Abstract
Chimeric antigen receptor (CAR)-T cell therapy has shown potential in improving outcomes for individuals with hematological malignancies. However, achieving long-term full remission for blood cancer remains challenging due to severe life-threatening toxicities such as limited anti-tumor efficacy, antigen escape, trafficking restrictions, and limited tumor invasion. Furthermore, the interactions between CAR-T cells and their host tumor microenvironments have a significant impact on CAR-T function. To overcome these considerable hurdles, fresh methodologies and approaches are needed to produce more powerful CAR-T cells with greater anti-tumor activity and less toxicity. Despite advances in CAR-T research, microbial resistance remains a significant obstacle. In this review, we discuss and describe the basics of CAR-T structures, generations, challenges, and potential risks of infections in CAR-T cell therapy.
Keywords
Adoptive cellular therapy, CAR-T, Immunotherapy, Infection, COVID-19
Introduction
Immunotherapy aims to fine-tune the immune system, which has evolved to strike a balance between eliminating harmful pathogens and protecting tissues from the unintended damage of an inflammatory response. Therapies that stimulate, enhance, or inhibit the immune system are widely available in this field of study. Immune response can be influenced by cytokines, immune-modulators, immune checkpoint inhibitors, and monoclonal therapeutic antibodies. Immunotherapy includes cell-based treatment strategies in addition to therapeutic chemicals and proteins. Adoptive cellular therapy (ACT), which delivers disease-targeting cells transiently, is a promising approach for cancer, autoimmune diseases, and infectious diseases [1,2]. Chimeric antigen receptor (CAR)-T cell therapy is the most popular ACT approach. It entails the transfer of autologous or allogeneic T cells that have been altered to express a CAR. The CAR, which was first described by Eshhar et al. in 1993, enables modified T cells to generate an immune response specific to an antigen against cells that carry the CAR target antigen independently of the major histocompatibility complex (MHC) [3].
Currently, the FDA has approved five CAR-T cell therapies for hematological malignancies that have CD19 antigen or BCMA targets. Up to 75% of patients treated with CD19 or CD22 CAR-T cells for hematological malignancies relapsed, indicating that lasting remission following CAR-T cell therapy is not guaranteed [4,5]. The development of antigen-negative tumors under CAR-T surveillance mainly attributed to antigen escape, becoming a hallmark of CAR-T cell therapy failure [6]. However, recurrence occurs in antigen-positive illness, implying that CAR-T cell-intrinsic variables can lead to inadequate antitumor response. Treatment of solid tumors is further hampered by the capacity of CAR-T cells to infiltrate into the tumor and effectively destroy target cells in an immunosuppressive milieu [7,8]. The tumor microenvironment includes a barrier of stromal cells and extracellular matrix, as well as immunosuppressive cells, which inhibit CAR-T cell effector function. Tumor-infiltrating immune cells, such as regulatory T cells, produce an environment hostile to CAR-T cells by secreting inhibitory cytokines and depleting IL-2. These factors result in inability to clear antigens in cancer and chronic viral infections [9].
Despite CAR-T cell therapy provides an excellent anti-malignancy impact, adverse effects are great concern. These include grade 3 or 4 infections (10–31%), neurotoxicity or neurological problems (40–64%), neutropenia (53–87%), and cytokine release syndrome (CRS, 77–93%) [10-12]. Some of these 'off-tumor' effects can be modified by improving the structure and function of CAR-T cells. Infectious diseases remain a major cause of morbidity and mortality worldwide. Despite improvements in CAR-T research, the emergence of microbial resistance remains a major challenge. Alternative therapeutic approaches are required for patients who do not respond or relapse. In this review, we will discuss the fundamentals of CAR-T structures, generations, challenges, and potential risks of infection in CAR-T cell therapy.
Structural Attributes of CAR-T Cells
The CAR-T cell design has advanced through the integration of preexisting immune cell elements to enable the direct identification of tumor antigens. The CAR receptor is a hybrid receptor designed with three distinct structural components: an extracellular domain (ECD), a transmembrane domain (TMD), and an intracellular domain (END). The scFv of CAR consists of the light and heavy chains of the antibody variable region, while the CAR amplitude CD3ζ is generated from the intracellular signaling domains of the TCR [13]. The detailed studies of the CAR components are discussed below.
Extracellular domain
The extracellular domain of the CAR is made up of an antigen binding domain (BD) and a hinge region. The BD scaffold often contains a single-chain variable fragment (scFv) made up of variable regions from an antibody's light (VL) and heavy (VH) chains. The binding domain is tailored to identify tumor antigens such as CD19, BCMA, CD20, and CD30, independent of antigen processing and presentation by HLA [14]. The antigen recognition domain can also be non-antibody based constructions, such as engineered binding scaffolds, nanobodies, and naturally occurring ligands and receptors, in addition to antibody-based binding domains (scFv) (Figure 1). A balance between supraphysiological T-cell activity and elevated TCR affinity or avidity to detect low epitope densities is essential to avoid potentially harmful cross-reactivities, as highlighted below the intricate relationship between receptors, TCR affinity, avidity, and epitope density.
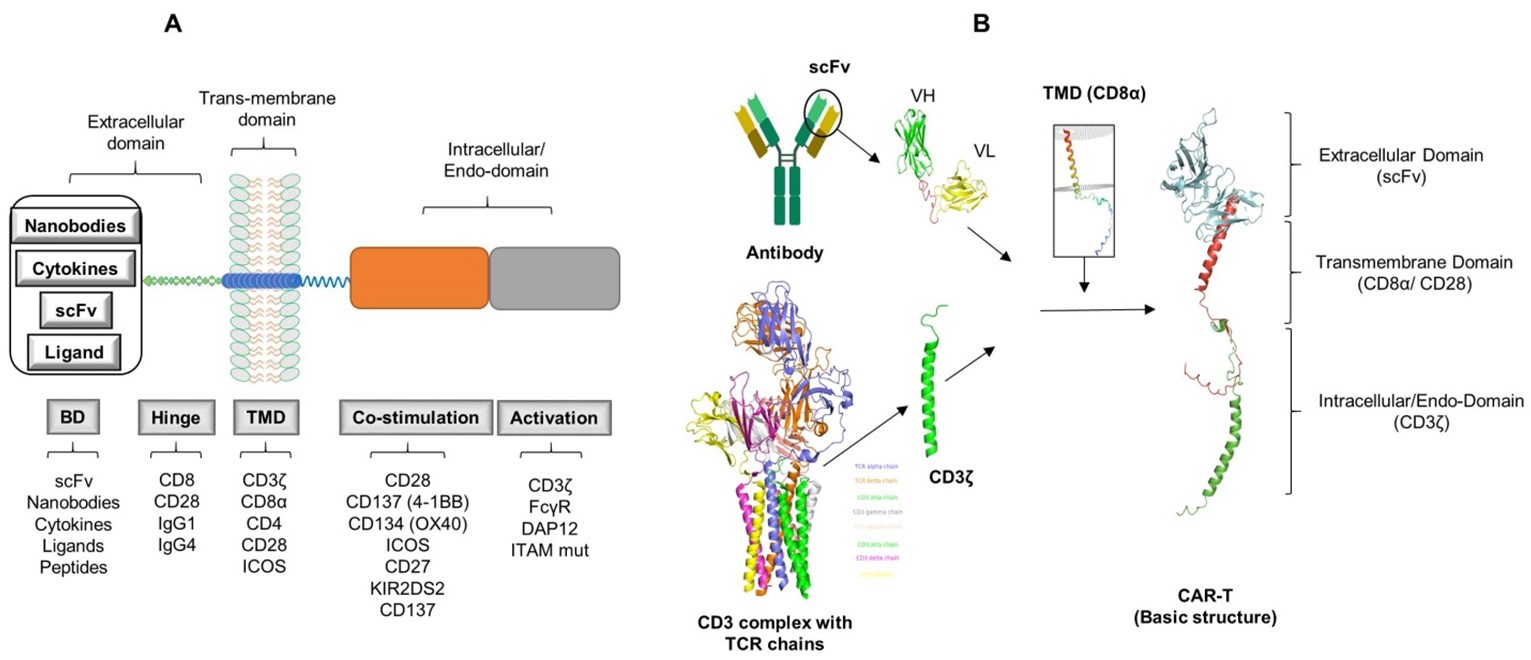
Figure 1. Basic structure of CAR-T. BD: Binding Domain; TMD: Trans-Membrane Domain; scFv: Single chain fragment of variable regions; VH: Variable region of Heavy chain; VL: Variable region of Light chain. Structure models were rendered by using PyMOL under the specific ID numbers from protein data bank (CD3 complex: 6JXR, CD8α: 8EW6, CD28: 7VU5, scFv model (CD19).
Affinity determination to improve CAR efficacy: Considering the widespread presence of the target antigen in healthy tissue, it is essential to regulate the scFv affinity to improve the CAR's specificity and reduce "on-target, off-tumor" adverse effects. CAR vectors with high affinity scFv sequences (KD <0.01 mM) displayed selective cytotoxicity towards highly expressing ErbB2 cells, whereas, anti-ErbB2 scFv with a low KD (dissociation constant) of 0.3 mM showed an opposite trend [15]. Similar to this, in a different study, affinity-modulated scFv sequences derived from monoclonal antibody mAb 4D5 were utilized to generate anti-ErbB2 CARs. Comparing CAR-T cells displaying a high-affinity 4D5 variant (KD ~0.6 nM) to CAR-T cells employing a lower-affinity 4D5 variant (KD ~1 mM), the latter exhibited an increased therapeutic efficacy in mice [16]. This was ascribed to low affinity scFv CARs' capacity to distinguish between tumors that express ErbB2 at higher densities than normal tissues, which is a characteristic of cancers. Caruso et al. evaluated the specificity of anti-EGFR CARs derived from Cetuximab against Nimotuzumab, which has 10 times less affinity than Cetuximab [17]. Unlike Cetuximab-based CARs, Nimotuzumab-based CARs demonstrated EGFR-density dependent activation in vitro but did not show potent affinity for low-density EGFR cells in vivo studies. In a different investigation, an anti-CD38 CAR with a low-affinity scFv (KD in the micromolar range) that was derived from an affinity-tuned antibody library was only cytotoxic to high-expression CD38 cells in vivo and in vitro, with no effect on normal CD38+ hematopoietic cells [18]. Similarly, thyroid cancer xenografts were cleared in mice without systemic damage by LFA-1 I domains with micromolar affinity to ICAM-1, which were more selective to cells expressing high levels of the target antigen (ICAM-1). While it has been shown that lowering affinity improves CAR-T cell specificity, there may be circumstances where it may decrease antitumor potency. CARs constructed from a 2A2 scFv with a 50-fold lower affinity showed less anti-tumor efficacy than anti-ROR1 CARs made from a higher affinity scFv (R12) [19]. Similarly, in mice models of acute myeloid leukemia, higher-affinity anti-FRb CARs (KD ~54.3 nM) demonstrated specific and complete elimination of tumors compared to lower affinity anti-FRb CARs (KD ~2.4 nM), which were ineffective against the disease [20]. However, non-specific off-tumor effects also led to significant neurotoxicity, despite the improved sensitivity and potency.
Affinity modification affects not only CAR signaling but also expansion, persistence, and cytokines production. In contrast to the conventional FMC63-based CARs (KD = 0.32 nM), which target similar epitopes on the CD19 antigen, low-affinity anti-CD19 CARs (CAT-CAR) (KD = 14.3 nM) showed higher proliferation and greater efficacy in vivo studies. While compared to the low-affinity CAT-CAR (both in vitro and in vivo), TNFα showed a slight increase, but IL-2 and IFNγ secretions were similar for both CARs [21]. Furthermore, it was observed that the scFv used in the CAT-CAR had a faster dissociation constant (Koff) (3 x 10-3s-1) than the FMC63 scFv (6.8 x 10-5s-1), suggesting might have contributed to its low affinity and limited the duration of receptor-ligand interactions. Faster Koff values may result in higher killing rates and consequently improve therapeutic effectiveness. It is anticipated that once the affinity is adequate, subsequent affinity improvements will not result in further CAR performance enhancement. Similar connections between signal strength and affinity factors such as Kon and Koff might impact how ligand-binding domain affinities influence CAR effectiveness. Therefore, ligand-binding affinities should be tuned by carefully evaluating the potential of on-target, off-tumor toxicity against the required potency of anti-tumor response.
Avidity implications in CAR expression: The affinity of the ligand-binding domain is an important parameter in CAR design. However, it remains a measure of monovalent receptor-ligand interactions. Multiple receptor-ligand interactions at the T cell-target cell interface, as well as receptor clustering at the immunological synapse, contribute to the overall effectiveness of interactions in both CAR-T cells and native T cells (Figure 2A) [22,23]. Avidity is a parameter that takes into account multiple interactions between the ligand and the receptor. It is influenced by affinity of particular ligand-binding domains, target cell ligand densities, and CAR expression levels. In one study, scFv sequences targeting HLA-A2-WT1 (Wilms tumor suppressor gene 1) peptide were used to develop CARs. Non-specific cross-reactivity with pMHCs exhibiting irrelevant peptides has been linked to both high affinity and avidity of CARs, which are co-expressed at high levels [23]. In another study, despite similar cytotoxicity against target cells, a high-affinity anti-CD123 CAR (KD = 2 nM) expressed at relatively low levels significantly reduced proliferation and cytokine production compared to a similarly high-affinity anti-CD123 CAR (KD = 1 nM) expressed at a much higher level, demonstrating avidity-related effects on effector functions [24].
