Abstract
Head and neck squamous cell carcinomas (HNSCCs) are a group of aggressive and genetically complex cancers, derived from the mucosal epithelium in the oral cavity, pharynx, and larynx. Radiotherapy, often combined with chemotherapy remains the mainstay treatment options for patients. But resistance to radiotherapy causes a critical hurdle in the treatment of HNSCC. Radiotherapy works by inducing the formation of DNA lesions, thus, resistance to irradiation involves the activation of the DNA damage response (DDR) pathways. Defects in DDR function accounts for eventual relapse and cancer progression. The receptor tyrosine kinase MET, and its sole ligand hepatocyte growth factor (HGF) are involved in a range of physiological and pathological processes in HNSCC. There is also compelling evidence suggesting that c-MET activation is crucial in conferring resistance to DNA-damaging agents such as irradiation in several tumour types via interaction with the DDR. Activation of the HGF/MET pathway has also been implicated in resistance to the only available targeted therapy in HNSCC, Cetuximab. Therefore, the HGF/MET axis represents a potentially valuable target for overcoming therapy resistance in HNSCC. Despite such strong evidence, the link between HGF/MET, DDR, and RT resistance in HNSCC has not been studied. This review aims to explore the crosstalk between c-MET and the DDR pathways in several tumour types and extend these findings specifically to HNSCC for exploiting HGF/MET in increasing radiosensitivity in HNSCC.
Introduction
Head and neck squamous cell carcinomas (HNSCCs) are a heterogeneous group of aggressive malignancies strongly linked with chronic tobacco exposure, excessive alcohol consumption, and infection with high-risk subtypes of Human Papilloma Virus (HPV). Molecularly, HNSCC is classified into HPV-positive and HPV-negative sub-types [1]. Approximately 600,000 new cases are diagnosed annually with 380,000 deaths worldwide [2]. Despite our increased understanding of the viral and genetic mechanisms underlying HNSCC, the 5-year overall survival rate remains around 50% [3]. Radiotherapy (RT), chemotherapy (CT), surgical eradication, or a combination of all modalities are the current therapeutic options but are highly toxic and cause psychological distress and severely compromised quality of life, and hence associated with both symptomology and treatment survivors of this cancer have the second-highest mortality rate of suicide (63.4 per 100,000; [2000-2014]) [4]. The functional and aesthetic features of the head and neck anatomy are factors that make HNSCCs difficult to treat as tumours are locate nearby critical anatomical structures which are sensitive to treatment. Radiotherapy (RT), chemotherapy (CT), surgical eradication, or a combination of all modalities are the current therapeutic options. Cetuximab is a monoclonal antibody (mAb) against the epidermal growth factor receptor (EGFR) and has been the only targeted FDA approved targeted therapy for HNSCC until the recent FDA approval of immunotherapy, but, both Cetuximab and immunotherapy clinical efficacy for HNSCC has been limited [5].
RT is the mainstay treatment in HNSCC and is a type of ionising radiation (IR) that induces an accumulation of irreversible DNA lesions in tumour cells, causing cell death. But RT is only effective in less than 50% of patients, and some patients will develop tumour recurrence [6,7]. A fundamental issue in radio-oncology is radioresistance (RR) – the underlying mechanisms by which tumour cells adapt to IR-induced changes are currently unknown. Multiple factors are implicated in the development of RR including tumour genetics (epi) genetics, tumour microenvironment (TME), the presence of cancer stem cells (CSCs), epithelial-to-mesenchymal transition (EMT), the immune system and microbiome [5]. Since RT induces DNA damage, alterations in intracellular DNA damage response (DDR) pathways contribute to how effectively cancers respond to RT. Defects in DDR account for locoregional recurrence, reduced quality of life, poor treatment response, and tumour progression [8-10]. Understanding these resistance mechanisms will help to develop effective radiosensitizers targeting functional components of the DDR is imperative to improve patient outcomes.
The receptor tyrosine kinase (RTK), c-MET (also known as mesenchymal-epithelial transition factor) and its sole ligand, hepatocyte growth factor (HGF) has gained considerable interest as a novel therapeutic target for cancer therapy in general and in HNSCC as they are both frequently overexpressed in HNSCC [11]. Aberrant HGF/ MET signalling has been associated with tumorigenesis in HNSCC as well as poor prognosis, lymph node metastasis, standard therapy resistance and possibly HPV-subtype [12]. Furthermore, activation of HGF/MET signalling has been implicated in ineffectiveness to EGFR-inhibition because of its crosstalk and consequent activation of different signal transduction pathways; this is critical as Cetuximab is frequently used in the management of HNSCC [11,12]. Additionally, apart from having a role in tumour pathogenesis, and the regulation of biological processes associated with growth factor signalling, MET is also relevant in the regulation of DDR [9]. This is important as dysregulated HGF/MET signalling could be a determinant in tumour sensitivity to DNA-damaging agents (DDAs). Aberrant MET signalling is associated with RR, and previous work has shown IR to upregulate the expression of MET and HGF in HNSCC [13,14] and other malignancies [15–17]. Taken together, because HGF and MET are overexpressed in HNSCC, and play a role in EGFR-resistance, the HGF/MET signalling pathway represents a potential therapeutic target also for radiosensitization in HNSCC. So far effects of targeting HGF/MET in combination with IR for the treatment of HNSCC has not been comprehensively explored, generating discordant findings [13,14,18]. Because of the importance of DDR in RT response, this review aims to summarise the relationship between the HGF/MET signalling pathway and DDR in different tumour types in general and to explore specifically how the current findings can be applied to HNSCC, exploiting the potential for increasing radiosensitivity.
HGF/MET Signalling in HNSCC
Structure and function
The MET proto-oncogene first identified in 1984, is located on chromosome 7q21–31 and encodes for the RTK, c-MET protein [19,20]. The MET receptor is an α-β disulphide-linked heterodimer with a molecular weight of 190 kDa. The 140-kDa β-chain spans the extracellular and cytoplasmic domain whilst the 50-kDa α-chain is extracellular (Figure 1); both forms are generated from proteolytic cleavage of a precursor protein at 170-kDa [21].
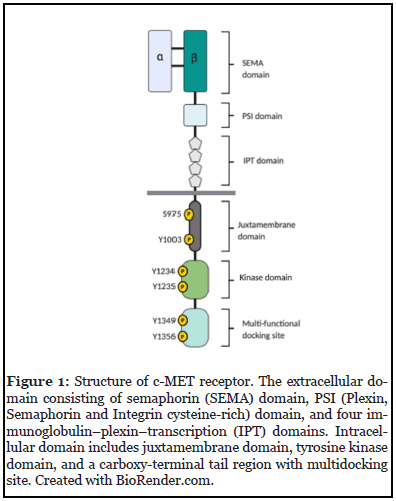
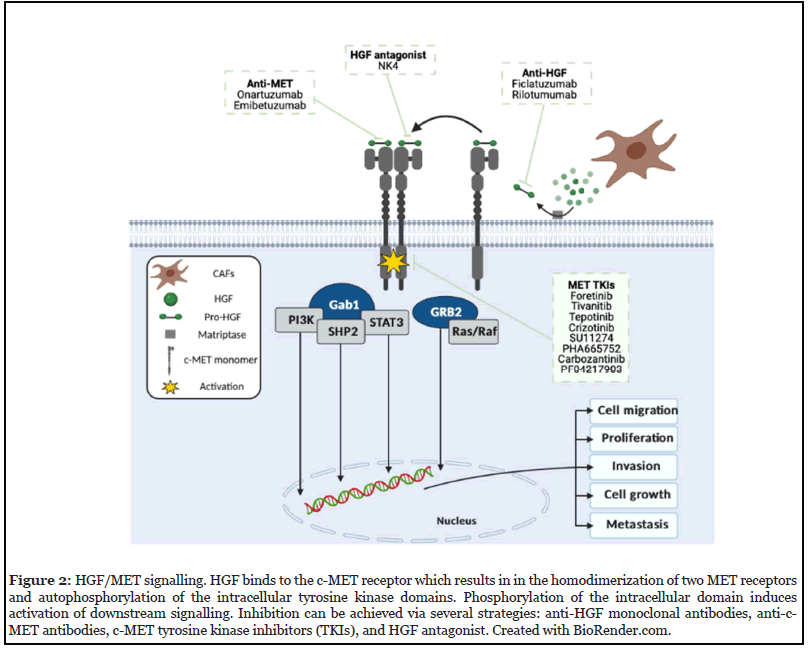
HGF/MET signalling is activated by the binding of HGF to the N-terminal region of c-MET. In HNSCC, HGF is predominantly secreted by cancer-associated fibroblasts (CAFs) within the TME in a paracrine manner as an inactive zymogen pro-HGF, which is then proteolytically cleaved by membrane-bound matriptase located on the tumour cell surface [22]. Cleavage allows the ligand to bind to both MET receptors. Upon binding, two MET receptors dimerize, leading to autophosphorylation of tyrosine residues (Y1230, Y1234, Y1235). This is followed by the phosphorylation of other tyrosine residues (Y1349 and Y1356), located on the carboxy-terminal site of the β-chain. Both tyrosine residues form a multi-docking site for the recruitment of adaptor proteins: growth-receptor-bound protein 2 (GRB2), and GRB2-associated binder 1 (Gab1), which recruits phosphoinositide-3-kinase (PI3K), Ras/ Raf (MAPK signalling), signal transducer and activators of transcription-3 (STAT3), and SH2-containing protein tyrosine phosphatase (SHP2) – promoting biological functions including cell survival, proliferation, invasion and angiogenesis, morphogenesis and stemness (Figure 2) [12,23].
