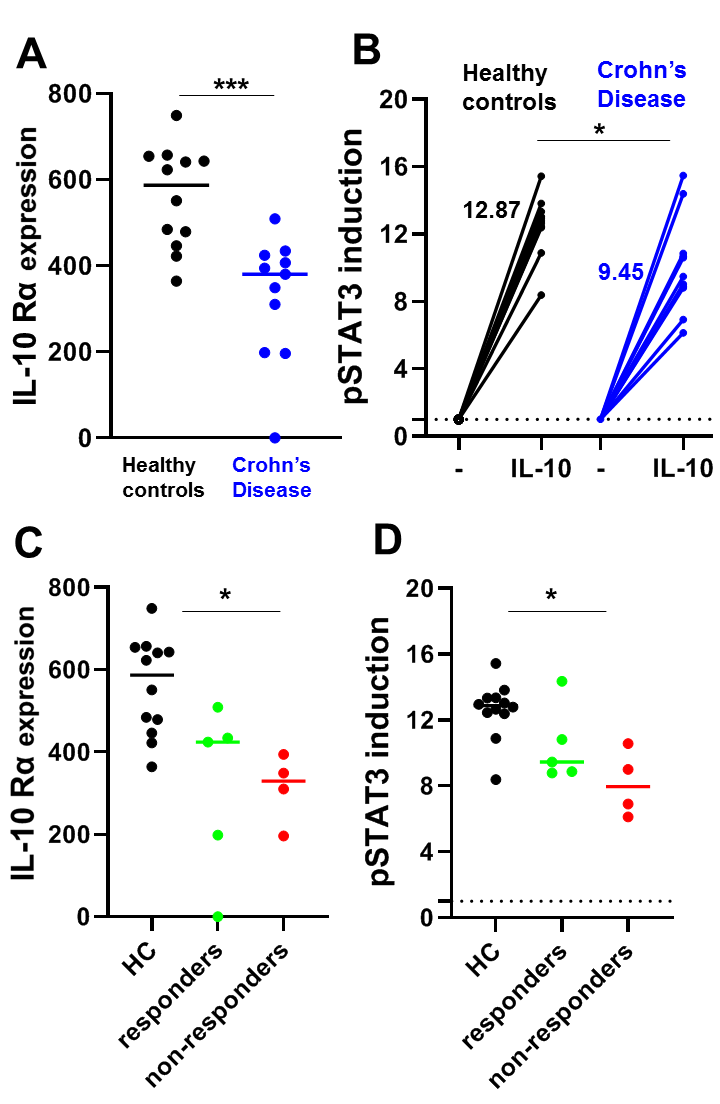
A study in pediatric CD patients showed that monocytes of patients were less responsive to IL-10 compared to healthy controls presumably due to the reduced expression of IL- 10 Rα [30]. We confirmed this finding in adult CD patients. We found that the expression of IL-10 Rα was lower in CD14+ monocytes of CD patients compared to healthy controls in peripheral blood obtained before the start of anti-TNF therapy (Figure 1A). Indeed, by performing flow cytometry for pSTAT3 in combination with CD14 in PBMCs with or without IL-10 stimulation, we also found a decreased IL-10 responsiveness in the monocytes of these CD patients compared to healthy controls (Figure 1B). Although the patient numbers were low, we found that both the pretreatment responsiveness to IL-10 and IL10 Rα expression levels of CD14+ monocytes from anti-TNF non-responding CD patients were significantly decreased compared to healthy controls, which was not the case for anti-TNF responsive CD patients (Figure 1C/D), indicating this could potentially be used as a predictive marker for anti- TNF response. Future research could focus on unravelling mechanisms that maintain IL-10 unresponsiveness.