Furthermore, numerous molecular engineering approaches are available to regulate CAR expression. In one study, it has been demonstrated that self-inactivating lentiviral vectors containing the EF1a promoter cause lower levels of CAR expression as compared to gammaretroviral vectors based on the LTR (long terminal repeat) promoter [25]. Moreover, compared to retrovirally integrated CARs, integrating CARs into the TRAC locus of T cells produced lower but dynamically controlled CAR surface expression. Additionally, T cells expressing CARs from the TRAC locus showed decreased tonic signaling and greater in vivo anti-tumor efficacy [26]. Given avidity considerations, ligand-binding domains must be evaluated in relation to the efficiency of CAR-T cells.
scFv aggregation results CAR tonic signals: Notably, in the absence of ligands, nonactivated T and B cells at quiescent state were shown to transmit a low-level constitutive signal characterized as a tonic TCR or B cell receptor (BCR) signal. Cell differentiation and continuation of cellular responses after antigen stimulation are mediated by tonic signaling from TCR or BCR (including pre-BCR) in lymphocytes [27,28]. In the case of CAR, self-aggregation of CAR, also known as the tonic signal, has been shown to produce different degrees of ligand-independent receptor signaling [29]. Remarkably, CAR-T cell exhaustion and malfunction have been associated with higher levels of tonic signaling. Tonic signaling is assumed to be caused by the structure of the CAR extracellular domain (Figure 2B). For instance, the tonic signal generated by CAR with the IgG1 CH2-CH3 region acting as a spacer between the transmembrane domain and scFv was greater than that of CAR with CH3 alone [30]. Aggregation of scFv has been related to tonic signaling and is involved in the regulation of CAR-T cell activity. An excessive amount of tonic signaling, or antigen-independent signaling, may ultimately lead to early T cell exhaustion.
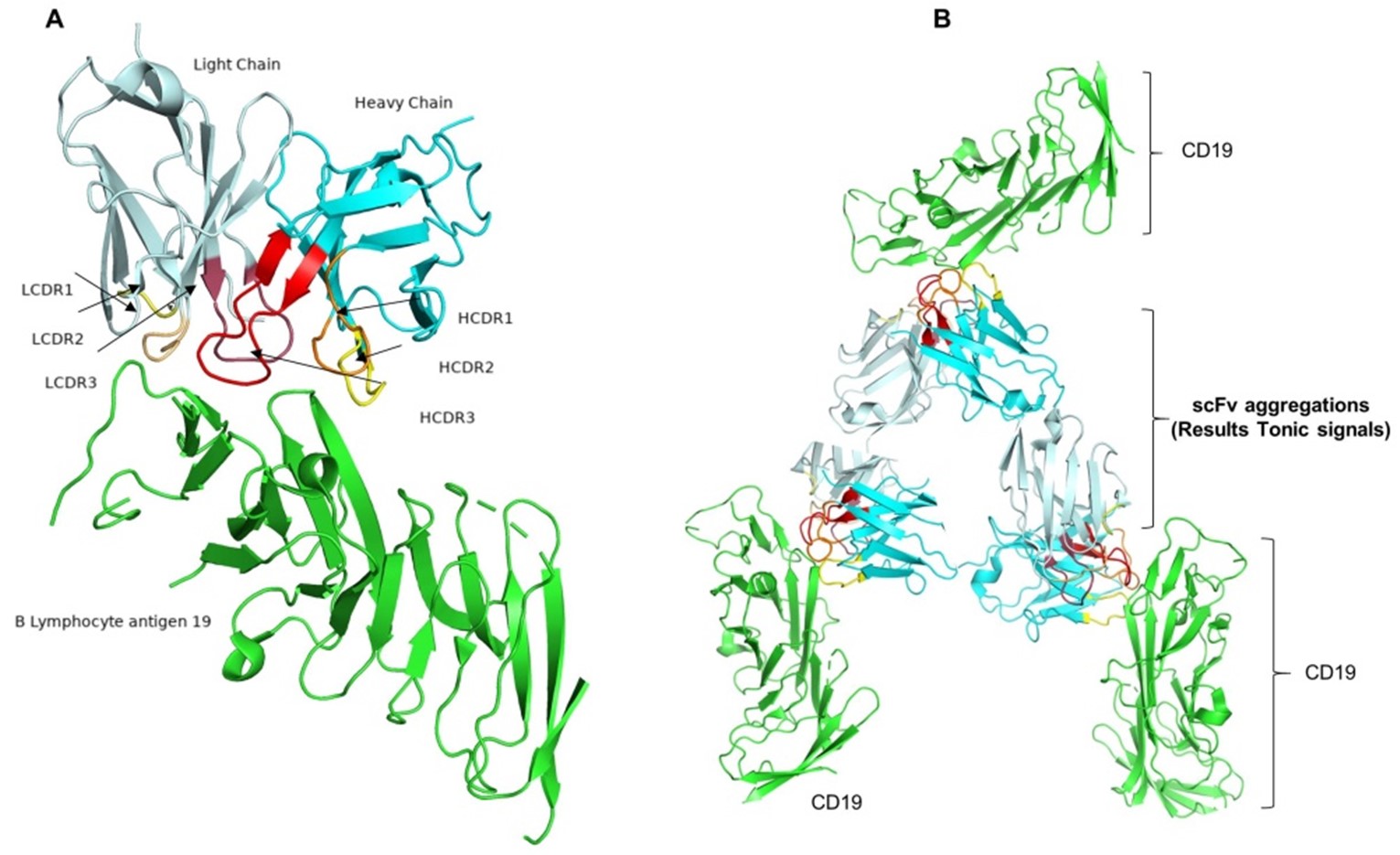
Figure 2. Structure and self-activation of the CAR are illustrated schematically. A: Fundamental structure of CAR. The extracellular domain of a CAR is the scFv, which has a variable region of heavy chain (VH) and variable region of light chain (VL), consisting of four framework regions (FR1-4) and three complementarity determining regions (CDR1-3) (red) (PBD: 6UUP). The green cartoon structure represents B lymphocytes antigen 19. B: Possible self-activation mechanism for CARs. CDRs are engaged in antigen binding, whereas FR regions are linked to self-aggregation. Self-activation can be prevented by developing a hybrid of CDRs from the self-activating CAR of interest and FRs from the non-self-aggregating CAR. The protein structure models were rendered by using PyMOL under the ID numbers (7URV and 6AL4) from protein data bank.
Long and colleagues reported that CAR surface aggregation leading to tonic signaling and exhaustion caused by framework areas of anti-GD2 14G2a scFv. Similarly, an anti-CD19 CAR (FMC63 scFv) was not found to exhibit tonic signaling. The authors discovered that substituting the anti-GD2 14G2a scFv framework regions with the anti-CD19 FMC63 CAR-scFv framework regions led to a higher degree of exhaustion [31]. It is challenging to determine whether removing scFv aggregative sequences will halt tonic signaling because anti-GD2 14G2a CAR modified with framework sections from FMC63 scFv did not express on the cell surface. Another study on tonic signaling found that CD28-CD3ζ second-generation anti-c-Met and anti-Mesothelin CAR-T cells exhibited antigen-independent proliferation without exogenous IL-2, but not CD28-CD3ζ FMC63-based anti-CD19 CAR-T cells [32]. The authors also reported a relationship between enhanced continuous antigen-independent proliferation and higher CAR expression. While scFv aggregation was not specifically addressed by the authors, the continuous proliferation phenotype observed may be due to the combinatory action of the scFv and CD28 costimulatory domain. Higher CAR expression levels may enhance aggregation potential on the cell surface and facilitate the dynamic switching of VH-VL domains between various CAR components [33]. Particularly when antigen densities on target cells need higher CAR expression, it is critical to carefully regulate the balance between high expression and aggregation propensities.
Antigen epitope location and accessibility: The adaptability of the CAR's modular structure makes it possible to target challenging epitopes, such as larger, thicker cell surface receptors, particularly tumor-associated molecules with heterogeneous glycosylation, such as MUC1 or mesothelin (MSLN). The reason of this anti-MUC1 SM3-scFv-based CARs’ poor efficacy was identified as glycosylation-independent steric hindrance [34]. When compared to CARs based on a membrane-distal epitope, CARs based on scFv that targeted the membrane-proximal region (Region III) of the MSLN molecule showed better functional response (cytotoxicity and cytokine production) both in vivo and in vitro. The researchers ascribed this to enhanced signal transmission enabled by the stiff structure of the membrane-proximal area. Additionally, the membrane-distal region of MSLN interacts functionally with proteins, such as CA125 (MUC16), which may impede the binding of CAR [35]. This implies that structural and functional characteristics of the target epitope, in addition to steric availability, should be taken into account when constructing CARs. Additional research in design may be necessary to determine suitable CAR structures that enable accessibility to both targets in novel CAR designs, such as bispecific CARs that target two antigens utilizing tandemly connected scFv sequences [36].
The hinge (spacer domain)
The extracellular domain comprises the hinge region, also known as the spacer domain. This region is a short portion of the ECD that is mainly derived from immunoglobulin G (IgG) and sporadically from the hinges of CD28 and CD8α. It provides a connection between the END and the ECD by bridging the gap between the TMD and the ECD. The main objectives are to increase target T-cell and CAR-T cell synapse formation, antigen attachment, and flexibility [37]. The CAR has more flexibility and access to membrane-proximal epitopes with longer hinges, whereas shorter spacers have less flexibility and target the antigen's distal epitopes [38,39]. Moreover, synaptic cleft lengths and consequently signaling events like kinetic segregation can be controlled via spacer domain modification. Generally, membrane-distal epitopes need shorter spacers to maintain the ideal synapse distance, while membrane-proximal epitopes need longer spacers (Figure 3A) [19]. Increasing the distance between two epitopes can reduce the exclusion of inhibitory phosphatases and also hinder the delivery of granzymes and perforins to the target cell, which reduces lytic efficiency. The extremely dense immune synapse in a physiological T-cell milieu prevents lytic granules from diffusing, which improves pore formation through perforins and granzymes delivery (Figure 3B) [40]. Although CAR-T immune synapses are not conventional, lytic granule transport and kinetic segregation are still assumed to be essential to CAR-T cell signaling and killing actions [41]. As a result, changing the spacer lengths can significantly impact the cytolytic activity and signaling of CAR-T cells. In a previous study, first-generation anti-CEA CARs were evaluated with or without an IgG1-Fc spacer [38]. The insertion of the IgG1-Fc spacer was shown to decrease IFNγ release without triggering a drop in lytic efficiency. The authors evaluated the same CARs in cell lines that expressed a shorter variant of the antigen in a membrane proximal site in an attempt to determine whether this impact was caused by epitope location. This did not, however, change the previously observed trend in IFNγ or lytic efficiency, which the authors had attributed to potential steric hindrances. The findings of this study emphasize the importance of considering ligand density and steric accessibility while designing spacer domains [38]. Spacer length has also been purported to affect mechano-transduction of ligand recognition. CARs with longer spacers (IgG4-Fc) that were generated against soluble homo-dimeric TGF-b showed decreased activation profiles compared to shorter (IgG4 hinge only) spacers [42]. IgG1-Fc and IgG4-Fc based spacers can be mutated to minimize FcγR interactions (e.g., by replacing the CH2 domain with an IgG2 CH2 domain and/or introducing mutations in other regions that minimize interactions with FcγR) for CARs where a long spacer is required to achieve optimal spacing between T cells and target cells. IgG2-based spacers have been employed sparingly in CARs because to their exceedingly low binding affinity to FcγR [43].
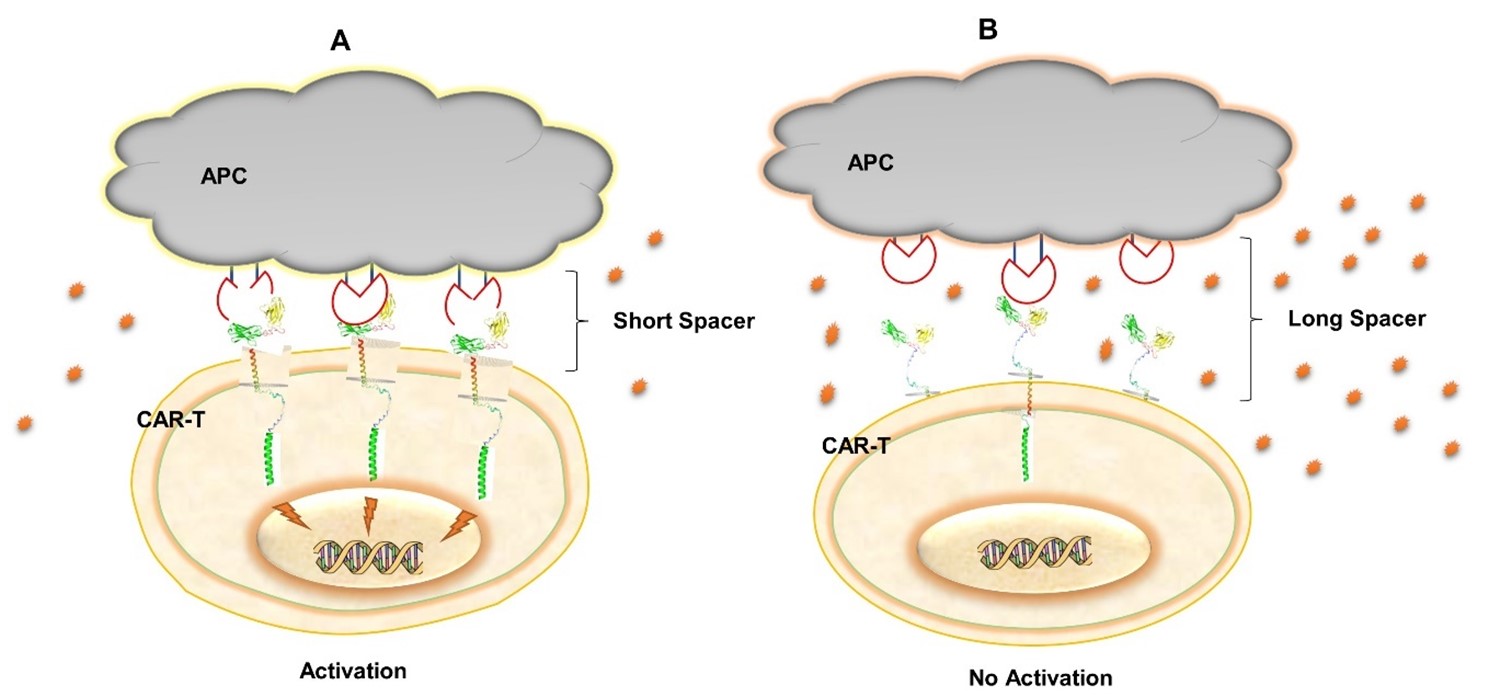
Figure 3. The spacing between synaptic clefts can be modulated using spacer design. (A) Short spacers shrink the synaptic gap when targeting membrane distal epitopes, allowing phosphatases to be excluded and hence increasing phosphorylation of cytoplasmic ITAMs, whereas long spacers (B) extend the synaptic cleft and may not exclude phosphatases.