HGF/MET alterations in HNSCC
A range of genetic abnormalities including overexpression of MET and HGF proteins, MET mutations, and amplification of the MET gene can increase HGF/MET pathway activation. The MET protein has been reported to be overexpressed in 80-90% of HNSCCs, alongside frequent mRNA overexpression [24-26]. Increased phosphorylated c-MET (p-MET) has been detected in HNSCC samples [24,26]. One recent study found increased p-MET expression in 30% of tumours and reported a significant correlation between p-MET and HGF overexpression. This implied that c-MET was activated in a paracrine manner in these HNSCC samples [24]. Data from The Cancer Genome Atlas (TCGA) denotes MET amplification and gain in copy number to be observed in approximately 20% of HNSCCs and linked with c-MET protein overexpression [27-29].
Overall mutational frequency of MET in primary tumours is low (<1%) [27,28], however, mutations have been identified in the MET tyrosine kinase, SEMA and juxtamembrane domains. For instance, a study found Y12353D mutation in 11% of 138 oral squamous cell carcinoma patients [30]. Whilst this is low, Di Renzo et al. suggested that constitutively activated MET somatic Y1253D mutation could undergo clonal selection during HNSCC progression and metastasis [31]. This emphasises the crucial role of activating MET mutations despite having low occurrence in primary tumours [11]. Notably, the Y123D mutation has demonstrated to affect the tumour’s response to IR, and correlate with tumour progression and recurrence [30]. This supports the concept that aberrant MET signalling has important potential for targeted therapy for radiosensitization in HNSCC.
Comparable to c-MET expression, HGF protein is overexpressed within the tumour-stroma in 50% of HNSCCs, and HGF expression has been reported to be in 58% of recurrent and metastatic (R/M) HNSCCs [24,26].
The importance of targeting the HGF/MET pathway in HNSCC
Cell migration/invasion in HNSCC: Most of the literature exploring MET in HNSCC has been associated with cell migration and invasion. For instance, in in vivo HNSCC models, Tao et al., reported silencing of MET or c-MET inhibition to inhibit cancer cell motility [32], and MET has been considered to promote a metastatic phenotype as MET expression has been reported to be higher in primary tumours with advanced lymph node metastasis compared to early-staged diseases [33]. In support, the epithelial marker, E-cadherin, of which its downregulation correlates with inferior prognosis and higher incidence of metastasis in HNSCC [34] and RR in other cancers [35] has been reported to be influenced by MET signalling, promoting EMT. In hypopharyngeal cancer, Kim et al., found HGF-induced activation of c-MET to reduce E-cadherin, leading to enhanced invasion [36]. HGF/MET signalling has also shown to aid HNSCC cells to evade apoptosis; Xi et al., showed MET inhibition to suppress proliferation and augment caspase-dependent apoptosis [37], and earlier work has reported HGF in HNSCC to inhibit anoikis by activating Akt and ERK pathways [38]. In other cancers, Akt has a prominent role in cell survival and anti-apoptosis and can be activated by HGF/MET signalling, triggering in the activation of anti-apoptotic proteins [39,40]. c-MET inhibition has been found to enhance IR-induced cell death measured by the clonogenic assay and doubled levels of active caspase-3, suggesting that MET-induced anti-apoptosis could contribute to RR – however, this was reported in glioblastoma [16]. The ability to bypass apoptosis is a strong indicator of a metastatic phenotype – further supporting the role of the MET pathway in HNSCC.
Resistance to EGFR-inhibition in HNSCC: Aside from HGF and MET being overexpressed in HNSCC tumours and a role in tumour progression, a crucial aspect of the HGF/MET signalling pathway is its implication in resistance to EGFR-inhibition. Despite EGFR being frequently overexpressed in HNSCC, the clinical efficacy of Cetuximab and other EGFR-inhibitors in recent clinical trials has been disappointing in both HPV positive and negative subtypes [41]. Around 87% of HNSCC patients treated with Cetuximab monotherapy were intrinsically (de nova) resistant, and the 13% of whom were originally responsive acquired resistance over a short period [42]. One established resistance mechanism against EGFRinhibition is the compensatory activation loop – the upregulation of alternative RTKs such as human epidermal growth factor receptors 2 and 3 (HER 2 & 3) reactivate PI3K and MAPK pathways, facilitating the capability to circumvent EGFR-inhibition [43-46]. As MET activates critical downstream signals which overlap with EGFRsignalling, activation of c-MET has also been implicated in such resistance. In a single-prospective study, the authors revealed a correlation between c-MET activation and poor prognosis in HNSCC patients treated with a Cetuximabbased regimen – supporting c-MET’s prognostic potential [24]. Yang et al., found c-MET activation by HGF to confer Cetuximab resistance by increasing phosphorylation of PI3K/Akt and MAPK pathways, and Cetuximab sensitivity was restored following MET inhibition [47]. In support, others reported activation of c-MET by HGF to reactivate the MAPK pathway in Cetuximab-resistant HNSCC cells [48]. Pertinent to IR, one study showed EGFR/PI3K/ Akt blockade in HNSCC to be inadequate to enhance radiosensitivity since tumour cells activated the MET/ MAPK pathway to evade IR-induced apoptosis [49]. These findings not only emphasise the significance of c-MET as a compensatory survival pathway for tumour cells to escape the EGFR-blockade in HNSCC but indicates that the horizontal blockade of EGFR and c-MET may be beneficial in cetuximab naïve or patients who have received Cetuximab. Pre-clinical data has shown promising results of this combinational approach in HNSCC [50,51]. Since Cetuximab plus RT is a standard of care treatment in RRM HNSCC, the HGF/MET axis represents a valuable target to overcome resistance. However, so far, no clinical trial nor pre-clinical study has explored combining EGFR- METinhibition as a radiosensitization approach.
Targeting the HGF/MET signalling pathway in HNSCC
Several agents have been developed to target both the HGF ligand and c-MET receptor. Three strategies have been established (Figure 2). Table 1 shows an overview of anti-MET/HGF agents in completed or ongoing clinical trials in HNSCC.
| Clinical trial Phase |
Treatment |
Class of HGF/MET agents |
Known pharmacological targets |
Study population |
Status |
Main findings; Estimated completion date; reason for termination | Clinical trial Identifier |
|---|---|---|---|---|---|---|---|
| II |
Foretinib |
Non-selective MET TKI |
c-Met, RON, VEGFR-1, VEGFR-2, VEGFR-3, PDGFR, kit, Tie-2, Flt-3 |
R/M HNSCC |
Completed |
Stable disease in 50% patients; minor tumour shrinkage in 43% patients; prolonged disease stabilisation ≥13 months in 14% * |
NCT00725764 |
| II |
Ficlatuzumab + Cetuximab |
Anti-HGF mAb | HGF |
R/M HNSCC |
Recruiting |
31st December 2020 |
NCT03422536 |
| Ib |
Ficlatuzumab + Cetuximab |
Anti-HGF mAb |
HGF |
R/M HNSCC |
Completed |
Median PFS: 5.4 [90% CI = 1.9–11.4] months; OS: 8.9 [90% CI = 2.7–15.2] months; no dose-limiting toxicities. |
NCT02277197 |
| Ib |
Ficlatuzumab + Cisplatin + IMRT |
Anti-HGF mAb |
HGF |
Intermediate or High Risk, Previously Untreated, LA HNSCC | Terminated |
Halted |
NCT02277184 |
| Ib |
INC280 (Cap- matinib) + Cetuximab |
Selective MET TKI |
c-MET |
MET positive HNSCC [determined by FISH, IHC]. |
Terminated |
Difficulties identifying patients who met eligibility. |
NCT02205398 |
| II |
Tivantinib + Cetuximab (TC) versus Cetuximab (C) |
Selective MET TKI* |
c-Met, PAK3, Flt4, Pim-1, tubulin, GSK1, GSK2 |
R/M HNSCC, or inoperable HNSCC. |
Completed |
Response rates in TC arm: 7.5%; C arm: 7.9%; median PFS and OS were 4 and 8 months respectively in both arms |
NCT01696955 |
| Ib/II |
E7050 (Golavitinib) + Cetuximab |
Non-selective MET TKI |
c-Met, VEGFR-2 | Platinum- resistant HNSCC | Completed |
No results posted |
NCT01332266 |
Abbreviations: R/M: Recurrent/Metastatic HNSCC; LA HNSCC: Locally Advanced HNSCC; FISH: Fluorescence in situ Hybridization; IHC: Immunohistochemistry; TC: Tivantinib plus Cetuximab; C: Cetuximab; IMRT: Intensity-Modulated Radiation Therapy; TKI: Tyrosine Kinase inhibitor.
NOTE: Data provided from https://clinicaltrials.gov/ct2/home
* This was a two-step trial initially designed to enrol a further 27 patients after observing one response from the first group (n=14).
But the trial did not meet the requirements and was consequently terminated.
Table 1: Ongoing or completed clinical trials examining HGF/MET targeted agents in HNSCC.
Anti-MET and anti-HGF biologics: Anti-MET/ HGF are mAbs that function by binding to c-MET and HGF respectively [52]. Ficlatuzumab is an HGF mAb shown to selectively bind and neutralising free HGF activity and has been reported to inhibit CAFinduced migration, invasion, proliferation, and MET phosphorylation in HNSCC cells cultured in CAF-cultured media [53]. Currently, Ficlatuzumab is the only mAb undergoing clinical development in HNSCC. In its first completed phase Ib trial, Ficlatuzumab combined with Cetuximab was found to have an acceptable safety profile and promising anti-tumour efficacy. The results from this trial were encouraging and have led to the current phase II trial examining Ficlatuzumab, with or without Cetuximab – this trial has just been completed (May 2021; NCT03422536). Other mAbs including Rilotumumab (AMG102), and Onartuzumab (MetMab), have been investigated pre-clinically and clinically in other cancers [54,55]. Hence, evaluation of other targeted biologics in HNSCC is advisable.