Materials & Methods
CD patient study
CD patients with complaints of active disease, scheduled for colonoscopy to determine disease activity, were asked to participate with written informed consent before new anti- TNF remission therapy was initiated. Endoscopic response to induction therapy was determined by endoscopy after 8 weeks. During colonoscopy the Crohn’s Disease Endoscopic Index of Severity (CDEIS) was determined and response was defined as >50 % reduction in CDEIS after 8 weeks of induction therapy with either adalumimab or infliximab. Two patients turned out to be therapy intolerant and therefore no response was determined. For patient information see Table 1. Prior to treatment initiation a blood sample was taken, and PBMCs were isolated by Ficoll Paque density-gradient centrifugation. After cell isolation, samples were cryopreserved with DMSO using Mr Frosty (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and afterwards stored in liquid nitrogen. Healthy volunteers were also asked to participate with written informed consent. The study protocol was approved by the Medical Ethical Committee of the AMC (Amsterdam UMC, METC09/113#09.17.0938)
| Patient | Gender | Age at inclusion (years) | CDEIS (start) | CDEIS (8 weeks) | CRP (start) | CRP (8 weeks) | Montreal classification* | anti-TNF | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| A1/A2/A3 | L1/L2/L3/L4 | B1/B2/B3/+p | |||||||||
| Responders | 1 | F | 52 | 16 | 1.67 | 20.2 | 4.1 | 2 | 1 | 1 | ADA |
| 2 | F | 30 | 16 | 0 | 27.2 | 1.1 | 2 | 2 | 1 | IFX | |
| 3 | F | 23 | 37 | 0 | 116.2 | 0.7 | 2 | 3 | 1 | IFX | |
| 4 | F | 51 | 10 | 0 | 4.4 | 0.7 | 2 | 3 | 2 | ADA | |
| 5 | M | 32 | 9.2 | 3.4 | 41.1 | 2.2 | 2 | 3 | 1 | IFX | |
| Non- responders | 6 | F | 31 | 5.2 | 6.4 | 17.4 | 15.8 | 2 | 1 | 1 | ADA |
| 7 | F | 23 | 19 | 19.8 | 90.0 | 43.3 | 2 | 3 | 2 | IFX | |
| 8 | M | 61 | 4 | 8 | 1.3 | 1.0 | 2 | 3 | 2 | ADA | |
| 9 | F | 56 | 8.4 | 7.8 | 2.5 | 1.9 | 3 | 1 | 2 | ADA | |
| 10 | M | 37 | 4.2 | - | 1.2 | - | 2 | 1 | 2 | IFX | |
| 11 | F | 23 | 5.2 | - | 4.2 | - | 1 | 1 | 1+p | IFX | |
|
CDEIS: Crohn’s Disease Endoscopic Index of Severity; CRP: C-reactive Protein (mg/L); * Montreal classification: Age at diagnosis: A1: <17 years, A2: 17–40 years, A3: >40 years; Maximal location of disease: L1: ileal, L2: colonic, L3: ileocolonic, L4: upper disease; Maximal disease behaviour: B1: non-stricturing, non-penetrating, B2: stricturing, B3: penetrating, p: perianal disease, IFX: Infliximab, ADA: Adalimumab |
|||||||||||
Flow cytometry
PBMCs were thawed and washed with prewarmed RPMI supplemented with 10% heat-inactivated FCS. After centrifugation @ 500 x g cell pellets were resuspended in 400 μl of medium and divided over two separate tubes for flow cytometric analyses of IL-10 Rα expression or pSTAT3 induction. For the IL-10 Rα cells were blocked with Fc block (1:20, BD Biosciences, 564220) in 0.5% BSA/PBS for 30 minutes at room temperature (RT) and subsequently stained with either CD14-PE-Cy7 (1:100, Clone 61D3, eBiosciences) alone (Fluorescence Minus One (FMO)) or in combination with IL-10-Rα-PE(1:20, clone REA239, Miltenyi Biotechnology). The mean fluorescence intensity (MFI) of IL10Rα was determined by subtracting the background MFI of unstained gated CD14+ cells (FMO). Other cells were either stimulated with 10 ng/ml rhIL10 (R&D, 217-IL-025) for 15 minutes or left untreated at 37°C. Afterwards cells were fixated by adding BD Cytofix/Cytoperm (BD Biosciences, 554722) in a 1:1 ratio and left for 30 min @ RT in the dark. After centrifugation cells were permeabilized using ice cold methanol for 30 min @ 4°C. After thorough washings with 0.5 % BSA/PBS cells were subsequently stained with CD14-PE-Cy7 and pSTAT3-APC (1:50, clone LUVNKLA, eBiosciences). Induction of pSTAT3 was calculated by dividing the IL-10-stimulated pSTAT3 MFI of the CD14+ gated population by the pSTAT3 MFI of the unstimulated CD14+ gated population. Flow cytometry was performed using a FACS Fortessa (BD) and FlowJo software (Treestar Inc., Ashland, OR) was used for analysis.
Conflict of Interest
The authors have no conflicts of interest related to this work.