Non-IgG-based spacers such as CD8 and CD28 hinge regions were utilized in clinically approved CAR-T cell therapies and have proven effective. Alabanza et al. discovered that inserting the CD28a hinge region into an anti-CD19 CAR increased activation-induced cell death (AICD) [44]. While there was no noticeable shift in cytotoxicity or in vivo tumor control, CD28 hinge-incorporating CAR-T cells exhibited increased levels of cytokine production (TNFα and IFNγ). The authors attributed this to the structure of the CD28 hinge, which is more prone to dimerization than the CD8α hinge and predicted that enhanced dimerization of CD28 hinge-CARs on the cell surface results in more activation signals, and thus greater AICD. Improved antigen-independent dimerization of CARs with S228P mutation in IgG4 hinge was also demonstrated to promote in vivo tumor regression and in vitro cytotoxicity [45].
Thus, these studies demonstrate that whereas structural characteristics of the hinge region can be utilized to alter CAR avidity and affinity, the functional implications of such modifications are not broadly applicable. It is possible to manipulate synaptic cleft distances through spacer length modification, which may then control signaling. Short spacers shrink the synaptic gap when targeting membrane distal epitopes, allowing phosphatases to be excluded and hence increasing phosphorylation of cytoplasmic ITAMs, whereas long spacers extend the synaptic cleft and may not exclude phosphatases.
Transmembrane domain (TMD): The TMD connects the extracellular domain to the intracellular domain, helps to express, anchor, and stabilize the CAR to the cell membrane, and allows effective CAR-T cell signaling [46,47]. Cellular adhesion and expression level are mostly regulated by the TMD of CAR. In comparison to other domains, TMDs have received less attention in systematic investigations of CAR design. TMD is typically designed to serve as the basis for the adjacent hinge or intracellular domains, including CD4, CD8α, CD28, CD4, ICOS, or CD3ζ (Figure 1). However, there is a significant possibility of natural receptor TMDs may interact with naive T-cell components that could hinder the efficaciousness and logical design of CARs. Elazar et al. recently identified programmable membrane proteins (proMPs), a class of de novo-designed receptor TMDs that can adjust the activities of modified CAR receptors. proMPs are revolutionary design tools that generate transmembrane homo-oligomers and contain entirely new sequences that are used to create novel constructions known as proCARs, or programmable CARs [48]. While compared to natural CD28 TMD containing CAR, the proCAR constructs significantly attenuate inflammatory cytokine production while presenting T-cells with a predictable range of in vivo functional potencies. Additionally, it has been demonstrated that TMD changes have no direct impact on the CAR's antigen-binding or signaling domains, indicating that this tactic may aid in the development of CAR-T cell therapies with the most favorable safety and efficacy characteristics.
Intracellular domain (END): An intracellular domain, also known as an endoplasmic domain or cytoplasmic tail, is the third CAR domain discovered in CAR-T-cells. CARs are constructed on the natural structure of TCRs, integrating various functional components. The TCR (CD3ζ) co-receptor utilizes three ITAMs for transmitting signals, making it a significant carrier of TCR signals [47]. The co-stimulatory molecules (CMs) included in this domain are CD28, CD27, CD134 (OX40), CD137, CD137 (4-1BB), and KIR2DS2, which influence metabolic cycles, apoptosis, and activation-induced cell death in addition to T cell differentiation pathways [49]. The large number of ITAMs likely contributes to signal amplification, as lowering the number of ITAMs impairs TCR-CD3 activity in murine models. Furthermore, even when the number of ITAMs remains constant, diversity is critical for signal transduction and T cell growth. Despite the ITAMs, the ICDs of each CD3 subunit have distinct molecular interactions. Basic rich stretches (BRSs), found on ζ and CD3ε, facilitate ionic interactions with the plasma membrane's inner leaflet [50]. CD3ε engages in contacts with the kinase Lck by noncanonical means involving the receptor kinase (RK) motif and the Lck SH3 domain, or through ionic interactions between the BRS and the acidic residues in the Lck unique domain [51]. Additionally, proline-rich sequences (PRS) found on CD3ε attract other proteins, including the adaptor Nck. TCR downregulation is facilitated by a membrane-proximal di-leucine motif present on CD3γ [52]. Velasco Cárdenas and colleagues recently generated novel CD3 CARs with only one of the CD3 intracellular domains [53]. CARs with CD3δ, CD3ε, or CD3γ cytoplasmic tails outperformed conventional ζ CAR-T cells in vivo. Transcriptomic and proteomic studies indicated variations in activation potential, metabolism, and stimulation-induced T cell dysfunctionality, which may explain the improved anti-tumor performance. Furthermore, dimerization of the CARs increased their overall functionality. Using these CARs as minimalistic and synthetic surrogate TCRs, the authors found the phosphatase SHP-1 as a novel interaction partner of CD3δ. SHP-1 binds to CD3δ-ITAM on phosphorylation of its C-terminal tyrosine. SHP-1 inhibits and restrains activation signals, perhaps preventing depletion and malfunction [53].
A significant amount of studies indicate that the synthetic component of the receptor design may be connected to CAR toxicity. Huang and colleagues developed a natural multi-chain immunoreceptor CAR based on the DNAX-activating protein of 12 kDa (Dap12) signaling domain for the first time in order to increase the safety of CAR-transduced T cells. This leads to antigen-specific cytotoxicity, cytokine production, and proliferation that is equivalent to CD3ζ-based CARs ex vivo/in vitro for hematological malignancies [54]. A transmembrane signaling adaptor protein known Dap12 includes a single immunoreceptor tyrosine-based activation motif (ITAM) with minimal homology to ITAMs found in the CD3ζ chain. Certain T cells, macrophages, and natural killer (NK) cells have been identified as the immune cells that were found to express Dap12, revealing that Dap12 may play an additional function in the immune response [55]. Dap12 was first discovered to activate NK cells upon ligation of its ligand with a corresponding receptor, thereby inducing phosphorylation of tyrosine residues in the ITAM and SRC-family kinase activation [46,56]. More than 20 Dap12-related receptors, including TREM1, TREM2, and KIRS, have currently been identified. Given the highly adverse biological features of the enrolled respondents B-ALL, such as a substantial tumor burden, rapid progressing disease, and high-risk genetics, the DAP12-BB CAR demonstrates a reduced toxicity profile and increased anti-tumor efficiency in B cell malignancies [57].
Incorporating co-stimulatory signaling domains, such as ICOS or 4-1BB, can improve CAR-T cell functions and, consequently, immune responses. These CAR-engineered T cells demonstrated greater persistence and effector functions, as well as better anticancer activity, paving the way for a novel approach to solid tumor treatment. Xiao Liang and colleague discovered that the CAR-T cells expressing dectin-1 have a distinctive phenotype and expression of an exhaustion signature [58]. The authors assessed the effects of the dectin-1 signaling domain on CAR-T cells both in vitro and in vivo. They further demonstrated that the incorporation of this dectin-1 signaling domain enhances the in vitro cytokine secretions by CD19 or HER2 specific CAR-T cells. Kagoya et al. developed 4-1BBζ and CD28ζ CARs with a truncated cytoplasmic domain of IL-2Rβ and a STAT3-binding (YXXQ) motif. The therapeutic effect of this modification was reported to be higher to that generated by CARs including only CD28 or 4-1BB costimulatory domains. It further enhanced CAR-T cell proliferation and prevented terminal effector cell differentiation [59]. In one study, Nair et al. compared CD28-based third-generation CARs with addition of 4-1BB, CD27, OX40, ICOS, or IL-15Rα to END. They found that the cytoplasmic domain of IL-15Rα showed greatest proliferation and rapid acquisition of effector cell function [60]. An unprecedented level of control over T cell fate and function becomes possible by the inclusion of multiple intracellular signaling domains into CARs. The identification of novel costimulatory pathways and the utilization of existing accessory molecules may prove advantageous for future CAR designs.
Generation of CAR-T
Since its development in the late 1980s, CAR-T cell therapies have made significant progress in enhancing activation, persistence, proliferation, safety, and efficacy. In the past thirty years, CAR-T cell therapies have undergone five generations, with changes to the endo-domain structure and the number of CMs utilized [61]. The intracellular domain of the CAR receptor has undergone considerable structural, molecular, and functional changes during these generations, despite their basic conformation and other domains remaining unchanged as shown in (Figure 4). The first generation of CARs had a simple structure with a single activation domain (the CD3ζ chains). The therapeutic efficacy of CD3ζ's basic structure was modest. The structure may stimulate T cells by sending signals, but it did not enhance cell growth, which is a vital step in the treatment of disease. In order to circumvent this issue, second-generation CAR-Ts were modified by adding a costimulatory signaling domain (such as CD28, 4-1BB, and OX40) to the intracellular region. This provided a dual-signal structure that significantly increased cell proliferation and enhanced activity [62]. The third-generation CARs were produced by introducing two costimulatory domains in addition to the activation domain CD3ζ chains, thereby increasing the survival of CAR-T cells. Fourth-generation CAR-T cells were then developed. TRUCKs are T cells that have been redirected for antigen-unrestricted cytokine-initiated killing. They are changed by inserting extra transgenes that cause the production of inducible cytokines (for example, interleukin (IL)-12). This resulted in the improvement of cell function as well as the regulation of the tumor microenvironment (TME) [63]. A fifth generation of CARs is being developed, based on the second generation, but with a reduced cytoplasmic IL-2 receptor β-chain domain and a binding site for the transcription factor STAT3. Antigen-specific activation of this receptor activates TCR (CD3ζ domains), co-stimulatory (CD28 domain), and cytokine (JAK-STAT3/5) signaling, providing all three synergistic signals needed for full T cell activation and proliferation [59].
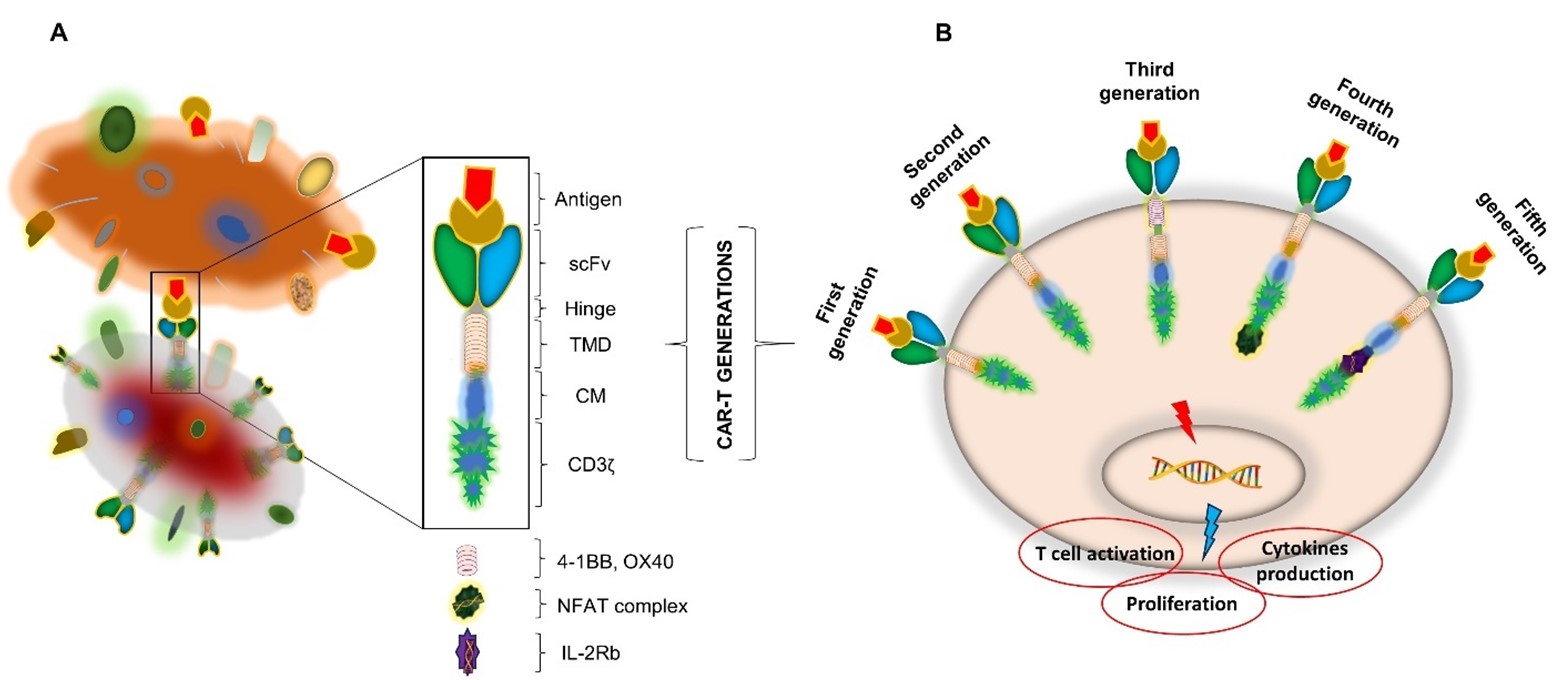
Figure 4. Basics of CAR-T and its generations. A: CAR-T cell interaction and basic components presentation. B: CAR-T cell therapies have undergone five generations, with changes to the endo-domain structure and the number of CMs (Co-stimulation molecule) utilized.