MET TKIs: Most of the MET TKIs are ATP competitive kinase inhibitor that competitively antagonises the occupancy of intracellular ATP binding, blocking the phosphotransferase activity of their targets [56,57]. TKIs are further subclassified into selective and non-selective TKIs; selective TKIs are directed at one specific pathway, whereas non-selective TKIs can target multiple pathways [58]. Foretinib (GSK36089; formerly known as XL880) is a multi-kinase inhibitor directed at c-MET and VEGF receptor 2 and is modestly potent in HNSCC cell lines (IC50 < 0.5 μm) [59]. Foretinib has been evaluated in a phase II HNSCC trial – this was a two-step trial initially designed to enrol a further 27 patients after observing one response from the first group (n=14) [60]. Nonetheless, this trial did not meet the requirements and was terminated. A possible explanation for this disappointing result could be that multi-kinase TKIs additionally target non-MET receptors, thus the dosage may have not significantly inhibited c-MET. Selective c-MET inhibitors solely target c-MET, and so can be dosed at a level that does not affect other kinases and causes minimal off-target effects – thereby increasing their efficacy. Tivantinib (ARQ197) is a potent selective inhibitor of catalytic c-MET activity and show to inhibit tumour proliferation and induce caspase-dependent apoptosis in in vitro HNSCC models [61]. However, in a recent phase II HNSCC trial, Tivantinib plus Cetuximab was not found to significantly enhance response rate or OS compared to Cetuximab alone but did increase toxicity [62]. However, it should be noted that this trial was performed in an unselected HNSCC population – emphasising the need for future trials to stratify patients based on the status of c-MET pathway dysregulation. Furthermore, Tivantinib is a non-competitive inhibitor and has emerged to be less selective compared to other c-MET inhibitors – this could be why Tivantinib failed in the recent phase II HNSCC trial mentioned [62]. A newer and highly potent selective TKI called Tepotinib used in MET exon14 skipping (a splice site mutation that leads to a loss of transcription of exon 14 of the MET gene) has been approved for MET-amplified NSCLC in Japan [63]. It is tolerated at its highest dose and reduce tumour burden [64,65]. Importantly, preclinical data has demonstrated Tepotinib to increase radiosensitivity in HNSCC [18]. Although exon14 skipping and MET amplification are rare in HNSCC, Tepotinib may offer a more selective way for either MET or MET/EGFR dual inhibition for HNSCC with or without RT and should be further explored.
Competitive HGF antagonism: A truncated, soluble MET receptor called NK4 decoy is a competitive HGF antagonist and another strategy for inhibiting HGF/ MET signalling. The N4 fragment is comprised of the HGF N-terminal hairpin and four Kringle domains. Its functions by binding to c-MET without activation, thereby competitively inhibiting HGF-depending on c-MET activation [66]. In gallbladder and pancreatic cancers, preclinical data has shown HGF antagonists to inhibit cancer cell invasion, proliferation, and angiogenesis [67]. As yet, NK4 hasn’t been evaluated in HNSCC.
The aforesaid paragraphs have provided a summary of strategies for inhibiting HGF/c-MET signalling in HNSCC. Given the importance of the DDR for genome stability, in the rest of this articles, we will focus on the relationship between c-MET signalling and the DDR by exploring the range of anti-MET/HGF agents targeting functional components of the DDR within the current literature.
Targeting Cancer’s Achilles’ Heel
A brief overview of the DDR
The rationale behind applying RT is based on the concept that rapidly proliferating tumour cells are more sensitive to IR compared to normal cells. RT destroys cancer by accumulating high physical energy of IR in tumour cells, damaging the DNA, and resulting in the functional impairment of cancer cells, and eventual cell death. IR causes a wide range of DNA lesions, amongst them, DNA double-stranded breaks (DSBs) have high lethality in DNA damage, chromosomal instability, and cancer cell death [68]. Nonetheless, activation of the DDR, a highly orchestrated network of interconnected and overlapping signalling pathways, is crucial for defence against continuous genetic insults by promoting cellular survival and maintaining genome stability and integrity.
DDR is orchestrated by sensors, transducers, and effector proteins and is divided into three acts: i) aberrant DNA structures are identified by sensors for the initiation of the DDR; ii) once DNA damage has been sensed, the signal is amplified by transducers; iii) lastly, effector proteins are activated and initiate a range of cellular processes including cell cycle control, DNA replication, DNA repair, transcription, and apoptosis [69].
The key-signalling components of the DDR are ATM (ataxia-telangiectasia mutated) and ATR (ataxiatelangiectasia and RAD3-related) – large serine/threonine kinases belonging to the phosphatidylinositol-3-kinaselike kinase family (PIKKs) [70,71]. ATM is predominantly activated by DSBs, whereas ATR responds to a wide array of damage including DSBs, and lesions which result in replication stress [72]. Hundreds of proteins are phosphorylated and activated by ATM/ATR in response to DNA damage. Checkpoint kinases 1 (Chk1), and 2 (Chk2), are the major downstream kinases of ATR and ATM respectively, critical for instigating cell cycle arrest through checkpoint activation by phosphorylating a range of effectors including p53 and cell cycle division 25 [CDC25] family phosphatases (isoforms: CDC25A/B/C) [73]. Cell cycle arrest allows clearance of damage before reentry into the cell cycle; if the damage is too significant and irreparable, different forms of cell death will occur [74]. There are two major repair pathways: non-homologous end-joining repair (NHEJ), which occurs throughout the whole cell cycle (predominantly in G1), or homologous recombination (HR), occurring during the S and G2 phases [75]. The former, re-joins the ends of DSBs with minimal homology and is highly error-prone since it often leads to the generation of insertion/deletion mutations [69]. HR is a high-fidelity process, requiring a second intact homologous DNA template for repair [76].
Because the curative intent of IR is to induce DNA damage, the DDR has significant relevance regarding RR and therapeutic response. Hence, targeting the ability of tumour cells to modulates DSB repair mechanisms and checkpoint activation – both of which constitute the DDR – is a credible radiosensitization approach.
Inhibition of HGF/MET signalling impairs DSB repair
The role of c-MET in DNA repair has been highlighted in previous radiobiological studies, with the majority demonstrating MET inhibition to impair DSB repair [13,15,77,78]. Cuneo et al., reported treatment with multikinase inhibitor, Crizotinib, to cause increased DSBs after IR as evident by the elevated γ-H2AX which persisted for 3-9 hours [78]. However, others found Crizotinib at any dose was ineffective to induce γ-H2AX foci formation and had no significant effect on DNA repair or radiosensitivity [79]. Although the majority of the literature shows suppression of DNA repair to increase DNA damage and consequently radiosensitivity when exposed to MET inhibition, only some have elucidated the repair mechanism implicated – which is HR. HR is an intricate process that involves a multiple of HR-related proteins and is divided into three acts: presynapsis, synapsis, and post-synapsis [80]. Because cells with compromised HR are sensitive to DDAs, HR can be targeted for radiosensitization [76,81].
Targeting the central component of HR – RAD51: RAD51 recombinase has a key role in orchestrating HR and is overexpressed in tumours [82]. Overexpressed RAD51 increases the efficiency of HR, thus correlates with a poorer prognosis, enhanced tumour cell survival, and resistance to DDAs [83-86] In HNSCC, a previous immunohistochemistry-based study found pre-treatment tumour biopsies with high RAD51 expression to have poorer cancer-specific survival following treatment compared to low RAD51 expression, suggesting RAD51 could influence the patient outcome [87]. Moreover, the reduction of RAD51 increases the accumulation of DSBs and sensitises tumours to DDAs [88,89]. Thus, RAD51 represents an attractive target for increasing radiosensitivity in HNSCC.
RAD51 activity can be regulated by post-translational modifications and protein-protein interactions which determines its role in DNA repair [90]. Studies have shown MET inhibition to impair DSB repair independent of cell cycle alterations by interacting with RAD51 regulatory proteins. c-ABL or its oncogenic fusion protein BCR/ Abl phosphorylates the Tyr315 and Tyr54 residues of RAD51 and modulates the recruitment of RAD51 to the DSB site and DNA strand exchange [91]. Earlier work has shown c-MET to phosphorylate peptide substrates of Abl, suggesting a potential signalling pathway between MET, Abl and RAD51 [92]. Based on this indication, Ganapathipillai and colleagues treated cells with SU11274 to target the novel MET/Abl/RAD51 pathway, impairing RAD51 expression by reducing phosphorylation of Abl [93]. This resulted in high persistent levels of γ-H2AX. In a follow-up study, the interplay between MET inhibition and HR-dependent DNA repair was supported. MET inhibition before IR reduced RAD51 levels by affecting BRCA2- RAD51 complex formation [94]. BRCA2 has an important role in the cell response to DNA damage since it regulates the DNA binding ability and intracellular localisation of RAD51 in DNA repair. Therefore, the loss of both proteins can harm genomic stability [95]. Despite these findings, the γ-H2AX levels weren’t examined post-IR, thus whether the treatment affected IR-induced DNA damage was unclear. Notably, the role of c-MET in BRCA2-dependent regulation of RAD51 has been supported. Recently, Chabot et al., found MET to directly regulate RAD51 by phosphorylating RAD51 on four tyrosine residues, which in turn was able to prevent binding of a BRCA4-28 peptide inhibitor, a derivative of BRCA2 which causes dissociation of the RAD51 nucleofilament [90]. To aid the translation, examining the effect of c-MET inhibition on RAD51 phosphorylation levels and DNA damage can help identify potential new combination therapeutic strategies.
The studies aforementioned have provided evidence demonstrating a relationship between MET and the HR machinery. This reinforces the role of c-MET in the DDR and indicates the clinical potential of MET inhibitors as radiation sensitisers by altering DNA repair kinetics. Although MET inhibition compromised the HR machinery [93,94], the effects on radiosensitivity weren’t examined [i.e. clonogenic survival of tumour cells]. The association between the mechanisms underlying MET-induced HR impairment and radiosensitivity should be investigated to permit these findings for therapeutic application. As MET inhibition elevates γ-H2AX persistently post-IR in HNSCC [13,18], the next step will be to explore these effects on DSB effector proteins, particularly related to HR. Furthermore, HPV status must be considered; HPVpositive cells possess defects in DSB repair compared to HPV-negative – a reason why HPV-positive HNSCCs have superior responses to RT [96-98]. Studying the effects of MET inhibition on DSB repair in both subtypes is required to open new perspectives for radiosensitization in HNSCC, and drive advancements in subtype-specific treatment.