References
2. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683-765.
3. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory Bowel Disease and Mutations Affecting the Interleukin-10 Receptor. New Engl J Med. 2009;361(21):2033-45.
4. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75(2):263-74.
5. Spencer SD, Di Marco F, Hooley J, Pitts-Meek S, Bauer M, Ryan AM, et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. The Journal of experimental medicine. 1998;187(4):571-8.
6. Franke A, Balschun T, Karlsen TH, Sventoraityte J, Nikolaus S, Mayr G, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40(11):1319-23.
7. Fiorentino DF, Bond MW, Mosmann TR. Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. The Journal of experimental medicine. 1989;170(6):2081-95.
8. Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity. 2014;40(5):720-33.
9. Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40(5):706-19.
10. van Deventer SJ, Elson CO, Fedorak RN. Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Crohn’s Disease Study Group. Gastroenterology. 1997;113(2):383-9.
11. Schreiber S, Fedorak RN, Nielsen OH, Wild G, Williams CN, Nikolaus S, et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease IL-10 Cooperative Study Group. Gastroenterology. 2000;119(6):1461-72.
12. Colombel JF, Rutgeerts P, Malchow H, Jacyna M, Nielsen OH, Rask-Madsen J, et al. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut. 2001;49(1):42-6.
13. Fedorak RN, Gangl A, Elson CO, Rutgeerts P, Schreiber S, Wild G, et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology. 2000;119(6):1473-82.
14. Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44(3):450- 62.
15. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274-88.
16. Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol. 2014;15(10):929-37.
17. Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative contextdependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6(3):498-510.
18. Smythies LE, Sellers M, Clements RH, Mosteller- Barnum M, Meng G, Benjamin WH, et al. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest. 2005;115(1):66-75.
19. Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol. 2014;122:177-210.
20. Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, et al. Single-Cell Analysis of Crohn’s Disease Lesions Identifies a Pathogenic Cellular Module Associated with Resistance to Anti-TNF Therapy. Cell. 2019;178(6):1493-508 e20.
21. Chapuy L, Bsat M, Sarkizova S, Rubio M, Therrien A, Wassef E, et al. Two distinct colonic CD14(+) subsets characterized by single-cell RNA profiling in Crohn’s disease. Mucosal Immunol. 2019;12(3):703-19.
22. Levin AD, Wildenberg ME, van den Brink GR. Mechanism of Action of Anti-TNF Therapy in Inflammatory Bowel Disease. Journal of Crohn’s & colitis. 2016;10(8):989-97.
23. Vos AC, Wildenberg ME, Duijvestein M, Verhaar AP, van den Brink GR, Hommes DW. Anti-tumor necrosis factor-alpha antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology. 2011;140(1):221-30.
24. McRae BL, Levin AD, Wildenberg ME, Koelink PJ, Bousquet P, Mikaelian I, et al. Fc Receptor-mediated Effector Function Contributes to the Therapeutic Response of Anti-TNF Monoclonal Antibodies in a Mouse Model of Inflammatory Bowel Disease. J Crohns Colitis. 2016;10(1):69-76.
25. Bloemendaal FM, Levin AD, Wildenberg ME, Koelink PJ, McRae BL, Salfeld J, et al. Anti-Tumor Necrosis Factor with a Glyco-engineered Fc Region Has Increased Efficacy in Mice with Colitis. Gastroenterology. 2017.
26. Vos AC, Wildenberg ME, Arijs I, Duijvestein M, Verhaar AP, de Hertogh G, et al. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflammatory bowel diseases. 2012;18(3):401-8.
27. Bloemendaal FM, Koelink PJ, van Schie KA, Rispens T, Peters CP, Buskens CJ, et al. TNF-anti-TNF Immune Complexes Inhibit IL-12/IL-23 Secretion by Inflammatory Macrophages via an Fc-dependent Mechanism. J Crohns Colitis. 2018;12(9):1122-30.
28. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143(2):347- 55.
29. Rennick DM, Fort MM, Davidson NJ. Studies with IL-10-/- mice: an overview. Journal of leukocyte biology. 1997;61(4):389-96.
30. Nunberg MY, Werner L, Kopylov U, Haberman Y, Lahad A, Weiss B, et al. Impaired IL-10 Receptor-mediated Suppression in Monocyte From Patients With Crohn Disease. J Pediatr Gastroenterol Nutr. 2018;66(5):779- 84.