The first generation of CAR-T was created in 1993 and had no additional CM. It had an extracellular domain that included scFv and a cytoplasmic domain that contained a CD3ζ (FcεRIγ) signaling domain (Figure 5) [3]. This triggers the TCR signaling pathway, which mediates the production of cytokines (such IL-2) without requiring the presence of HLA. Major signaling motifs that generate the activating signal (signal 1) after ligand binding are commonly designed into first-generation CARs. After ligand binding, second- and third-generation CARs are designed to release one or more co-stimulatory signals (signal 2) in addition to the activation signal (signal 1). The majority of first-generation CAR-T cells lacked in vivo proliferation and persistence and primarily used the intracellular CD3ζ domain as their primary signaling motif [61]. The predominant activation region of Fc receptors was also the g-chain in early investigations [64]. First-generation anti-CEA CARs with the FcεR1γ chain were compared to those with the CD3ζ cytoplasmic domain in another study conducted by Haynes et al. As a result of having three ITAMs in monomeric CD3ζ compared to one in FcεR1γ, this study demonstrated that CD3ζ -based CARs produced more IFNγ and triggered more cytotoxicity than FcεR1γ -based CARs [65]. First generation CAR-T cells were found to exhibit impaired T-cell proliferation, minimal cytokine release, and poor in vivo persistence of T-cell responses due to the lack of CM and cytokine-mediated interactions. Because of this decreased anticancer efficacy, First generation CARs are now considered as outdated [66].
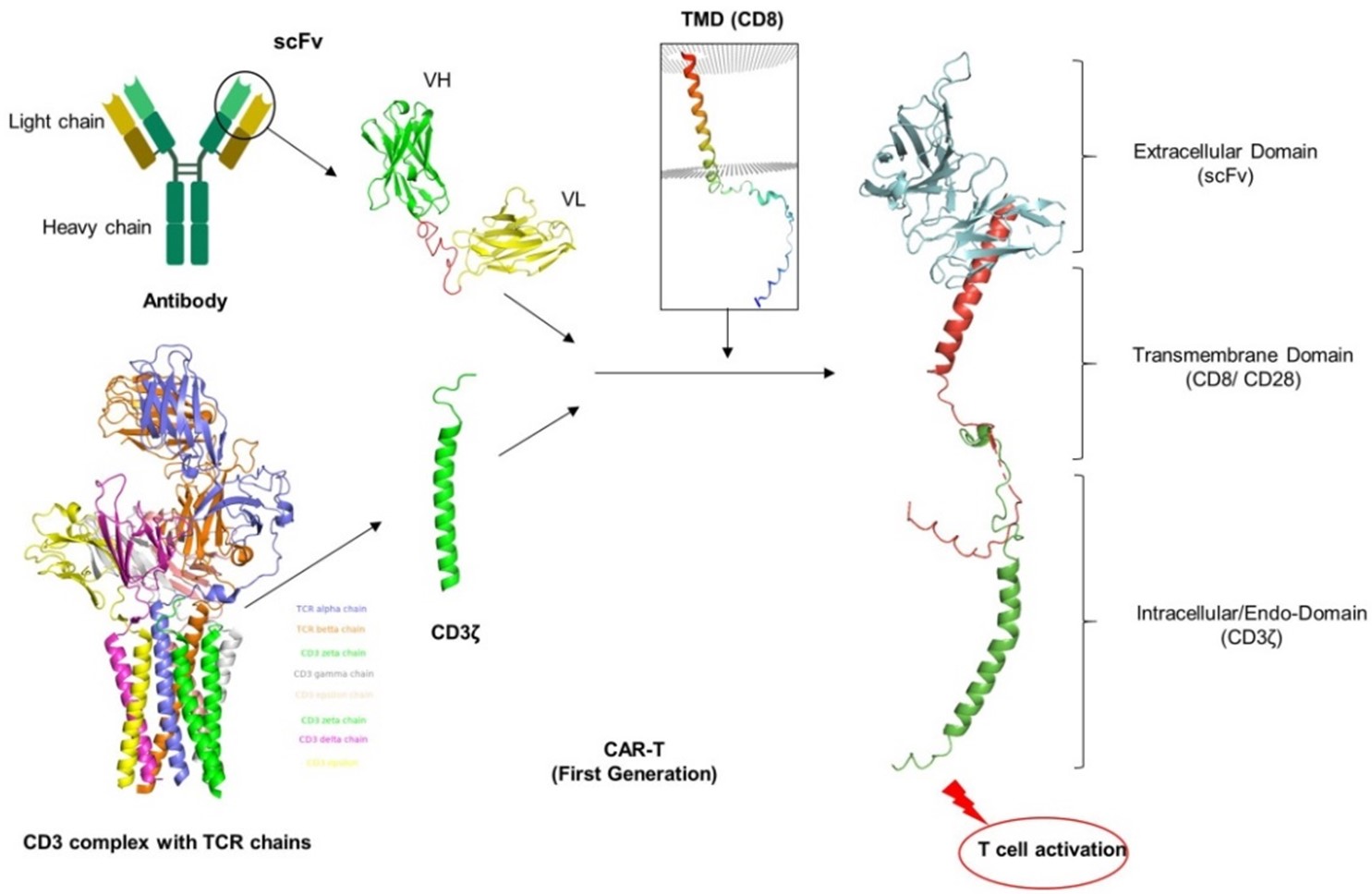
Figure 5. First generation of CAR-T development. Structure models were rendered by using PyMOL under the specific ID numbers from protein data bank (CD3 complex: 6JXR, CD8α: 8EW6, CD28: 7VU5. The variable region of heavy chain (VH) and light chain (VL) from scFv (single chain fragments of variable region) were simply conjugated with CD3ζ of TCR complex via CD8 or CD28 transmembrane domain to trigger T cell activation and cytokine production with the involvement of HLA molecule.
Second generation CAR-T cells have CMs such as CD28, CD134 (OX-40), or CD137 (4- 1BB) in addition to intracellular CD3ζ domains. This results in two distinct signaling pathways mediated by CD3ζ and CMs (Figure 6) [67]. Currently in the market, all FDA-approved drugs are 2G CAR-T-cells, which possess CM in addition to CD3ζ. Because 2G CAR-T cells are more resistant to apoptosis and exhibit a greater ability to survive in vivo, the presence of CMs in these cells improves T-cell activation, proliferation, survival, cytokine secretion, cytotoxicity, and sustained response. Studies have shown that CAR-T cell constructs with CD137 have a weaker tonic signaling than those with CD28 or CD134, but they are more persistent and have a prolonged response due to its delayed activation [68]. On the other hand, CD28-based CAR-T cell therapy is associated with increased T-cell proliferation, survival, memory cell formation, and phosphorylation, which leads to strong signaling and a faster response [33]. The very successful 2G CAR-T cell treatments that target CD19 are currently being used in clinical trials to treat B cell malignancies [69]. More recently, clinical trials are being conducted on obecabtagene autoleucel (obe-cel), a novel kind of CD19 CAR genetically modified with CAT-41BB-Z, and it is exhibiting remarkable effects in certain adult patients with recurrent B-cell acute lymphoblastic lymphoma (ALL) [70].
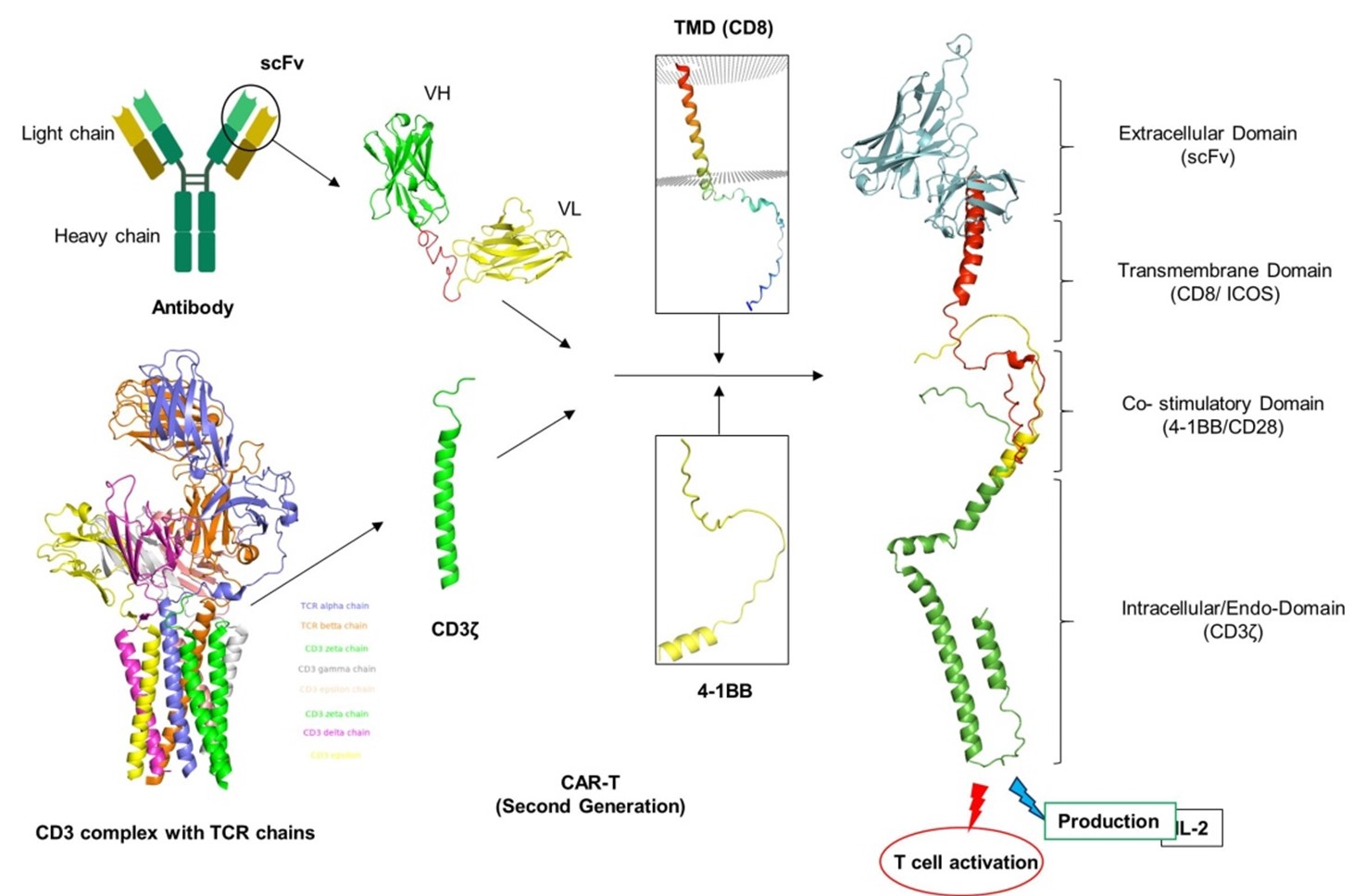
Figure 6. Second generation of CAR-T development. The second generation of the CAR-T development is simply upgradation of first-generation CAR-T by addition of CMs such CD28, CD134 (OX-40), or CD137 (4-1BB) to intracellular CD3ζ domain. Structure models were rendered by using PyMOL under the ID numbers from protein data bank (CD3 complex: 6JXR, CD8α: 8EW6, 4-1BB: 7YXU.
Second-generation CARs, including those based on CD28 and 4-1BB, are appealing for their capacity to provide long-lasting effects and improved effectiveness. They are currently used in authorized medicines like Kymriah and Yescarta [71]. 4-1BB-based CARs have lower in vitro performance than CD28-based CARs, but they often lead to enhanced long-term persistence. The clinical effectiveness of the two co-stimulatory domains is uncertain because to the absence of direct clinical comparisons [72]. Xiong et al. found that 4-1BB-based CARs form stronger immunological synapses compared to CD28-based CARs and suggested that synapse quality could serve as a predictor of in vivo effectiveness [73]. Another study examined the signaling of second-generation CARs based on CD28 and 4-1BB utilizing phosphoproteomic mass spectrometric analysis [74]. Remarkably, phosphorylation of CARs with CD28 co-stimulatory domains was demonstrated to occur more quickly and intensely, suggesting stronger signals than CARs with a 4-1BB domain. It's interesting to note that varied phosphorylation pathways were not found, indicating that the various functional effects of the CD28 and 4-1BB second-generation CARs may be explained by variations in signaling kinetics and intensity rather than the kinds of signaling pathways triggered. The signal strength in CAR-T cells is determined by all of their structural components, each of which influences signal transduction. TCR signal intensity is essential for determining positive and negative T cell selection, differentiation phenotypes, and cytokine release. Similar effects on signal strengths can affect CAR functioning and ultimately their effectiveness [75]. Although there have been notable advancements, the issue of persistence and relapse in CAR-T cells with a single co-stimulatory molecule remains unresolved, leading to the development of third-generation CARs.
Third generation CAR-T involves the combination of CD3ζ with multiple CMs, including CD28, CD137 (41BB), CD134 (OX-40), NKG2D, CD27, TLR2, or inducible T-cell co-stimulator (ICOS). This results in the generation of integrated CAR-T cell constructs, such as CD3ζ-CD28-OX40, CD3ζ-CD28 -41BB, CD3ζ-ICOS-4-1BB, and CD3ζ-TLR2-CD28, as shown in Figure 7 [62,76]. The most widely utilized construct of third generation CAR-T cell products at the moment is CD3ζ-CD28-41BB-based CAR-T-cells. The limitations of each CM utilized in 2G CARs must be overcome by many CMs used in 3G CAR-T cells. As a result, 3G CARs include two CMs that may be effective in the short term with significant and efficient tumor clearance, like CD28, as well as long-lasting clinical responses, like in 4-1BB [77]. Preclinical studies showed 3G CARs outperformed 2G CAR-T-cells in the treatment of certain cancer types, exhibiting greater safety profiles, in vivo proliferation, persistence, and anticancer potential [62]. A study conducted by Ramos et al. consistently demonstrated that 3G CAR-T cells directed against CD19 showed greater expansion and longer persistence than 2G CAR cells targeting CD19 [69]. However, 3G CAR-T cells exhibit an increased risk of severe adverse effects and a faster CAR-T cell exhaustion than 2G CAR-T cells, due to over activation of multiple CM-mediated signals.