The role of c-MET in DNA repair of CSCs: Cancer stem cells (CSCs) have a crucial role in the progression and development of HNSCC [99-101]; they’re a subpopulation within the heterogeneous tumour, with the capability to selfrenew and generate more mature, differentiated tumour cells, and therefore contribute to tumour recurrence and metastasis [102,103]. Of note, CSCs has shown to play a major role in the expression and maintenance of RR in HNSCC. As the majority of anti-cancer therapies target the tumour bulk rather than CSCs, targeting their self-renewal pathways may be beneficial [103]. c-MET expression has shown to promote self-renewal and stemness in HNSCC and confers resistance to DDAs [103,104]. A possible explanation is that CSCs have heightened DNA repair, and higher levels of RAD51 compared to differentiated cells [105]. Thus, targeting c-MET to inhibit DSB repair in CSCs could be a plausible radiosensitisation strategy in HNSCC.
Work by De Bacco et al., reported MET to promote RR in glioblastoma stem-like cells (GSCs) by inducing ATMhyperactivation via a novel MET-Akt-Aurora-A signalling pathway [106]. MET inhibition reduced Akt activation, ATM phosphorylation, and RAD51 expression in irradiated GSCs, resulting in accumulated DNA damage. Because MET inhibition prevented the phosphorylation of cyclin-dependent kinase (CDK) inhibitor, p21 by Akt, the protein translocated into the nucleus. Therefore, it can be speculated that p21 inhibited CDK1 (which mediates BRCA2-RAD51 formation), impairing DSB repair [106,107]. Translationally, this study demonstrates the clinical potential of MET inhibitors for increasing radiosensitivity in CSCs which could be applied to HNSCC since Crizotinib has shown to inhibit CSC-like populations and impair metastasis in patient-derived xenograft (PDX) HNSCC models [103]. It would be interesting to evaluate if EGFR uses the same signalling cascade identified in this study to drive RR; this could be exploited with anticancer combinational therapies in HNSCC involving cotargeting c-MET and EGFR, given that EGFR is frequently overexpressed in HNSCC, and is demonstrated to activate DNA repair [108,109].
Targeting the cell cycle with MET/HGF inhibitors
In the literature, MET inhibition has shown to enhance radiosensitivity interestingly in two opposing manners: to arrest or not arrest the cell cycle. Recent work by Nisa et al., found Tepotinib to increase radiosensitivity in HNSCC cells by prolonging G2 arrest, which increased the G2/M population [18]. While it can be argued that initiation of cell cycle arrest would’ve allowed tumour cells time to remove any DNA damage induced by IR, the levels of γ-H2AX increased for two hours post-IR. This suggests that Tepotinib impaired DNA repair and accumulated DNA damage independent of cell cycle alterations. The genotoxic effects of IR vary at different points of the cell cycle. Because there are active DNA segregation and cell division within the G2 and M phases, IR can cause multiple DNA damages. As a result, the G2 and M phases are most sensitive to IR, followed by G0 and G1 phases, whilst the S phase is most radioresistant [73,110]. Therefore, an increase in the G2/M population led to radiosensitization. Likewise, others reported Foretinib to promote IR-induced G2/M arrest by increasing phosphorylation of Cdc2 (CDK1) and degradation of cyclin B1 [111]. Additionally, there was a reduction in DNA repair which was expected since Foretinib has previously shown to exert antitumour effects by inhibiting DSB repair regulated by MET [112]. Because of the role of cyclin B1-Cdc2 complex in G2-M transition, it can be assumed that degradation of cyclin B1 in this study reduced cellular proliferation and increased G2/M-fraction and consequently resulting in increased radiosensitivity [113]. Notably, it would’ve been interesting to examine the effects on CDC25 since Cdc2 is also activated by dephosphorylation of the inhibitory Tyr14 and Tyr15 site by CDC25C [114,115]. In HNSCC, cyclin B1 overexpression correlates with poorer clinical outcome. Earlier work by Hassan et al., found HNSCC cells with overexpressed cyclin B1 to be resistant to IR and proposed to stratify patients based on cyclin B1 expression as a marker of sensitivity [116]. Additionally, previous work showed cyclin B1 downregulation to suppress proliferation and sensitise breast cancer cells to DDAs [117]. Due to this, administering MET inhibition in HNSCCs with overexpressed cyclin B1 for increasing radiosensitivity could be explored given the relationship between c-MET and cyclin B1 demonstrated in other malignancies.
Because activation of cell cycle checkpoints halts cellular progression to ensure effective DNA damage repair, impairing these pathways to prevent DNA repair and accumulate detrimental chromosomal aberrations could make DNA-damaging treatments more effective. Yu et al., reported pre-treatment of SU11274 to eliminate IR-induced G2/M cell cycle arrest and suppress DNA repair, resulting in radiosensitization [118]. Whilst there was a reduction in the most radiosensitive phases, low concentrations of SU11274 alone increased the G1 fraction, and decreased the percentage of S-phase cells, still placing cells in a radiosensitive state. Although the mechanism underlying how MET inhibition abrogated G2/M arrest wasn’t elucidated, Chk1 has shown to regulate the G2-M and S-phase checkpoints [119]. Upon DNA damage, Chk1 is activated and inhibits CDC25 phosphatases by phosphorylation, making them unable to dephosphorylate and activate CDKs, resulting in cell cycle arrest (reviewed by [120]). Others reported the direct relationship between the ATR/Chk1 and HGF/MET pathways since HGF induced Chk1 phosphorylation; this can be exploited therapeutically [121]. Medova et al., found c-MET inhibitor, PHA665752 to release affected cells from DNA-damaged S-phase arrest by destabilising the ATR-CHK1-CDC25B pathway [77]. This decreased the number of cells within S-phase and permitted further cell cycle with damaged DNA. Notably, PHA665752 also maintained elevated pATM and γ-H2AX levels. Since ATR has been implicated in HR [122-124], it can be speculated that reduced activation of ATR by MET inhibition also compromised DSB repair independent of the cell cycle. Chk1 has been reported to be an important therapeutic target in HNSCC [125]. The following section will explore how the crosstalk between c-MET and ATR pathways shown in other tumours could be applied to HNSCC for promoting sub-type specific treatment.
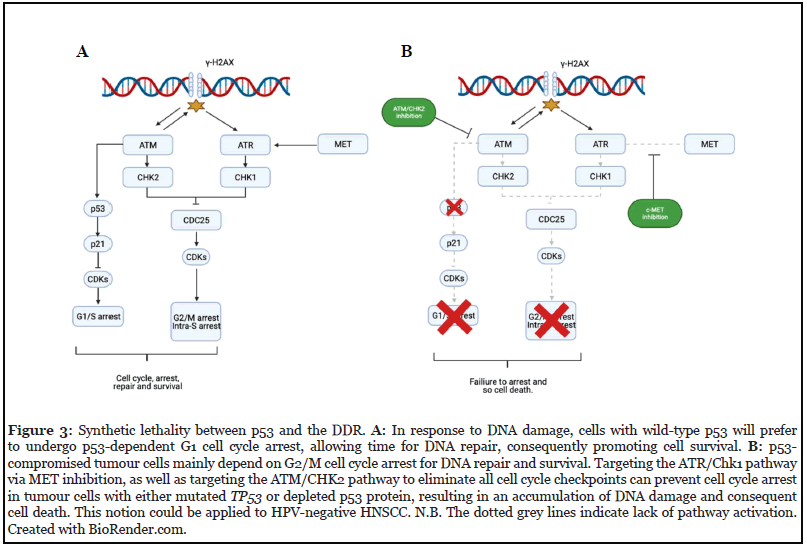
Hitting cancer’s weak spot: synthetic lethality in HPV-negative HNSCC: The most common genetic lesion in HPV-negative HNSCC mutation of the tumour suppressor TP53. In a recent TCGA study, HPV-negative tumours were reported to harbour >84% of TP53 mutations which disrupts components of the G1/S checkpoint machinery [126]. Conversely, most HPV-positive tumours had inactivated p53 due to HPV E6 oncoprotein [127]. Although there is crosstalk between Chk1/2 pathways, unlike the G2/M checkpoint which depends on multiple pathways, the G1/S checkpoint is thought to rely solely on the ATM-CHK2-p53 pathway [73,128]. When ATM phosphorylates Chk2, both kinases phosphorylate and stabilises p53. Consequently, p21 expression increases and induces G1 arrest by inhibiting G1 and early S-phase cyclin-CDK complexes [129,130]. In response to IR, p53 is activated; however, as p53 is mutated in HPV-negative HNSCC, the cells have reduced G1/S checkpoint-induced cell cycle arrest and strongly depend on the G2/M checkpoint for facilitating the growth arrest required for DNA repair and survival from genotoxic stress. On the other hand, in HPV-positive HNSCC, there is a low yet functional basal concentration of p53 which can be induced by IR and capable of inducing cell cycle arrest – the low level of wild-type p53 is another reason why HPV-positive HNSCCs demonstrate to have a superior response to RT compared to HPV-negative [131-133]. Despite differences in clinical and molecular characteristics, this hasn’t altered treatment guidelines for specific subtypes [134].
Conceptually, the loss of wild type p53 can be exploited therapeutically by inducing synthetic lethality [135]. By abrogating the G2 checkpoint, mutant or depleted TP53 cells could be sensitised to DDAs – this is because the absence of both G1 and G2 checkpoints cause the segregation of chromosomal aberrations, forcing cells into the M phase to die [136,137]. As stated, in the previous section, in 2010 Medova et al., reported c-MET inhibition to attenuate the ATR-CHK1-CDC25 pathway – responsible for intra-S and G2-M arrest [77]. The same group then examined the effects of MET inhibition on p53-status in their following study and found Tepotinib plus IR to increase cytotoxicity, phopsho-histone H3, and DNA damage in p53-deficient cells compared to wild-type p53 [138]. Due to this, the authors proposed that targeting c-MET in MET-driven tumours with a p53-compromised background may be a potential checkpoint abrogator. Interestingly, this notion can be extrapolated to HPV-negative HNSCC; therefore, it can be hypothesised that inhibiting c-MET to attenuate CHK1 activation and consequently eliminate the G2 checkpoint in HNSCCs harbouring depleted p53 protein or TP53 mutation, could increase the cytotoxicity of IR.