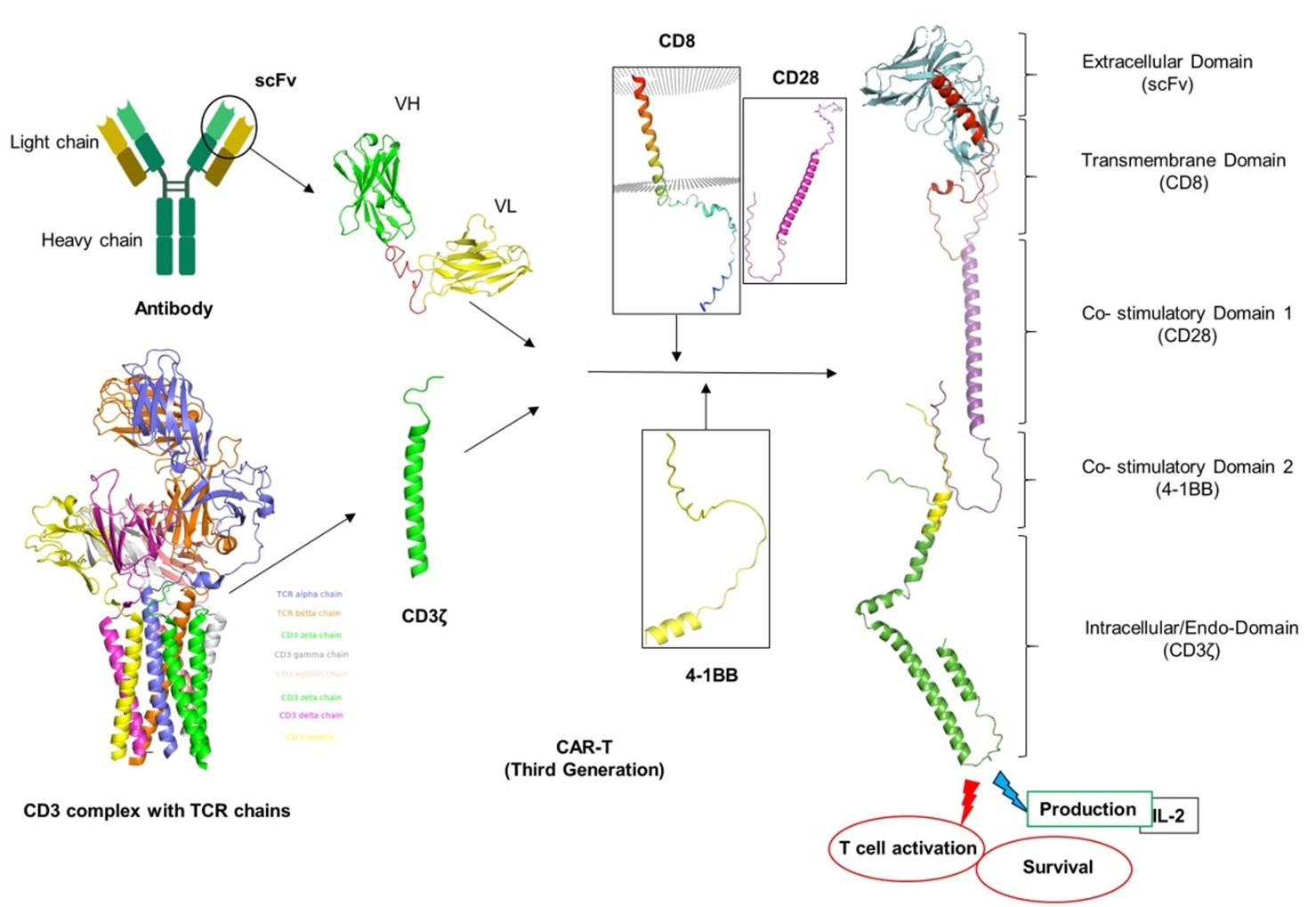
Figure 7. Third generation of CAR-T. This construct involves the combination of CD3ζ with multiple CMs, including CD28, CD137 (41BB), CD134 (OX-40), NKG2D, CD27, TLR2, or inducible T-cell co-stimulator (ICOS) to make it more effective. PyMOL graphics tool was utilized to render the structure models under their specific ID numbers from protein data bank (CD3 complex: 6JXR, CD8α: 8EW6, CD28: 7VU5, 4-1BB: 7YXU.
Third-generation CARs containing the CD28 and 4-1BB domains have been tested against a variety of targets including CD19, PSMA, GD2, and mesothelin [72]. In one study, third-generation CARs with 4_1BB and CD28 co-stimulatory domains were compared with anti-CD19 second-generation CARs with a CD28 co-stimulatory domain to assess the variations in intracellular signaling between the two groups. This study found that the phosphorylation status of signaling proteins increased generally in third-generation CAR-T cells, suggesting that third-generation CARs may have stronger signals than second-generation CARs [78]. Third-generation anti-PSMA and anti-mesothelin CD28-4-1BB-CD3ζ CARs showed better tumor eradication and more persistence in preclinical trials when compared to their second-generation counterparts [79]. A similar increase in anti-tumor potency and persistence was observed in third-generation ICOS-4-1BB-CD3ζ based anti-mesothelin CARs [77]. A direct comparative clinical study between second-generation CD28-based CARs and third-generation 4-1BB-CD28-based CARs revealed that the third-generation CARs were more persistent and prolonged than the second-generation CAR-T in B-cell malignancies, especially in cases where the disease burden was low [80].
Abate-Daga et al. compared the effectiveness of third-generation CD28-4-1BB based anti-PSCA CARs and second-generation CD28-based CARs. The results showed that although third-generation CARs' in vivo persistence was generally improved in preclinical mouse xenograft models of pancreatic cancer, the anti-tumor potency of the second-generation CARs still outperformed the third-generation formats [81]. In a different study, third-generation anti-GD2 CARs with CD28-OX40-CD3ζ domains produced better in vitro cytokine secretion (IL-2, TNFα) and proliferation than second-generation (CD28-CD3ζ or OX40-CD3ζ) and first-generation (CD3ζ) forms [82]. Apart from structural variations in co-stimulatory domains, patient heterogeneity and different therapies may potentially contribute to the dearth of advantages of third-generation CARs. Hombach et al. found that third-generation, anti-CEA CD28-CD3ζ-OX40 CARs were not effective compared to second-generation CD28-CD3ζ CARs [83]. The use of cytokine-induced killer cells (CIKs) in this investigation makes it challenging to generalize the findings to conventional CAR-T cell engineering. Concurrent clinical comparisons and comprehensive mechanistic investigations on various co-stimulatory designs will be necessary to confirm the clinical effectiveness of each design. Furthermore, Zhao et al. examined the structures of seven chimeric antigen receptors and demonstrated that a second-generation CD28-based CAR that was co-expressed with the 4-1BB ligand (4-1BBL) outperformed a third-generation CAR that was both CD28 and 4-1BB. According to this study, co-stimulatory module type and spatial arrangement have an impact on CAR function [84]. However, the advantage of third-generation CARs over their second-generation equivalents remains debatable.
Fourth generation CAR-T is also known as T-cell redirected for universal cytokine-mediated killing (TRUCK), universal CAR (UniCAR-T), or armored CAR-T cells [85]. Fourth generation CAR essentially resembles 2G CAR-T cell designs, with significant modifications to the intracellular signaling domain (Figure 8). This entails incorporating a nuclear factor of the activated T-cell (NFAT)-responsive cassette carrying transgenic immune modifiers (proteins) such as cytokines (IL-2, IL-5, IL-12, IFN-γ) and CMs (CD28, OX-40, or 4-1BB) [86]. NFATs are designed transcription factors that control constitutive or inducible expression of transgenic proteins and their transportation to the intended tumor site upon CAR-T cell activation. This improves the tumor microenvironment's ability to support immune responses. The NFAT promoter sequence is activated during antigen-induced CAR-signaling, which triggers the innate immune cells to produce cytokines that eliminate cancer [87]. As a result of overcoming the difficulty of antigen loss within tumor cells, 4G CAR-T cells have been shown to have a significant role in modifying the tumor microenvironment. Armored CAR-T cells significantly reduce systemic toxicity while improving T-cell proliferation, persistence, memory cells, and anti-tumor activity [88]. They also help patients' post-infusion immune systems to recover. Notwithstanding these significant advancements, 4G CARs remain notably less effective against solid tumors and are associated with certain unfavorable outcomes because of TRUCK T-cell activation off-tumor on-target and transgenic cytokine production in healthy tissues [88].
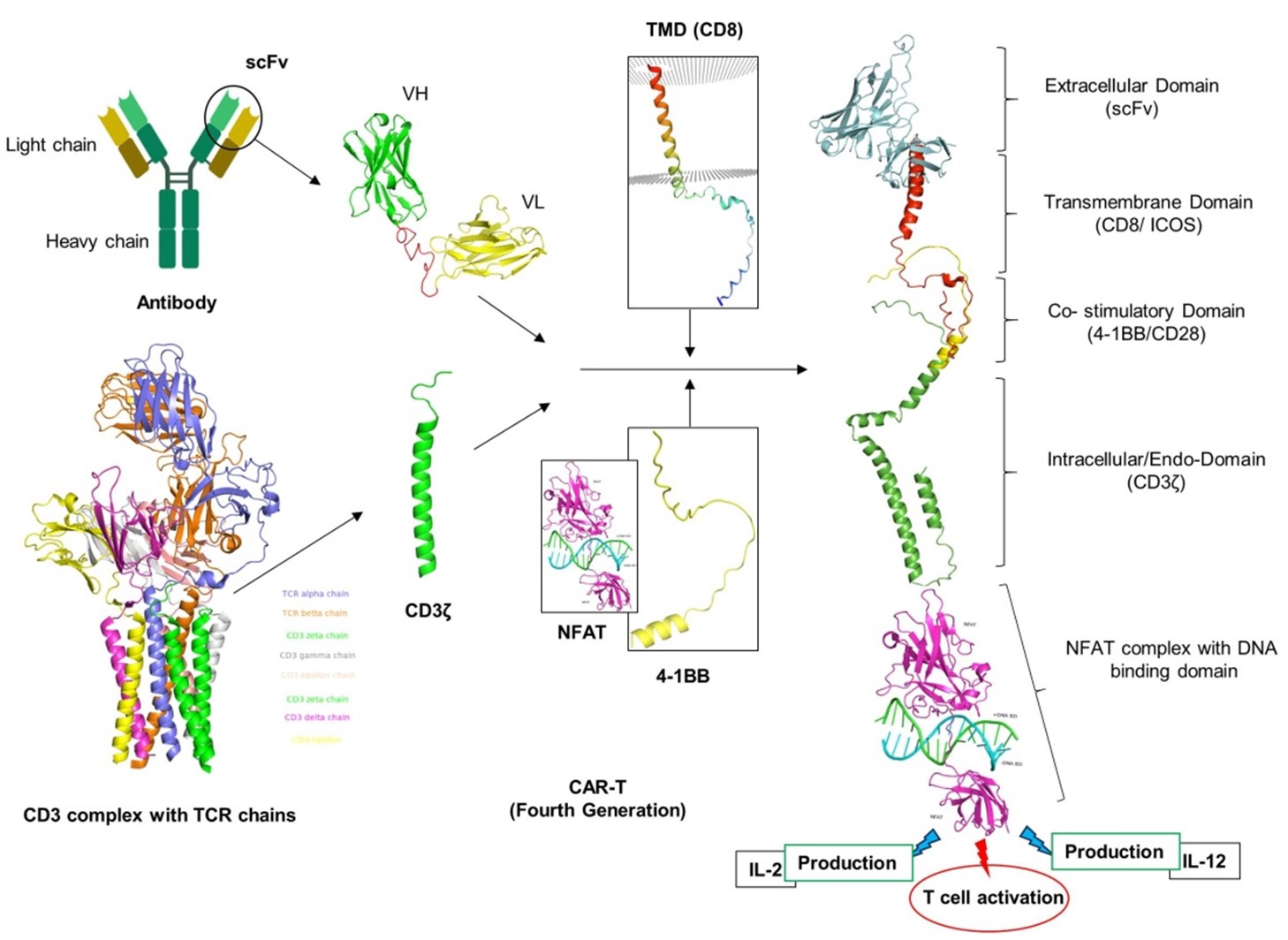
Figure 8. Fourth generation of CAR-T. Fourth generation CAR essentially resembles 2G CAR-T cell designs with modification of CM regions by inserting nuclear factor of the activated T-cell (NFAT) (PDB:1OWR).
The fifth-generation CAR-T-cell, also referred to as the next generation, is presently in active development with the intention of resolving the limitations of previous generations of CAR-T cell therapy. The structure of CAR-T cells is continuously being improved. This generation comprises advanced CARs that go beyond conventional CARs by adding extra structures like cytokine receptors (IL-2Rb) with a motif for binding transcription factors such as STAT-3/5 to optimize CAR function by binding with multiple antigens or targets with low antigen density [89]. Conventional CARs, on the other hand, are significantly improved monovalent CARs that only target one specific antigen (Figure 9). With a broader therapeutic window and an improved safety profile, 5G CAR-T cells are the most developed generation of CAR-T cells. It is equally effective as fourth generation CARs in establishing a favorable tumor microenvironment and boosting a patient's immunity following infusion [54].