Because CHK1 has been proposed to be a promising target in HNSCC cell cycle regulation, and recent HNSCC studies have shown ATR inhibition to sensitise HPVnegative cells to DDAs, our assumption is encouraging [125,139]. Moreover, other studies have shown synthetic lethal targeting to sensitize HNSCC cells to DDAs with other inhibitors [137,140,141]. Because of this, applying c-MET inhibitors in a synthetic lethal manner could provide an alternate approach for HPV-negative HNSCCs. Thus, evaluating the relationship between ATR and c-MET in HNSCC is important. However, despite the loss of wildtype p53, at the same time, tumour cells may use the ATMCHK2- CDC25 pathway as compensation [142,143] (Figure 3). Therefore, additional inhibitors targeting ATM/ CHK2 may be used concurrently with MET inhibition to ensure complete abrogation of cell cycle checkpoints. Nonetheless, using combinational therapy reduces the clinical translatability of our conjecture since increasing the number of inhibitors could increase the likelihood of toxicity. Furthermore, others have shown inhibition of CHK1 to potentiate effects of DDAs irrespective of p53 status [144,145]. This indicates that other determinants aside from p53 including (epi) genetics, Rb, p16, and Wee1 will influence treatment. Although MET is overexpressed in HNSCC, the differences in MET expression and mutations between both HPV-subtypes is unresolved [12]. Recent work by Alsahafi and colleagues demonstrated EGFR to have a distinct oncogenic role in HPV-negative HNSCC cell lines when compared to their HPV-positive counterpart – this raises the question as to whether similar findings would be observed with c-MET given the crosstalk between both transduction pathways [146]. This must be elucidated since our concept is only clinically applicable to patients where c-MET is aberrantly expressed, and G1 checkpoint-related genes including TP53 are mutated.
The Translational Relevance of MET Inhibitor Therapy for HNSCC
There is a lack of sufficient pre-clinical and clinical studies investigating the combination of targeted drugs in combination with RT. Additionally, most available data is based on in vitro, cell line studies which do not reflect the tumour architecture and microenvironment. Physiological model systems such as PDXs, ex vivo patient-derived organotypic tissue culture, or genetically engineered mouse models are necessary to evaluate the pre-clinical radiosensitization studies to gain insight into the drug response and kinetic, thereby improving clinical trial design and decision-making.
Despite a relatively large number of anti-HGF/MET targeted drugs available, none has so far been approved for clinical use yet. One possible explanation may be the lack of stratification biomarker to select the patient population that may benefit for such intervention. The detection of MET-positive patients has demonstrated to be difficult, as apparent by the terminated trial involving Capmatinib (see Table 1). Hence, identifying diagnostic tools for better patient selection is essential for the success of future clinical trials. Recently, a validated phospho-MET immunoassay has been made commercially available and is assumed to be an accurate biomarker for c-MET pathway dysregulation, however, this assay awaits evaluation within a clinical setting [147,148]. Overall, the genomic, as well as TME and other molecular characteristics of each patient’s cancer, needs to be considered to successfully develop the concept of precision-based medicine.
Conclusions and Future Perspectives
Although previous reviews have demonstrated the role of MET in tumour progression regarding growth and metastasis, this review has emphasised the significance of MET signalling in regulating responses to DNA damage which could aid tumour cells evade IR-induced cytotoxicity. Because of this assumption, there is a strong rationale for targeting HGF/MET signalling for enhancing IR-based therapies in MET-driven tumours. But additional studies are required to identify the specific pathways by which MET is signalling with the DDR to open new treatment perspectives in HNSCC.
In HNSCC, only a select number of preclinical studies have evaluated MET inhibitors as a radiosensitizer. Although recent in vivo data from Nisa and colleagues reported the potential of Tepotinib for radiosensitization [18], work by Baschnagel et al., reported negative findings using Crizotinib, making the role of MET inhibition with IR unresolved [14]. A probable explanation for this discordance could be due to differences in drugs, experimental design, and HNSCC models. However, because of the MET-DDR link demonstrated in other malignancies, the role of HGF/MET in Cetuximab resistance, and c-MET overexpression correlating with aggressive disease in HNSCC tumours, c-MET still represents a valuable target in HNSCC for radiosensitization and should be evaluated further. Importantly, future radiosensitization studies should concentrate on inhibiting combinations of RTKs such as EGFR and c-MET as well as inhibiting common downstream signalling proteins such as c-SRC and PI3/ Akt/mTOR.
Author Contributions
MT and AV formulated the topic of the review. AV wrote the manuscript and prepared the table and figures. MT critically revised the manuscript and the figures.
Declaration of Competing Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
We would like to thank Dr Danupon Nantajit for helpful discussions. We would like to thank the Rosetrees Trust and Stoneygate Trust for supporting this study through grant reference M117-F2 to MT.
Contribution to Field Statement
Head and neck squamous cell carcinomas (HNSCC) are an aggressive, genetically intricate, and difficult to treat group of cancers. Despite our increased understanding of the genetic and viral mechanisms underlying this dreadful disease, the survival rate of HNSCC patients has only increased modestly over the past three decades, and the treatment options available are limited. Cetuximab, an EGFR-directed monoclonal antibody is the only approved targeted therapy for HNSC but has been proven to be extremely inefficient, emphasising the dire need for the discovery better druggable targets.
In this review we provide an update on the role of MET receptor and its ligand HGF in the development and progression of HNSCC. We highlight the crosstalk between MET/HGF and EGFR signalling pathways and explore the interchange between the cancer associated fibroblast (CAF), through secretion of HGF, causing activation and oncogenesis through MET signalling in cancer cells. Importantly, this review explores the relatively unidentified role of MET/HGF in reprogramming the DNA damage response (DDR) which is a key player in determining therapy response/resistance in cancers. The knowledge provided here will help to shift the focus of targeted therapy for HNSCC towards combination multimodal treatment strategies targeting MET/HGF and DDR.
References
2. Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015. JAMA Oncology. 2017 Apr 1;3(4):524.
3. Kyrgias G, Hajiioannou J, Tolia M, Kouloulias V, Lachanas V, Skoulakis C, et al. Intraoperative radiation therapy (IORT) in head and neck cancer. Medicine. 2016 Dec;95(50):e5035.
4. Osazuwa-Peters N, Simpson MC, Zhao L, Boakye EA, Olomukoro SI, Deshields T, et al. Suicide risk among cancer survivors: Head and neck versus other cancers. Cancer. 2018 Oct 15;124(20):4072-9.
5. Alsahafi E, Begg K, Amelio I, Raulf N, Lucarelli P, Sauter T, et al. Clinical update on head and neck cancer: molecular biology and ongoing challenges. Cell Death & Disease. 2019 Aug 15;10(8):540.
6. Mendenhall WM, Dagan R, Bryant CM, Fernandes RP. Radiation Oncology for Head and Neck Cancer. Oral and Maxillofacial Surgery Clinics of North America. 2019 Feb;31(1):31-8.
7. Dietz A, Rudat V, Dreyhaupt J, Pritsch M, Hoppe F, Hagen R, et al. Induction chemotherapy with paclitaxel and cisplatin followed by radiotherapy for larynx organ preservation in advanced laryngeal and hypopharyngeal cancer offers moderate late toxicity outcome (DeLOSI- trial). European Archives of Oto-Rhino-Laryngology. 2009 Aug 30;266(8):1291-300.
8. Bhardwaj V, Cascone T, Cortez MA, Amini A, Evans J, Komaki RU, et al. Modulation of c-Met signaling and cellular sensitivity to radiation. Cancer. 2013 May 15;119(10):1768-75.
9. Medová M, Aebersold D, Zimmer Y. The Molecular Crosstalk between the MET Receptor Tyrosine Kinase and the DNA Damage Response—Biological and Clinical Aspects. Cancers. 2013 Dec 19;6(1):1-27.
10. Huang R-X, Zhou P-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduction and Targeted Therapy. 2020 Dec 1;5(1):60.
11. Hartmann S, Bhola NE, Grandis JR. HGF/Met Signaling in Head and Neck Cancer: Impact on the Tumor Microenvironment. Clinical Cancer Research. 2016 Aug 15;22(16):4005-13.
12. Rothenberger N, Stabile L. Hepatocyte Growth Factor/ c-Met Signaling in Head and Neck Cancer and Implications for Treatment. Cancers. 2017 Apr 24;9(12):39.
13. Liu T, Li Q, Sun Q, Zhang Y, Yang H, Wang R, et al. MET inhibitor PHA-665752 suppresses the hepatocyte growth factor-induced cell proliferation and radioresistance in nasopharyngeal carcinoma cells. Biochemical and Biophysical Research Communications. 2014 Jun;449(1):49-54.
14. Baschnagel Andrew M, Galoforo Sandra, Thibodeau Bryan J, Ahmed Samreen, Nirmal Sonali, Akervall Jan, et al. Crizotinib Fails to Enhance the Effect of Radiation in Head and Neck Squamous Cell Carcinoma Xenografts. Anticancer Research. 2015;35(11):5973-82.
15. Bhardwaj V, Zhan Y, Cortez MA, Ang KK, Molkentine D, Munshi A, et al. C-Met Inhibitor MK-8003 Radiosensitizes c-Met–Expressing Non–Small-Cell Lung Cancer Cells With Radiation-Induced c-Met–Expression. Journal of Thoracic Oncology. 2012 Aug;7(8):1211-7.