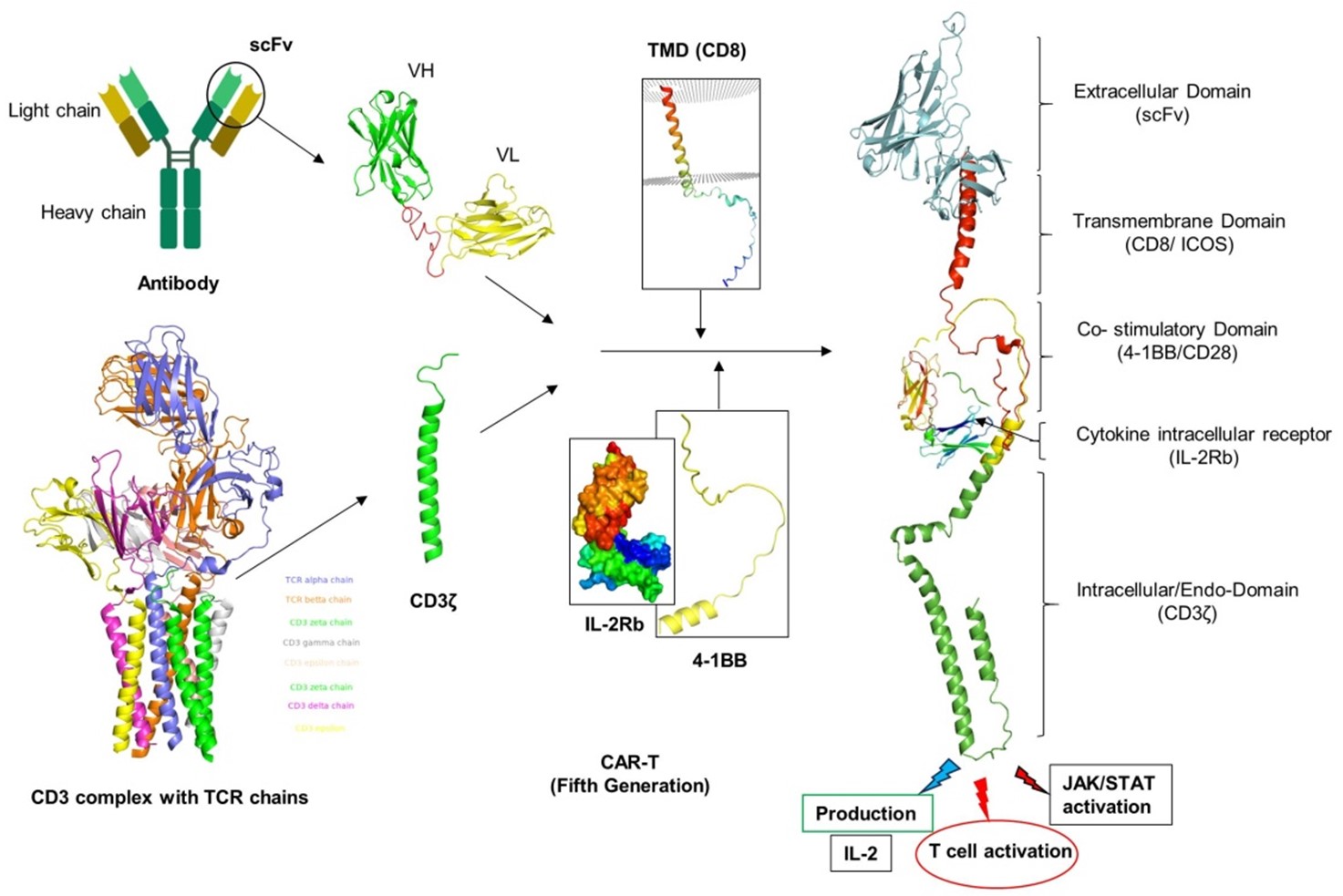
Figure 9. Fifth generation of CAR-T. Fifth generation of CAR contains an extra domain IL-2Rb of cytokine receptor via JAK/STAT signaling pathway (PDB IDs: 6DG5, 7S2S and 2B51).
Recently, more sophisticated CAR-T cells known as Boolean logic gated CAR-T cells have been created to enhance the specificity of CAR-T cells, regulate their actions, and get around some of the limitations of conventional CARs [90]. These are contemporary CAR technologies designed to increase the cancer-specificity of CAR-T cells, which will improve the efficacy of the therapy and lessen its adverse effects. Logic gating can take many different forms, the most popular ones being AND-, OR- NOT and IF-Better logic gates [91]. However, the effectiveness of next-generation conventional CARs in penetrating and trafficking into solid tumors is limited, and the issue of adverse reactions remains unclear.
Mechanism of Action of CAR-T Therapy
CAR-T cell therapy is an immune-modulatory approach that utilizes CARs to guide T-cells towards tumor cells that display particular surface proteins. Consequently, this activates intracellular signaling, which stimulates T lymphocytes and eliminates cancer cells [92]. Unlike previous anti-tumor cells (ACTs) like TIL and TCR treatments, CAR-T cells are killer cells with engineered CAR receptors that can identify antigens and eliminate cancer cells that express particular surface antigens without requiring the usage of HLA. Furthermore, CARs can bind to and target a wide spectrum of antigens, including proteins, gangliosides, carbohydrates, and any other substances found on cancer cells, regardless of the HLA presentation on the cell. By overcoming the immunosuppressive environment, this makes more cancer cells susceptible to CAR-T cell attacks, giving it a more adaptable therapy than previous HLA-dependent ACTs [14,93]. The signaling mechanisms of normal T-cells share similarities to the cytotoxic mechanism of CAR-T cells. The scFv of CAR receptors enable CAR-T cells to encounter tumor antigens after being infused into a patient. Tumor surface antigens such as CD19, B cell maturation antigen (BCMA), CD20, CD30, and many more can be targeted by CAR-T cells; among these, CD19 is the most extensively investigated antigen target, followed by BCMA [94,95]. Activation and conformational changes occur in CAR-T cells immediately upon interacting with tumor antigen. In particular, the intracellular domain's constituents form micro-clusters through centripetal movement, forming the immunological synapse's core region. This process enables the recruitment and phosphorylation of the cascade proteins downstream, such as CD3ζ and CMs. Following activation, the CAR-T cells go through a proliferative and differentiating process that is necessary for the effector functions or the ability of the CAR-T cells to kill cancer [96].
Using a number of complementary mechanisms, including the recruitment of additional immune system components, the perforin-granzyme system, and death ligand-death receptors, CAR-T cells mediate tumor-killing actions (Figure 10). The cytolytic mechanism mediated by perforin-granzyme is the primary means by which CAR-T cells eliminate cancer cells. Fast calcium-mediated degranulation or the release of the cytotoxic effector proteins (granzymes and perforin), from the lytic granules of CAR-T cells, takes place upon detection of surface antigens on a target T-cell and activation of CAR-T cells. Cytotoxic granzymes can enter the cytoplasm of target T-cells through transmembrane holes created by perforin on the target T-cells' plasma membranes after they are released. Serine proteases called granzymes are key components in CAR-T cells' ability to lyse cancer cells. Through the stimulation of both caspase-dependent and caspase-independent apoptotic pathways, these enzymes eliminate antigen-positive cancer cells. The surrounding phagocytic cells will eventually rapidly eliminate dead cancer cells [97,98].
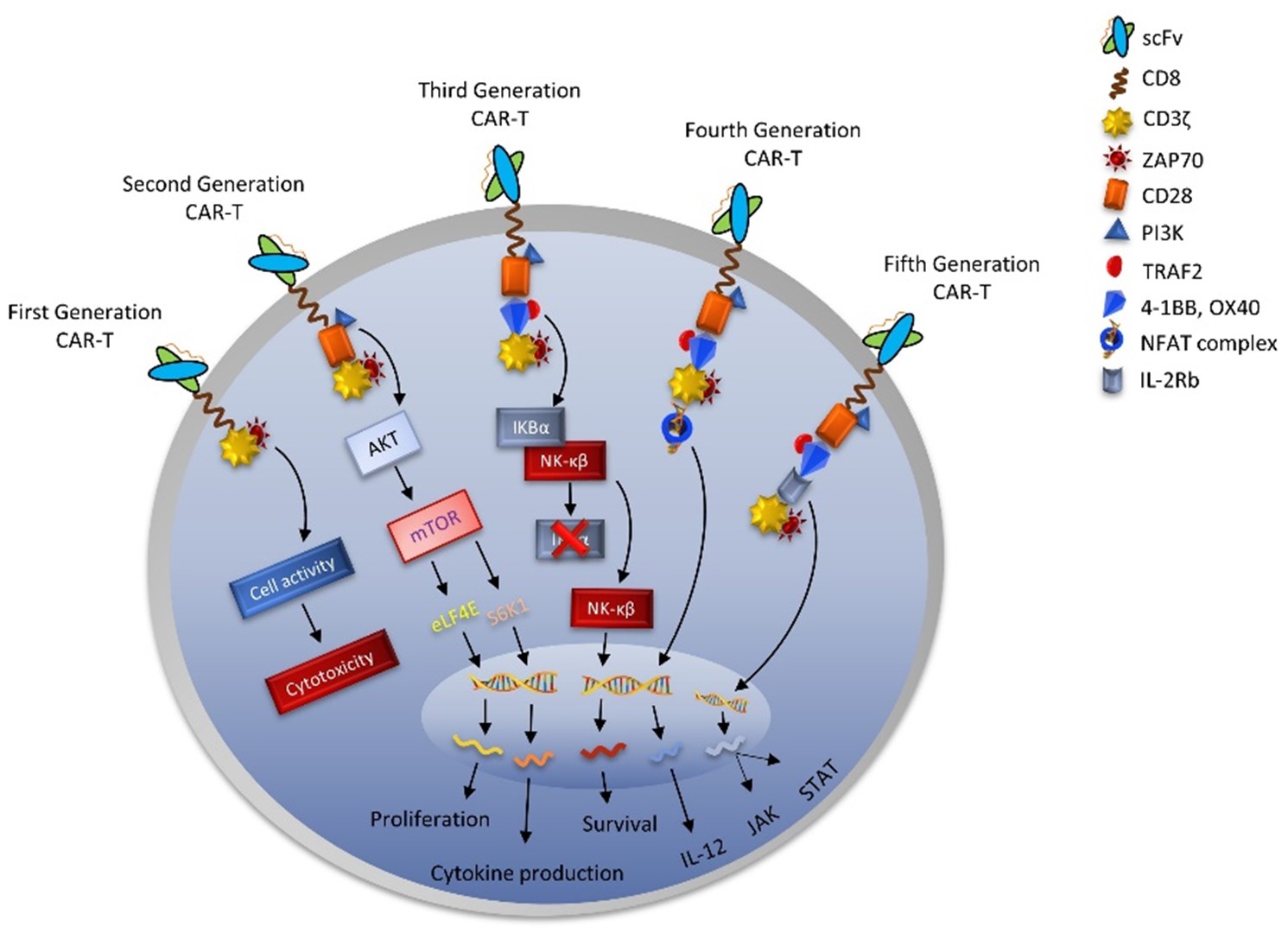
Figure 10. Mechanism of action of CAR-Ts and structural differences.
Additionally, the CAR-T cells mediate their cytolytic effector actions through death ligand–death receptors such the TNF-related apoptosis-inducing ligand (TRAIL) systems and the Fas–Fas ligand axis. The cytotoxic mechanism known as Fas-FasL occurs without the need for perforin and is triggered when the target T-cell membrane's Fas binds to the activated CAR-T cells' FasL. These interactions between Fas and FasL lead to the production of caspase 8, which triggers the apoptotic pathway's downward stream to kill cancer cells [99]. Targeting antigen-negative tumor cells inside the antigen-positive tumor microenvironment requires the slow-moving Fas-FasL system, compared to the perforin-granzyme axis. However, the evidence that is currently available suggests that human CAR-T cell treatment is capable of killing cancer cells without requiring for the Fas–FasL death pathway. Surprisingly, CAR-T cell-mediated tumor eradication appears to be significantly aided by the TRAIL effector system. A significant amount of research revealed that CAR-T cells cause cancer cells to undergo TRAIL-induced apoptosis in order to carry out their anti-tumor action [100].
CAR-T cell therapy targets tumor cells by recruiting immune system components to eliminate tumors [101]. Therefore, additional growth factors and cytokines must be created in order to penetrate tumor cells and cause inflammation, which in turn destroys cancer cells. Cytokines are another way that dead cancer cells can propagate CAR-T cells to kill more cancer cells. Additionally, through cytokine-mediated recruitment, CAR-T cells can increase the effectiveness of their anti-tumor response by recruiting more immune cells, like B cells and NK cells, to the tumor site [102,103]. Permanent or temporary elimination of all cancer cells is possible using CAR-T cells. For certain blood cancers, CAR-T cells may trigger a long-term remission and remain in the body for months after the infusion has ended, preventing cancer from recurring.
CAR-T Approval and Challenges
The US Food and Drug Administration (FDA) has approved several CAR-T cell therapies, allowing them to become a common cancer treatment after years of arduous studies. In the treatment of blood malignancies, CD19- and BCMA-directed CAR-T cell treatments achieved the most favorable results and approvals. The FDA has approved six CAR-T cell therapies, all of which are second-generation CAR-T cell products, to treat patients with a variety of severe hematological malignancies. Refractory and relapsed (R/R) blood cancers, including B-cell lymphomas, leukemia, and multiple myeloma (MM), have shown noteworthy clinical responses when treated with CAR-T [104]. Patients with B-cell malignancies have demonstrated substantial and long-lasting benefit from CD19-directed autologous CAR-T cell treatments, such as tirabelecleucel, axicabtagene ciloleucel, brexucabtagene autoleucel, and lisocabtagene maraleucel [105]. For patients with leukemia and malignant lymphomas, these are currently regarded as conventional therapies. It is approved to treat patients with R/R high-grade B-cell lymphoma (HGBCL) with three CAR-T-cell products: tiraselecleucel, axicabtagene ciloleucel, and lisocabtagene ciloleucel [106,107]. In contrast, brexucabtagene autoleucel and tisagenlecleucel have approval for adults with R/R mantle cell lymphoma (MCL) and patients under the age of 25 with R/R B-cell acute lymphoblastic leukemia (B-ALL). These four CAR-T cells have demonstrated remarkable activity and are currently approved for R/R non-Hodgkin lymphoma (NHL) [108,109]. Despite CAR-T therapy improving the survival rate of patients with R/R malignancies, the accompanying on-target off-tumor toxicities and particularly infections, limit the efficacy of this curative therapy. FDA-approved CAR-T cell therapies are briefly covered below, divided into two categories: CD19- and BCMA-targeted CAR-T cell therapies. In addition, Table 1 summarizes all FDA-approved CAR-T cell products.
|
CAR-T Cell |
Brand Name/ Nick Name |
Date |
Target |
SD |
|
Tiagenlecleual |
tisa-cel /Kymriah |
08/30/2017 |
CD19 |
41BB-CD3ζ |
|
Axicabtgene ciloleucel |
axi-cel /Yescarta |
10/18/2017 |
CD19 |
CD28-CD3ζ |
|
Brexucabtagene maraleucel |
Brexu-cel /Tecartus |
07/24/2020 |
CD19 |
CD28-CD3ζ |
|
Lisocabtagene maraleucel |
liso-cel /Breyanzi |
02/05/2021 |
CD19 |
41BB-CD3ζ |
|
Idecabtagene vicleucel |
ide-cel /Abecma |
03/26/2021 |
BCMA |
41BB-CD3ζ |
|
Ciltacabtagene autoleucel |
cilta-cel /Carvykti™ |
02/28/2022 |
BCMA |
41BB-CD3ζ |
Challenges with CD-19 directed CAR-T
Tisagenlecleucel, also referred to as KymriahTM or tisa-cel, is a second-generation autologous CART-19 therapy that uses 4-1BB as CM. As of August 30, 2017, Novartis's Tisa-cel is the first CAR-T cell therapy to receive FDA approval for commercial use [110]. It is being used to treat adults and children with a variety of advanced-stage lymphomas. It is indicated for the treatment of R/R large B-cell lymphoma following two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified or emerging from follicular lymphoma (FL), or second-line DLBC and HGBCL [111]. However, Tisagenlecleucel is linked to a higher incidence of cytokine release syndrome (CRS) in 22% of patients, as well as neurotoxicity in 12% [112]. Another longitudinal clinical research comprising 115 individuals who had received this CAR-T cell therapy revealed a 53% overall response rate (ORR) and a 39% complete response (CR), with 27% developing CRS [113]. Despite its promising efficacy and safety, it has seen limited use in the treatment of individuals with primary central nervous system (CNS) lymphoma and chronic lymphocytic leukemia (CLL).
Axicabtagene ciloleucel, also known as YescartaTM or axi-cel, is a CD-19 CAR-T cell with CD28 as the CM. FDA approved Axi-cel from Kite Pharma on October 18, 2017, as the second-approved CAR-T cell for treating adult patients with large B-cell lymphoma that is not responding to first-line chemotherapy, relapses within a year of first-line chemotherapy, or relapses or is resistant to two or more lines of systemic therapy [114]. These patients include those with DLBCL not otherwise specified or resulting from indolent lymphoma, primary mediastinal large B-cell lymphoma, and HGBCL. With a 78% CR, 89% ORR, and a tolerable safety profile, axicabtagene ciloleucel is an extremely effective first-line treatment for high-risk DLBCL [115]. Additionally, this medication, which has a 92% and 76% ORR and CR rate, respectively, has just been approved for adult patients with R/R FL following two or more lines of systemic therapy. Although 8% and 21% of patients experienced CRS and neurotoxicity, respectively, the median length of remission was determined to be 18 months or more [116]. For the treatment of patients with primary CNS lymphoma and MCL, axi-cel is still undergoing evaluation and has not yet received approval.
Brexucabtagene autoleucel, commonly known as TecartusTM or Brexu-cel is the third-approved CAR-T cell for the treatment of certain patient subgroups with leukemia and lymphoma [117]. The FDA approved the autologous anti-CD19 CAR-T product on July 24, 2020, for the treatment of adult patients with R/R MCL. It had a 93% ORR and a 67% CR rate when it was licensed under fast approval due to positive responses in clinical trials. Furthermore, following a year of follow-up, the patients' durability of response showed amazing results, with 61% progression-free survival (PFS) and 83% overall survival (OS). Brexu-cel was authorized on October 1, 2021, as the initial CAR-T cell treatment for adults with R/R B cell-ALL diagnosis [118]. At about 16 months of follow-up, the 71% CR rate was seen, along with median remission duration of 12.8 months and an OS of 18.2 months. The main adverse effects of brexu-cel therapy included CRS, neurotoxicity syndrome, cytopenia, and infection [118].
Lisocabtagene maraleucel, referred to as BreyanziTM or liso-cel, is a CD-19 CAR-T that incorporates the 4-1BB and CD3ζ molecules. On February 5, 2021, the FDA approved liso-cel from Juno Therapeutics for the first time as a therapeutic agent for adult patients with large B cell lymphoma who had received two or more lines of systemic therapy. These patients included those with grade 3b FL, HGBCL, primary mediastinal large B-cell lymphoma, DLBCL non-specified or arising from indolent lymphoma [119]. Liso-cel is also demonstrating an amazing response for MCL, CLL, and primary CNS lymphoma, although not having FDA approval yet [118]. The cellular makeup and phenotype of adoptively transferred T-cells, including T-cell subtypes and subpopulations, are important components of immunotherapy efficacy. Liso-cel is given in a predetermined combination including a particular proportion of CD4+ and CD8+ CAR-T cells [120]. A proper balance between CD4+ and CD8+ T-cells can improve a product's capacity to destroy a tumor. Research suggesting that B-ALL patients could experience high remission rates from treatment with CAR-T cells that have a consistent CD4:CD8 ratio (1:1). Finding the variables linked to CAR-T cell proliferation, persistence, and toxicity is made possible by immunotherapy using a CAR-T-cell product with a specified composition. Finding the factors associated with CAR-T cell proliferation, persistence, and toxicity is made possible by immunotherapy using a CAR-T-cell product with a specified composition. In order to reduce toxicity and increase disease-free survival, it also makes the development of CAR-T cell dosage and lymphodepletion techniques easier [121]. In one study, lisocabtagene maraleucel was administered to 61 participants. 33 people (54%) had refractory disease, 13 people (21%) experienced a relapse within a year of starting first-line therapy, and 15 people (25%) experienced a recurrence after 12 months. Leukopenia (13 [21%]), thrombocytopenia (12 [20%]), and neutropenia (29 [48%] patients) were the most frequent grade 3 or worse treatment-emergent adverse events. Thirteen patients (21%) experienced significant adverse events associated with lixocabtagene maraleucel during treatment. Among those studied, 19 (31%; grade 3 in three) had neurological instances and 23 (38%; grade 3 in one) had cytokine release syndrome; there were no grade 4 events or deaths [122]. Another study found that 268 patients who received liso-cel as second-line therapy for large B-cell lymphoma experienced cytokine release syndrome (45%; Grade 3, 1.3%), as well as CAR-T cell-associated neurologic toxicities (27%) [123]. These findings urge for further assessment and preventive measures.
Challenges with BCMA directed CAR-T
Idecabtagene vicleucel, also known as AbecmaTM or ide-cel, is a BCMA-targeted CAR-T cell treatment that the FDA approved on March 26, 2021. Following four or more previous lines of therapy, including proteasome inhibitors, immune-modulatory drugs, and anti-CD38 monoclonal antibodies, ide-cel was the first CAR-T cell product used to treat patients with R/R MM [124]. With a 73% ORR and a 33% CR rate, patients with R/R MM who had extensive ide-cel treatment demonstrated noticeably better responses. Additionally, these patients' survival improved as their PFS and OS were 8.8 and 19.4 months, respectively. CRS and neurotoxicity were among the less common side events, with 5% and 3% of cases, respectively [125]. Idecabtagene vicleucel was used in another trial to investigate potential treatments for relapsed/refractory multiple myeloma (RRMM) [126]. The authors found that although the safety profile was satisfactory and the overall response rate (ORR) was 69%, chronic hematologic toxicity continued to be a major challenge. Furthermore, the immunological effector-cell associated neurotoxicity syndrome (ICANS) affected 1 patient (6%), febrile neutropenia affected 11 patients (69%), infections affected 5 patients (31%), and cytokine release syndrome (CRS) affected 15 (94%) patients. 14 (25%) persons, experienced prolonged hematologic toxicities. Similarly, another study was conducted on older and high-risk patients showed an ORR ≥ 50%. Cytopenias (97%) and cytokine release syndrome (CRS; 84%) were the most frequent grade toxicities [127].
Ciltacabtagene autoleucel (CarvyktiTM or CLTa-cel) was approved by the FDA on February 28, 2022, making it the sixth CAR-T cell treatment [128]. Legend Biotech and Johnson & Johnson's Janssen Pharmaceutical Companies first disclose it. The CarvyktiTM possesses strong avidity against human BCMA due to its structural composition, which consists of two Ilama (camelid) heavy chains (VH) combined into a single chain variable fragment (scFv) that binds with two BCMA epitopes. Conversely, the cilta-cel endodomain is intended to contain the costimulatory domain 4-1BB and the T-cell activation domain CD3ζ [129]. One study examined subsets of individuals with R/R MM who had received four or more prior lines of treatment. According to the results, patients with R/R MM who were treated with cita-cel had a 98% ORR and an 80% CR rate [130]. Furthermore, after a longer period of patient follow-up, its safety profile remains similar. When it comes to MM efficacy, Cilta-cel typically works better than Idecel, but it also exhibits comparable adverse effects. Early, profound, and long-lasting responses were seen in CARTITUDE-1, a phase Ib/II trial assessing the safety and effectiveness of ciltacabtagene autoleucel (cilta-cel) in severely pretreated patients with relapsed/refractory multiple myeloma. One study includes evaluations of patient subgroups at high risk and presents updated data two years after the final patient enrolled (median follow-up [MFU] around 28 months). Hematologic grade 3/4 treatment-emergent adverse events (TEAEs) accounted for the majority (≥ 25%); grade 3/4 nonhematologic TEAEs (≥ 5%) included pneumonia (10.3%), hypophosphatemia (7.2%), elevated gamma-glutamyl transferase (6.2%), hypertension (6.2%), weariness (5.2%), and increased AST (5.2%). Before the report, no CRS was noticed [131].
Risk of infection and exhaustion with CAR-T therapy
CAR-T cell therapy is a more diverse approach to acquired cell therapy usually produces a long-term remission in patients with blood cancer. Modified T-cells can only recognize antigens that are typically expressed on the cell surface, which narrows the pool of possible target antigens. For several reasons, (summarize in Figure 11A) the use of CAR-T cell therapy typically yields lower results than expected. Its low therapeutic efficacy, adverse effects, high cost, and practical considerations are the main obstacles limiting CAR-T cell therapy from assuming advantage of conventional therapy with a larger impact [54]. Further limiting CART therapy for solid tumors is the ability of CAR-T cells to infiltrate solid tumors and effectively destroy target cells within an immunosuppressive milieu. CAR-T cells cannot penetrate tumor cells due to the extracellular matrix and stromal cell barrier present in tumor microenvironment [132]. Immunosuppressive cells also proliferate in the tumor microenvironment, further restricting the function of CAR-T effector cells. Regulatory T cells and other immune cells that penetrate tumors develop hostile environment to CAR-T cells by secreting inhibitory cytokines and depleting IL-2 [9]. In cases of chronic viral infection and malignancy, these factors ultimately result in an antigen-clearing failure. T cell exhaustion in CAR-T cell treatment leads to resistance and relapse because it suppresses T cell proliferation and effector activity as a result of continuous antigen stimulation. Repetitive exposure to antigens specific to a disease condition T cells into a dysfunctional state characterized by reduced proliferative capacity and effector activity [133]. Although CD8+ T cells have been shown to characterize exhaustion in the most comprehensive way, CD4+ T cells have also been shown to exhibit dysfunctional states as a result of continuous antigen stimulation. Acute antigen stimulation causes naive CD8+ T (Tn) cells to differentiate into effector T (Teff) cells [134]. Cells go through significant clonal expansion during differentiation, along with functional and metabolic changes, leading to a population of effector T cells that are specific to an antigen [135]. The antigen density exposed to Tn cells determines the amount of this reaction, with greater antigen resulting in increased T cell proliferation [136]. Antigen density and T cell antigen sensitivity, however, are adversely correlated. Most effector T cells disappear after antigen clearance. Memory T (Tm) cells are produced from the few remaining cells, and they remain in the host even when the stimulatory antigen is not present. Prolonged antigen stimulation, however, can disrupt CD8+ T cell development to an exhausted state marked by a reduction in proliferative capacity and effector function during persistent infection or cancer [137]. Thus, improving CAR-T cell therapy necessitates the development of strategies to prevent T cell exhaustion (Figure 11B).
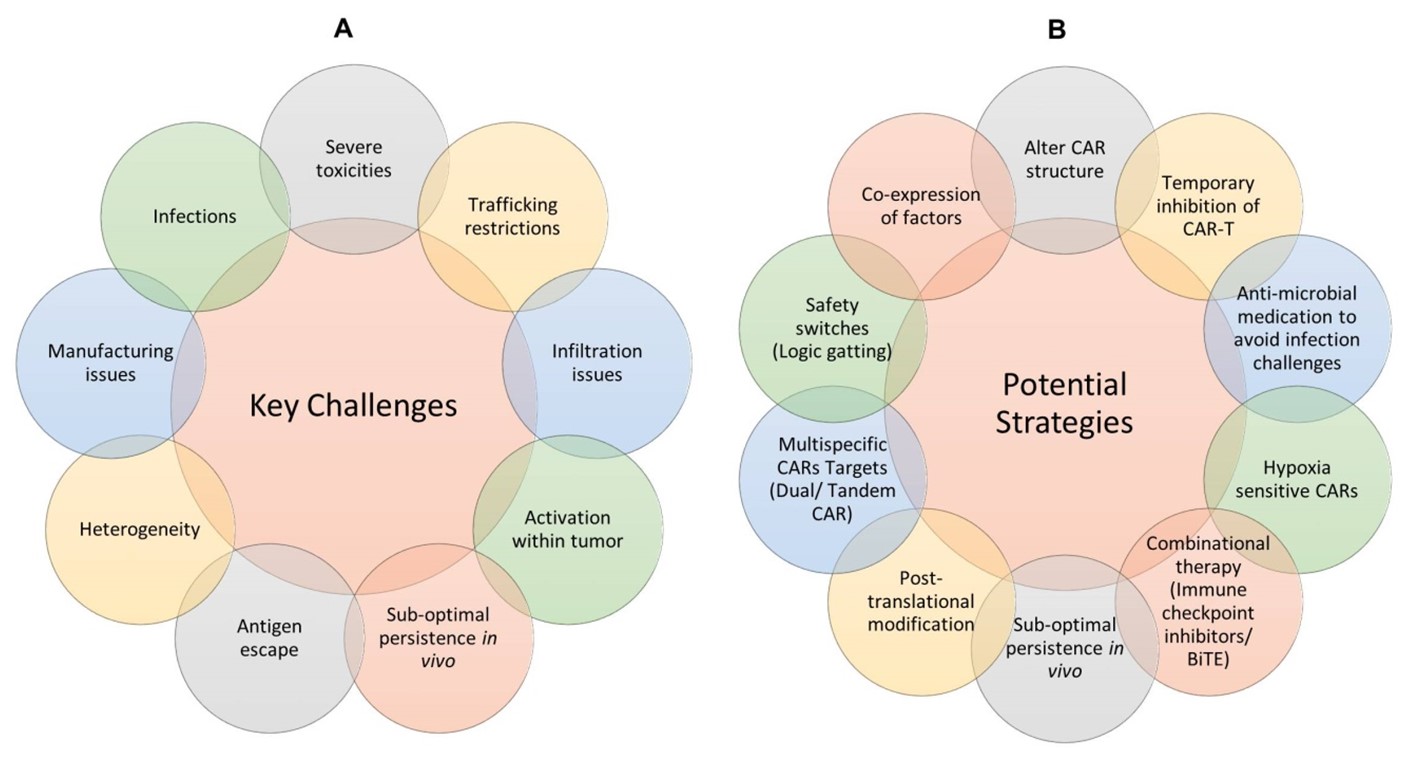
Figure 11. Summary of CAR-T challenges and potential strategies to improve CAR-T efficacy.