16. de Bacco F, Luraghi P, Medico E, Reato G, Girolami F, Perera T, et al. Induction of MET by Ionizing Radiation and Its Role in Radioresistance and Invasive Growth of Cancer. JNCI: Journal of the National Cancer Institute. 2011 Apr;103(8):645-61.
17. Qian L-W, Mizumoto K, Inadome N, Nagai E, Sato N, Matsumoto K, et al. Radiation stimulates HGF receptor/c- Met expression that leads to amplifying cellular response to HGF stimulation via upregulated receptor tyrosine phosphorylation and MAP kinase activity in pancreatic cancer cells. International Journal of Cancer. 2003 May 1;104(5):542-9.
18. Nisa L, Francica P, Giger R, Medo M, Elicin O, Friese-Hamim M, et al. Targeting the MET Receptor Tyrosine Kinase as a Strategy for Radiosensitization in Locoregionally Advanced Head and Neck Squamous Cell Carcinoma. Molecular Cancer Therapeutics. 2020 Feb;19(2):614-26.
19. Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984 Sep;311(5981):29-33.
20. Park M, Dean M, Kaul K, Braun MJ, Gonda MA, vande Woude G. Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proceedings of the National Academy of Sciences. 1987 Sep 1;84(18):6379-83.
21. Ma PC. c-Met: structure, functions and potential for therapeutic inhibition. Cancer and Metastasis Reviews. 2003;22(4):309-25.
22. Szabo R, Rasmussen AL, Moyer AB, Kosa P, Schafer JM, Molinolo AA, et al. c-Met-induced epithelial carcinogenesis is initiated by the serine protease matriptase. Oncogene. 2011 Apr 10;30(17):2003-16.
23. Jeon H-M, Lee J. MET: roles in epithelialmesenchymal transition and cancer stemness. Annals of Translational Medicine. 2017 Jan;5(1):5-5.
24. Madoz-Gúrpide J, Zazo S, Chamizo C, Casado V, Caramés C, Gavín E, et al. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. Journal of Translational Medicine. 2015 Dec 29;13(1):282.
25. Morello S, Olivero M, Aimetti M, Bernardi M, Berrone S, di Renzo MF, et al. MET receptor is overexpressed but not mutated in oral squamous cell carcinomas. Journal of Cellular Physiology. 2001 Dec;189(3):285-90.
26. Seiwert TY, Jagadeeswaran R, Faoro L, Janamanchi V, Nallasura V, el Dinali M, et al. The MET Receptor Tyrosine Kinase Is a Potential Novel Therapeutic Target for Head and Neck Squamous Cell Carcinoma. Cancer Research. 2009 Apr 1;69(7):3021-31.
27. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Science Signaling. 2013 Apr 2;6(269):pl1-pl1.
28. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discovery. 2012 May;2(5):401-4.
29. Cho YA, Kim EK, Heo SJ, Cho BC, Kim HR, Chung JM, et al. Alteration status and prognostic value of MET in head and neck squamous cell carcinoma. Journal of Cancer. 2016;7(15):2197-206.
30. Aebersold DM, Landt O, Berthou S, Gruber G, Beer KT, Greiner RH, et al. Prevalence and clinical impact of Met Y1253D-activating point mutation in radiotherapy-treated squamous cell cancer of the oropharynx. Oncogene. 2003 Nov 20;22(52):8519-23.
31. di Renzo MF, Olivero M, Martone T, Maffe A, Maggiora P, de Stefani A, et al. Somatic mutations of the MET oncogene are selected during metastatic spread of human HNSC carcinomas. Oncogene. 2000 Mar 16;19(12):1547- 55.
32. Tao X, Hill KS, Gaziova I, Sastry SK, Qui S, Szaniszlo P, et al. Silencing Met receptor tyrosine kinase signaling decreased oral tumor growth and increased survival of nude mice. Oral Oncology. 2014 Feb;50(2):104-12.
33. Galeazzi E, Olivero M, Gervasio FC, de Stefani A, Valente G, Comoglio PM, et al. Detection of MET oncogene/hepatocyte growth factor receptor in lymph node metastases from head and neck squamous cell carcinomas. European Archives of Oto-Rhino-Laryngology. 1997 Jan;254(S1):S138-43.
34. Ren X, Wang J, Lin X, Wang X. E-cadherin expression and prognosis of head and neck squamous cell carcinoma: evidence from 19 published investigations. OncoTargets and Therapy. 2016 Apr;2447.
35. Theys J, Jutten B, Habets R, Paesmans K, Groot AJ, Lambin P, et al. E-Cadherin loss associated with EMT promotes radioresistance in human tumor cells. Radiotherapy and Oncology. 2011 Jun;99(3):392-7.
36. Kim C-H, Kim J, Kahng H, Choi EC. Change of E-Cadherin by Hepatocyte Growth Factor and Effects on the Prognosis of Hypopharyngeal Carcinoma. Annals of Surgical Oncology. 2007 May 16;14(5):1565-74.
37. Xi W-H, Yang L-Y, Cao Z-Y, Qian Y. Tivantinib (ARQ- 197) exhibits anti-tumor activity with down-regulation of FAK in oral squamous cell carcinoma. Biochemical and Biophysical Research Communications. 2015 Feb;457(4):723-9.
38. Zeng Q, Chen S, You Z, Yang F, Carey TE, Saims D, et al. Hepatocyte Growth Factor Inhibits Anoikis in Head and Neck Squamous Cell Carcinoma Cells by Activation of ERK and Akt Signaling Independent of NFκB. Journal of Biological Chemistry. 2002 Jul;277(28):25203-8.
39. Hervieu A, Kermorgant S. The Role of PI3K in Met Driven Cancer: A Recap. Frontiers in Molecular Biosciences. 2018 Oct 24;5.
40. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003 Dec 8;22(56):8983-98.
41. Rieckmann T, Kriegs M. The failure of cetuximabbased de-intensified regimes for HPV-positive OPSCC: A radiobiologists perspective. Clinical and Translational Radiation Oncology. 2019 Jul;17:47-50.
42. Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz- Rubio E, Rolland F, et al. Open-Label, Uncontrolled, Multicenter Phase II Study to Evaluate the Efficacy and Toxicity of Cetuximab As a Single Agent in Patients With Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck Who Failed to Respond to Platinum- Based Therapy. Journal of Clinical Oncology. 2007 Jun 1;25(16):2171-7.
43. Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008 Jun 25;27(28):3944-56.
44. Wang D, Qian G, Zhang H, Magliocca KR, Nannapaneni S, Amin ARMR, et al. HER3 Targeting Sensitizes HNSCC to Cetuximab by Reducing HER3 Activity and HER2/ HER3 Dimerization: Evidence from Cell Line and Patient- Derived Xenograft Models. Clinical Cancer Research. 2017 Feb 1;23(3):677-86.
45. Leonard B, Brand TM, O'Keefe RA, Lee ED, Zeng Y, Kemmer JD, et al. BET Inhibition Overcomes Receptor Tyrosine Kinase–Mediated Cetuximab Resistance in HNSCC. Cancer Research. 2018 Aug 1;78(15):4331-43.
46. Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biology & Therapy. 2011 May 27;11(9):777-92.
47. Yang H, Mo C, Liu G L, Li W, Guan J, Liu J, et al. Combination of cetuximab with met inhibitor in control of cetuximab-resistant oral squamous cell carcinoma. American Journal of Translational Research. 2019;11(4).
48. Novoplansky O, Fury M, Prasad M, Yegodayev K, Zorea J, Cohen L, et al. MET activation confers resistance to cetuximab, and prevents HER2 and HER3 upregulation in head and neck cancer. International Journal of Cancer. 2019 Aug 1;145(3):748-62.
49. Ettl T, Viale-Bouroncle S, Hautmann MG, Gosau M, Kölbl O, Reichert TE, et al. AKT and MET signalling mediates antiapoptotic radioresistance in head neck cancer cell lines. Oral Oncology. 2015 Feb;51(2):158-63.
50. Xu H, Stabile LP, Gubish CT, Gooding WE, Grandis JR, Siegfried JM. Dual Blockade of EGFR and c-Met Abrogates Redundant Signaling and Proliferation in Head and Neck Carcinoma Cells. Clinical Cancer Research. 2011 Jul 1;17(13):4425-38.
51. Wang D, Lu Y, Nannapaneni S, Griffith CC, Steuer C, Qian G, et al. Combinatorial approaches targeting the EGFR family and c-Met in SCCHN. Oral Oncology. 2021 Jan;112:105074.
52. Kim JH, Kim BJ, Kim HS. Clinicopathological impacts of high c-Met expression in head and neck squamous cell carcinoma: a meta-analysis and review. Oncotarget. 2017 Dec 22;8(68):113120-8.
53. Kumar D, Kandl C, Hamilton CD, Shnayder Y, Tsue TT, Kakarala K, et al. Mitigation of Tumor-Associated Fibroblast-Facilitated Head and Neck Cancer Progression With Anti–Hepatocyte Growth Factor Antibody Ficlatuzumab. JAMA Otolaryngology–Head & Neck Surgery. 2015 Dec 1;141(12):1133.
54. Hari SB, Merritt EA, Maly DJ. Conformation- Selective ATP-Competitive Inhibitors Control Regulatory Interactions and Noncatalytic Functions of Mitogen- Activated Protein Kinases. Chemistry & Biology. 2014 May;21(5):628-35.
55. Wu Y-L, Soo RA, Locatelli G, Stammberger U, Scagliotti G, Park K. Does c-Met remain a rational target for therapy in patients with EGFR TKI-resistant nonsmall cell lung cancer? Cancer Treatment Reviews. 2017 Dec;61:70-81.
56. Broekman F, Giovanetti E, Peters J G. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World Journal of Clinical Oncology. 2011;2(2):80.
57. Liu L, Shi H, Liu Y, Anderson A, Peterson J, Greger J, et al. Synergistic Effects of Foretinib with HER-Targeted Agents in MET and HER1- or HER2-Coactivated Tumor Cells. Molecular Cancer Therapeutics. 2011 Mar;10(3):518- 30.
58. Bauman JE, Ohr J, Gooding WE, Ferris RL, Duvvuri U, Kim S, et al. Phase I Study of Ficlatuzumab and Cetuximab in Cetuximab-Resistant, Recurrent/Metastatic Head and Neck Cancer. Cancers. 2020 Jun 11;12(6):1537.
59. Munshi N, Jeay S, Li Y, Chen C-R, France DS, Ashwell MA, et al. ARQ 197, a Novel and Selective Inhibitor of the Human c-Met Receptor Tyrosine Kinase with Antitumor Activity. Molecular Cancer Therapeutics. 2010 Jun;9(6):1544-53.
60. Kochanny SE, Worden FP, Adkins DR, Lim DW, Bauman JE, Wagner SA, et al. A randomized phase 2 network trial of tivantinib plus cetuximab versus cetuximab in patients with recurrent/metastatic head and neck squamous cell carcinoma. Cancer. 2020 May 15;126(10):2146-52.
61. Markham A. Tepotinib: First Approval. Drugs. 2020 Jun 2;80(8):829-33.
62. Falchook GS, Kurzrock R, Amin HM, Xiong W, Fu S, Piha-Paul SA, et al. First-in-Man Phase I Trial of the Selective MET Inhibitor Tepotinib in Patients with Advanced Solid Tumors. Clinical Cancer Research. 2020 Mar 15;26(6):1237-46.
63. Friese-Hamim Manja, Bladt Friedhelm, Locatelli Guiseppe, Stammberger Uz, Blaukat Andree. The selective c-Met inhibitor tepotinib can overcome epidermal growth factor receptor inhibitor resistance mediated by aberrant c-Met activation in NSCLC models. American Journal of Cancer Research. 2017;7(4).
64. Sakai K, Nakamura T, Kinoshita T, Nakamura T, Matsumoto K. HGF-Antagonists: Structure, Activities, and Anti-cancer Approach. Current Signal Transduction Therapy. 2011 May 1;6(2):191-9.
65. Mizuno S, Nakamura T. HGF–MET Cascade, a Key Target for Inhibiting Cancer Metastasis: The Impact of NK4 Discovery on Cancer Biology and Therapeutics. International Journal of Molecular Sciences. 2013 Jan 7;14(1):888-919.
66. Vítor AC, Huertas P, Legube G, de Almeida SF. Studying DNA Double-Strand Break Repair: An Ever- Growing Toolbox. Frontiers in Molecular Biosciences. 2020 Feb 21;7.
67. Begg K, Tavassoli M. Inside the hypoxic tumour: reprogramming of the DDR and radioresistance. Cell Death Discovery. 2020 Dec 18;6(1):77.
68. Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair. 2009 Sep 2;8(9):1004-8.
69. Lempiäinen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. The EMBO Journal. 2009 Oct 21;28(20):3067-73.
70. Marechal A, Zou L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harbor Perspectives in Biology. 2013 Sep 1;5(9):a012716-a012716.
71. Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. International Journal of Radiation Oncology*Biology*Physics. 2004 Jul;59(4):928-42.
72. Sia J, Szmyd R, Hau E, Gee HE. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Frontiers in Cell and Developmental Biology. 2020 Feb 13;8.
73. Scully R, Panday A, Elango R, Willis NA. DNA doublestrand break repair-pathway choice in somatic mammalian cells. Nature Reviews Molecular Cell Biology. 2019 Nov 1;20(11):698-714.
74. Budke B, Lv W, Kozikowski AP, Connell PP. Recent Developments Using Small Molecules to Target RAD51: How to Best Modulate RAD51 for Anticancer Therapy? ChemMedChem. 2016 Nov 21;11(22):2468-73.
75. Medova M, Aebersold DM, Blank-Liss W, Streit B, Medo M, Aebi S, et al. MET Inhibition Results in DNA Breaks and Synergistically Sensitizes Tumor Cells to DNADamaging Agents Potentially by Breaching a Damage- Induced Checkpoint Arrest. Genes & Cancer. 2010 Oct 1;1(10):1053-62.
76. Cuneo KC, Mehta RK, Kurapati H, Thomas DG, Lawrence TS, Nyati MK. Enhancing the Radiation Response in KRAS Mutant Colorectal Cancers Using the c-Met Inhibitor Crizotinib. Translational Oncology. 2019 Feb;12(2):209-16.
77. Tumati V, Kumar S, Yu L, Chen B, Choy H, Saha D. Effect of PF-02341066 and radiation on non-small cell lung cancer cells. Oncology Reports. 2013 Mar;29(3):1094-100.
78. Li X, Heyer W-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Research. 2008 Jan 1;18(1):99-113.
79. Huang F, Mazin A v. Targeting the homologous recombination pathway by small molecule modulators. Bioorganic & Medicinal Chemistry Letters. 2014 Jul;24(14):3006-13.
80. Klein HL. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair. 2008 May;7(5):686-93.
81. Qiao G-B, Wu Y-L, Yang X-N, Zhong W-Z, Xie D, Guan X-Y, et al. High-level expression of Rad51 is an independent prognostic marker of survival in non-smallcell lung cancer patients. British Journal of Cancer. 2005 Jul 14;93(1):137-43.
82. Balbous A, Cortes U, Guilloteau K, Rivet P, Pinel B, Duchesne M, et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer. 2016 Dec 5;16(1):604.
83. Hansen LT, Lundin C, Spang-Thomsen M, Petersen LN, Helleday T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. International Journal of Cancer. 2003 Jul 1;105(4):472-9.
84. Nagathihalli NS, Nagaraju G. RAD51 as a potential biomarker and therapeutic target for pancreatic cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2011 Dec;1816(2):209-18.
85. Connell P P, Jayathilaka K, Haraf D, Weichselbaum R, Vokes E, Lingen M. Pilot study examining tumor expression of RAD51 and clinical outcomes in human head cancers. International Journal of Oncology. 2006;28(5).
86. King HO, Brend T, Payne HL, Wright A, Ward TA, Patel K, et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Reports. 2017 Jan;8(1):125-39.
87. Short SC, Giampieri S, Worku M, Alcaide-German M, Sioftanos G, Bourne S, et al. Rad51 inhibition is an effective means of targeting DNA repair in glioma models and CD133+ tumor-derived cells. Neuro-Oncology. 2011 May 1;13(5):487-99.
88. Chabot T, Defontaine A, Marquis D, Renodon-Corniere A, Courtois E, Fleury F, et al. New Phosphorylation Sites of Rad51 by c-Met Modulates Presynaptic Filament Stability. Cancers. 2019 Mar 23;11(3):413.
89. Popova M, Shimizu H, Yamamoto K, Lebechec M, Takahashi M, Fleury F. Detection of c-Abl kinasepromoted phosphorylation of Rad51 by specific antibodies reveals that Y54 phosphorylation is dependent on that of Y315. FEBS Letters. 2009 Jun 18;583(12):1867-72.
90. Bardelli A, Longati P, Gramaglia D, Basilico C, Tamagnone L, Giordano S, et al. Uncoupling signal transducers from oncogenic MET mutants abrogates cell transformation and inhibits invasive growth. Proceedings of the National Academy of Sciences. 1998 Nov 24;95(24):14379-83.
91. Ganapathipillai SS, Medova M, Aebersold DM, Manley PW, Berthou S, Streit B, et al. Coupling of Mutated Met Variants to DNA Repair via Abl and Rad51. Cancer Research. 2008 Jul 15;68(14):5769-77.
92. Medová M, Aebersold DM, Zimmer Y. MET inhibition in tumor cells by PHA665752 impairs homologous recombination repair of DNA double strand breaks. International Journal of Cancer. 2012 Feb 1;130(3):728- 34.
93. Davies AA, Masson J-Y, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, et al. Role of BRCA2 in Control of the RAD51 Recombination and DNA Repair Protein. Molecular Cell. 2001 Feb;7(2):273-82.
94. Rieckmann T, Tribius S, Grob TJ, Meyer F, Busch C-J, Petersen C, et al. HNSCC cell lines positive for HPV and p16 possess higher cellular radiosensitivity due to an impaired DSB repair capacity. Radiotherapy and Oncology. 2013 May;107(2):242-6.
95. Weaver AN, Cooper TS, Rodriguez M, Trummell HQ, Bonner JA, Rosenthal EL, et al. DNA double strand break repair defect and sensitivity to poly ADP-ribose polymerase (PARP) inhibition in human papillomavirus 16-positive head and neck squamous cell carcinoma. Oncotarget. 2015 Sep 29;6(29):26995-7007.
96. Bhide SA, Thway K, Lee J, Wong K, Clarke P, Newbold KL, et al. Delayed DNA double-strand break repair following platin-based chemotherapy predicts treatment response in head and neck squamous cell carcinoma. British Journal of Cancer. 2016 Sep 1;115(7):825-30.
97. Sun S, Wang Z. Head neck squamous cell carcinoma c-Met+ cells display cancer stem cell properties and are responsible for cisplatin-resistance and metastasis. International Journal of Cancer. 2011 Nov 15;129(10):2337- 48.
98. Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman JI, Prince ME, et al. Endothelial Cell-Initiated Signaling Promotes the Survival and Self-Renewal of Cancer Stem Cells. Cancer Research. 2010 Dec 1;70(23):9969-78.
99. Clay MR, Tabor M, Owen JH, Carey TE, Bradford CR, Wolf GT, et al. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head & Neck. 2010 Sep;32(9):1195-201.
100. Batlle E, Clevers H. Cancer stem cells revisited. Nature Medicine. 2017 Oct 1;23(10):1124-34.
101. Sun S, Liu S, Duan SZ, Zhang L, Zhou H, Hu Y, et al. Targeting the c-Met/FZD8 Signaling Axis Eliminates Patient-Derived Cancer Stem-like Cells in Head and Neck Squamous Carcinomas. Cancer Research. 2014 Dec 15;74(24):7546-59.
102. Lim YC, Kang HJ, Moon JH. c-Met pathway promotes self-renewal and tumorigenecity of head and neck squamous cell carcinoma stem-like cell. Oral Oncology. 2014 Jul;50(7):633-9.
103. Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Seminars in Cancer Biology. 2015 Apr;31:16-27.
104. de Bacco F, D'Ambrosio A, Casanova E, Orzan F, Neggia R, Albano R, et al. MET inhibition overcomes radiation resistance of glioblastoma stem-like cells. EMBO Molecular Medicine. 2016 May 4;8(5):550-68.
105. Esashi F, Christ N, Gannon J, Liu Y, Hunt T, Jasin M, et al. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature. 2005 Mar;434(7033):598-604.
106. Hochhauser D, Hartley JA. The interaction of EGFR and repair of DNA damage following chemotherapy and radiation. Drug Discovery Today: Disease Models. 2012 Jun;9(2):e69-73.
107. Bai J, Guo X-G, Bai X-P. Epidermal Growth Factor Receptor-Related DNA Repair and Radiation-Resistance Regulatory Mechanisms: A Mini-Review. Asian Pacific Journal of Cancer Prevention. 2012 Oct 31;13(10):4879- 81.
108. Tian Y, Tang L, Yi P, Pan Q, Han Y, Shi Y, et al. MiRNAs in Radiotherapy Resistance of Nasopharyngeal Carcinoma. Journal of Cancer. 2020;11(13):3976-85.
109. Chen G-Z, Dai W-S, Zhu H-C, Song H-M, Yang X, Wang Y-D, et al. Foretinib Enhances the Radiosensitivity in Esophageal Squamous Cell Carcinoma by Inhibiting Phosphorylation of c-Met. Journal of Cancer. 2017;8(6):983-92.
110. Welsh JW, Mahadevan D, Ellsworth R, Cooke L, Bearss D, Stea B. The c-Met receptor tyrosine kinase inhibitor MP470 radiosensitizes glioblastoma cells. Radiation Oncology. 2009 Dec 22;4(1):69.
111. Norbury C, Nurse P. Animal Cell Cycles and Their Control. Annual Review of Biochemistry. 1992 Jun;61(1):441-68.
112. Liu K, Zheng M, Lu R, Du J, Zhao Q, Li Z, et al. The role of CDC25C in cell cycle regulation and clinical cancer therapy: a systematic review. Cancer Cell International. 2020 Dec 3;20(1):213.
113. Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cellular and Molecular Life Sciences. 2013 Nov 19;70(21):4009-21.
114. Hassan Khaled A, El-Nagger Adel K, Soria Jean- Charles, Liu Dian, Hong Wuan K, Mao Li. Clinical significance of cyclin B1 protein expression in squamous cell carcinoma of the tongue. Clinical Cancer Research. 2001;7(8).
115. Androic I, Krämer A, Yan R, Rödel F, Gätje R, Kaufmann M, et al. Targeting cyclin B1 inhibits proliferation and sensitizes breast cancer cells to taxol. BMC Cancer. 2008 Dec 29;8(1):391.
116. Yu H, Li X, Sun S, Gao X, Zhou D. c-Met inhibitor SU11274 enhances the response of the prostate cancer cell line DU145 to ionizing radiation. Biochemical and Biophysical Research Communications. 2012 Oct;427(3):659-65.
117. Zhou B-BS, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nature Reviews Cancer. 2004 Mar;4(3):216-25.
118. Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25 activity in the cell cycle: clinical implications of CDC25 overexpression and potential treatment strategies. Molecular and Cellular Biochemistry. 2016 May 2;416(1-2):33-46.
119. Song N, Che X, Xu L, Qu J, Zheng H, Hou K, et al. A novel function of hepatocyte growth factor in the activation of checkpoint kinase 1 phosphorylation in colon cancer cells. Molecular and Cellular Biochemistry. 2017 Dec 1;436(1-2):29-38.
120. Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Molecular Cell. 2017 Jan;65(2):336- 46.
121. Kim D, Liu Y, Oberly S, Freire R, Smolka MB. ATRmediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells. Nucleic Acids Research. 2018 Sep 19;46(16):8311-25.
122. Wang H, Wang H, Powell SN, Iliakis G, Wang Y. ATR Affecting Cell Radiosensitivity Is Dependent on Homologous Recombination Repair but Independent of Nonhomologous End Joining. Cancer Research. 2004 Oct 1;64(19):7139-43.
123. van Harten AM, Buijze M, van der Mast R, Rooimans MA, Martens-de Kemp SR, Bachas C, et al. Targeting the cell cycle in head and neck cancer by Chk1 inhibition: a novel concept of bimodal cell death. Oncogenesis. 2019 Jul 17;8(7):38.
124. The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015 Jan 28;517(7536):576-82.
125. Leemans CR, Braakhuis BJM, Brakenhoff RH. The molecular biology of head and neck cancer. Nature Reviews Cancer. 2011 Jan 16;11(1):9-22.
126. Pellegata NS, Antoniono RJ, Redpath JL, Stanbridge EJ. DNA damage and p53-mediated cell cycle arrest: A reevaluation. Proceedings of the National Academy of Sciences. 1996 Dec 24;93(26):15209-14.
127. Goto H, Natsume T, Kanemaki MT, Kaito A, Wang S, Gabazza EC, et al. Chk1-mediated Cdc25A degradation as a critical mechanism for normal cell cycle progression. Journal of Cell Science. 2019 Jan 15;132(2):jcs223123.
128. Morgan MA, Lawrence TS. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clinical Cancer Research. 2015 Jul 1;21(13):2898-904.
129. Rischin D, Young RJ, Fisher R, Fox SB, Le Q-T, Peters LJ, et al. Prognostic Significance of p16INK4A and Human Papillomavirus in Patients With Oropharyngeal Cancer Treated on TROG 02.02 Phase III Trial. Journal of Clinical Oncology. 2010 Sep 20;28(27):4142-8.
130. Lindel K, Beer KT, Laissue J, Greiner RH, Aebersold DM. Human papillomavirus positive squamous cell carcinoma of the oropharynx. Cancer. 2001 Aug 15;92(4):805-13.
131. Fakhry C, Westra WH, Li S, Cmelak A, Ridge JA, Pinto H, et al. Improved Survival of Patients With Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma in a Prospective Clinical Trial. JNCI Journal of the National Cancer Institute. 2008 Feb 20;100(4):261-9.
132. Perri F, Longo F, Caponigro F, Sandomenico F, Guida A, della Vittoria Scarpati G, et al. Management of HPV-Related Squamous Cell Carcinoma of the Head and Neck: Pitfalls and Caveat. Cancers. 2020 Apr 15;12(4):975.
133. Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Frontiers in Oncology. 2015 Dec 21;5.
134. Ashwell S, Zabludoff S. DNA Damage Detection and Repair Pathways--Recent Advances with Inhibitors of Checkpoint Kinases in Cancer Therapy. Clinical Cancer Research. 2008 Jul 1;14(13):4032-7.
135. Deneka AY, Einarson MB, Bennett J, Nikonova AS, Elmekawy M, Zhou Y, et al. Synthetic Lethal Targeting of Mitotic Checkpoints in HPV-Negative Head and Neck Cancer. Cancers. 2020 Jan 28;12(2):306.
136. Mikami K, Medova M, Nisa L, Francica P, Gluck AA, Tschan MP, et al. Impact of p53 Status on Radiosensitization of Tumor Cells by MET Inhibition- Associated Checkpoint Abrogation. Molecular Cancer Research. 2015 Dec 1;13(12):1544-53.
137. Dok R, Glorieux M, Bamps M, Nuyts S. Effect of ATR Inhibition in RT Response of HPV-Negative and HPVPositive Head and Neck Cancers. International Journal of Molecular Sciences. 2021 Feb 3;22(4):1504.
138. Gadhikar MA, Sciuto MR, Alves MVO, Pickering CR, Osman AA, Neskey DM, et al. Chk1/2 Inhibition Overcomes the Cisplatin Resistance of Head and Neck Cancer Cells Secondary to the Loss of Functional p53. Molecular Cancer Therapeutics. 2013 Sep 1;12(9):1860-73.
139. Osman AA, Monroe MM, Ortega Alves M v., Patel AA, Katsonis P, Fitzgerald AL, et al. Wee-1 Kinase Inhibition Overcomes Cisplatin Resistance Associated with High- Risk TP53 Mutations in Head and Neck Cancer through Mitotic Arrest Followed by Senescence. Molecular Cancer Therapeutics. 2015 Feb 1;14(2):608-19.
140. Thanasoula M, Escandell JM, Suwaki N, Tarsounas M. ATM/ATR checkpoint activation downregulates CDC25C to prevent mitotic entry with uncapped telomeres. The EMBO Journal. 2012 Aug 15;31(16):3398-410.
141. Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004 Nov 17;432(7015):316-23.
142. Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010 Nov 23;29(46):6149-59.
143. Bartucci M, Svensson S, Romania P, Dattilo R, Patrizii M, Signore M, et al. Therapeutic targeting of Chk1 in NSCLC stem cells during chemotherapy. Cell Death & Differentiation. 2012 May 25;19(5):768-78.
144. Alsahafi EN, Thavaraj S, Sarvestani N, Novoplansky O, Elkabets M, Ayaz B, et al. EGFR overexpression increases radiotherapy response in HPV-positive head and neck cancer through inhibition of DNA damage repair and HPV E6 downregulation. Cancer Letters. 2021 Feb;498:80-97.
145. Srivastava AK, Hollingshead MG, Weiner J, Navas T, Evrard YA, Khin SA, et al. Pharmacodynamic Response of the MET/HGF Receptor to Small-Molecule Tyrosine Kinase Inhibitors Examined with Validated, Fit-for- Clinic Immunoassays. Clinical Cancer Research. 2016 Jul 15;22(14):3683-94.
146. Hughes VS, Siemann DW. Failures in preclinical and clinical trials of c-Met inhibitors: evaluation of pathway activity as a promising selection criterion. Oncotarget. 2019 Jan 4;10(2):184-97.