Infection occurred in 12–55% of patients within the first year of the tisagenlecleucel, axicabtagene ciloleucel, and lisocabtagene maraleucel registration trials, with 23–33% of the infections were severe [138]. Surprisingly, infection had a low fatality rate (≤ 3%), with most deaths caused by recurrent cancer. One study found that the occurrence of all infection episodes within the initial 30 days varied from around 27% to 36% of patients [139-141]. Another study calculated the entire 28-day infection incidence rate to be 2.35 infections per 100 days at risk [142]. Most infections complicating CAR-T cell treatment have been identified in registration clinical studies. Although it might be higher in patient groups who are more vulnerable, the incidence of central line-associated infection has been reported as events per 100,000 patients per years [139].
Several studies have demonstrated a link between higher risk of infections with the extent of CRS [143]. A study with 133 CD19+ CAR-T patients that examined infection density and pre- and post-treatment risk factors for infection within the first three months found that infection density was higher in the first thirty days compared to days 31 to 90. The most prevalent infections during the first 30 days were bacterial (23% of patients), followed by viral (17%) and fungal (3%) infections [143]. According to Park et al., in a phase-I trial of CD19+ CAR-T therapy in 53 patients with B-cell ALL, CRS grade ≥ 3 was strongly associated with risk of infection, particularly bloodstream infections [144]. After receiving CAR-T infusion, 42% of the patients had 26 infections (30% bacterial, 10% viral and 8% fungal) within 30 days. Another study reported that bacterial infections were the most prevalent cause of subsequent infections in 60 patients with diffuse large B-cell lymphoma (DLBCL) receiving CD19-directed CAR-T therapy. At one year, the cumulative incidence of all bacterial, viral, and fungal infections was 63%, 57%, 45%, and 4%, respectively [139]. Furthermore, it was demonstrated that the use of systemic corticosteroids to treat immune effector cell-associated neurotoxicity syndrome (ICANS) or CRS is independently linked to a higher risk of infections [139].
Risk of bacterial infection: Overall, bacterial infections are somewhat common in patients undergoing CAR-T cell treatment. Prior to receiving CAR-T cell infusion, a significant number of patients have antimicrobial treatment and several rounds of chemotherapy, which significantly impacts the composition of their microbiome. In the neutropaenic phase, this would enhance the possibility of invasive infection and multidrug-resistant microbial colonization. The majority of infections that occurred within the first 90 days were caused by bacteria, with about 40% of those classified as severe and 6% as potentially fatal. In a particular study, bacteria accounted for 22 out of 36 (61%) early infections that occurred within the first 30 days following CAR-T cell infusion [141]. This involved typical locations such the circulation, genitourinary system, lungs, and soft tissue; Clostridioides difficile infection was also observed. High prevalence of C. difficile infection within this group was observed with cohort infection rates varying from 12.5% to 20% [138,145]. In another study, 17% of patients had bacterial infection during the first 28 days following infusion; bloodstream infections accounted for half of these infection incidents, with a smaller proportion resulting from gram-negative bacteria that are resistant to drugs [143].
In additional research, the incidence of early and late bacterial infection was 31% and 15%, respectively, in 85 patients with R/R B-cell lymphoma [140]. One death from a bloodstream infection caused by Streptococcus mitis was noted. During a 12-month follow-up period, a total of 101 infection events occurred in a cohort of 40 patients, comprising 60 bacterial infections [139]. A significant indicator of a serious bacterial infection seems to be the lack of response to CAR-T cell treatment. Treatment response is correlated with both the amount of risk and the duration of the first severe bacterial infections. Another risk factor for a serious bacterial infection was a history of infections within 30 days prior to CAR-T cell therapy (HR 3.98) [139].
A factor in the development of B-cells without the need for antigen and the activation of B-cells through immunoglobulin, CD19 is expressed only on B-cells in earlier stages and on follicular DCs [146]. CD19 plays a role in B-cell immunoglobulin activation and antigen-independent development. It is expressed exclusively on follicular DCs and earlier-stage B-cells. On the other hand, terminal PCs stop producing CD19, unlike BCMA [147]. Additionally, surface CD19 expression is absent in malignant PCs. Other research has demonstrated that a mutation in the CD19 gene causes a decrease in memory B-cells but an acceptable development of precursor and early B-cells, which results in hypogammaglobulinemia. Especially in the early post-CAR-T period, recipients of CD19-directed CAR-T treatments may be more susceptible to bacterial infections due to the reduction of CD19 on nonmalignant cells [148]. The globally impaired immune repertoire with BCMA-directed CAR-T may result in more viral infections than bacterial infections with CD19-directed CAR-T. Thus, patients with R/R MM who get anti-BCMA CAR-T treatment may be at a significantly increased risk of developing severe COVID-19. As a result, frequent review of immunological response and booster doses may be required [149,150]. Although sparse, there is some information available on BCMA expression in neurons, primarily in the basal ganglia and cerebellum. Although the findings require validation and proof of concept, upcoming clinical trials with anti-BCMA CAR-T and bispecific T-cell engagers will reveal any non-T-cell-mediated neurotoxicity caused by BCMA expression. Anti-BCMA therapy should be used with caution because of the increased risk of central nervous system infections.
Risk of viral infection: CAR-T cell therapy has shown efficient in treating HIV and chronic hepatitis B virus (HBV) infection [145]. Patients infected with HIV, HBV, or the hepatitis C viruses were routinely excluded from CAR-T cell therapy clinical studies. This mostly addresses reactivation and uncontrolled viral replication. Patients with chronic HBV infections appear to respond to antiviral treatment well. A group of 70 patients in China receiving CAR-T cell therapy revealed no significant difference in toxicity or response between those with and without HBV [151]. Human herpesvirus 6 (HHV-6), Ebstein Barr virus (EBV), and CMV herpesvirus appear to be rare. There are no available studies on the course of CMV and EBV infection in absence of CAR-T cell therapy. One year following CAR-T cell therapy, a patient with relapsed large B-cell lymphoma developed progressive multifocal leukoencephalopathy [152]. Risk factors for viral infection in this population were investigated. Interestingly, CD4 and CD8 T-cell counts evaluated at 30 days were not significantly lower in individuals who were to eventually acquire viral infection [140]. According to one study, following CAR-T cell therapy, patients with low immunoglobulin G (IgG) prior to lymphocyte-depleting chemotherapy were more likely to develop a virus (HR 5.7). Replacing intravenous immunoglobulin (IVIG) failed to change the infection rate. The development of viral-specific neutralizing antibodies may be more critically impacted by baseline reduction of plasma cells and antibodies before CAR-T cell treatment [139]. A group of 39 adult patients with B-cell malignancies has shown persistent retention of antiviral antibodies following CD19 CAR-T cell treatment. Moreover, 95% of patients maintained anti-measles IgG levels [153].
Unsurprisingly, viral infections are widespread in CAR-T treated population, especially in the late phase, as many of these patients have received lymphocyte-depleting chemotherapy and have significant hypogammaglobulinaemia. Typical viral infections included respiratory syncytial virus, cytomegalovirus, influenza, and polyomaviruses. Overall, the most common late infectious cause in this sample appears to be viral respiratory tract infection [142]. In several analyzed groups, respiratory viruses accounted for the majority of late infections (>28 days post infusion) [144]. This was verified in a second small study of influenza A-related mortality occurred and the majority of virus infections occurred after 30 days [139]. Patients who underwent CAR-T cell therapy responded quickly to treatment and transitioned back into the community while still at risk, which may account for a high frequency of respiratory virus infection during the late phase. Between 9.2% and 28% of people had a viral infection during this time. In one group during the early and late phases of CAR-T cell therapy, viral infections affected 14% and 22% of patients, respectively [140].
The most common viral pathogen was infection with rhinovirus. In one study, the frequency of respiratory viruses that cause sickness was equal in the early and late post infusion periods of CAR-T therapy, occurring at roughly 8%. Prophylaxis against CMV reactivation is not recommended in several guidelines, although it seems to be rare [143,144]. Pneumonitis and viremia affected just 2/88 (2.3%) of the individuals in one trial [141]. A patient who had received CAR-T cell therapy that targeted the BCMA recently developed a prolonged concerning of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The patient had persistently increased viral RNA during sickness for more than two months before succumbing to the infection, even after receiving convalescent plasma and the antiviral drug remdesivir [154,155].
By August 1, 2021, SARS-CoV-2 had infected over 200 million individuals and resulted in over 4 million deaths worldwide. Individuals with cancer, particularly those with hematologic malignancies, have a greater chance of severe COVID-19 and death, with mortality rates ranging from 13% to 39% [156-158]. CAR-T and hematopoietic cell transplant (HCT) are more susceptible to significant COVID-19 challenges. A recent study from the Center for International Blood and Marrow Transplant Research registry of 318 HCT recipients discovered a 30% death rate in 30 days following the emergence of COVID-19. The New York study included five patients with B cell NHL (B-NHL) who had received CAR-T products targeting CD19, four with axi-cel and one with tisa-cel [159]. In a different case study, a 73-year-old R/R MM patient experienced significant COVID-19 12 days after receiving CAR-T treatment that targets BCMA [154]. After experiencing continuous viremia for more than two months, the patient died. In another trial, which comprised two CAR-T recipients among twenty cancer patients, it was proven that there was significant immunosuppression, ongoing viral shedding, and viable virus by cell culture [160]. A second study from the Dana-Farber Cancer Institute examined the results of 27 CAR-T recipients among the 127 patients treated with cellular therapy during the initial COVID-19 wave. One DLBCL patient died from COVID-19-related complications 121 days after receiving CAR-T infusion, while another patient developed COVID-19 51 days after receiving tisa-cel during the trial period [161].
There is more information regarding the general infection risk associated with CAR-T therapy than there is for COVID-19 outcomes for recipients of CAR-T therapy. Numerous patient- and disease-related factors influence the risk of infections linked to CAR-T. Some of the major variables that may increase the risk of infections include the use of a lymphodepletion (LD) chemotherapy regimen, the interval between cell collection and infusion, bridging therapy, the dose of CAR-T cells, fresh versus cryopreserved cells, single versus fractionated dosing, signaling and costimulatory domains, TAA (CD19 versus BCMA), and the length of lymphopenia and hypogammaglobulinemia [162]. These observations raise questions about the COVID-19 outcomes for patients receiving CAR-T cell therapy.
Risk of fungal infection: Fungal infection has been reported rarely in patients receiving CAR-T cell therapy, despite the high level of multifactorial immune suppression. There are still a few studies offering in-depth analyses of fungal infection. Fluconazole prophylaxis has generally been advised in this population according to institutional guidelines. Most fungal infections develop during the first 30 days and usually occur in combination with CRS or neutropaenia [163]. It is rare and could be connected to the length of the neutropaenia. According to several studies, the incidence of invasive fungal infections ranges from 1% to 15% overall; 0–10% and 0–7% of these are related to yeast and mold infections, respectively [164]. The use of echinocandin or fluconazole is the basis for the majority fungal infections. The risk of infection-related mortality is low among patients who have CAR-T cell treatment complications due to an invasive fungal infection [165]. Patients undergoing fluconazole prophylaxis frequently experienced early Candida spp. infection, primarily in the bloodstream. Many types of mold infections (Aspergillus, Fusarium, Mucorales, Cunninghamella) have been reported to cause illness, mostly in the lungs. Only two adult patients in a group of 85 with refractory B-cell lymphoma were found to have fungal infections (caused by Candida krusei and Fusarium spp.), and both of these patients passed away [140]. There has been one reported case of Coccidioides infection that happened more than a year after CAR-T cell therapy [139]. Only one fungal infection was found in another group of 85 adult and pediatric patients between 30 and 60 days after therapy [141].
Patients with long-term risk factors, such as corticosteroid use and neutropaenia, were more likely to develop late-invasive fungal infections. Out of 40 patients with R/R DLBCL who underwent CAR-T cell therapy and were followed up for a year, only two cases of fungal infections were reported [139]. Following thirty days, two patients developed invasive pulmonary Aspergillosis and Pneumocystis jirovecii pneumonia (PJP). Despite the fact that prophylactic measures are widely used and successful, only three patients have been documented to have experienced PJP in the literature to date [143,163,165]. Due to severe Aspergillus fumigatus lung infection and Candida glabrata pancolitis, one patient died away. It is most likely the result of prolonged high-dose corticosteroids for CRS together with severe granulocyte colony stimulating factor (G-CSF) refractory neutropenia lasting over 50 days [166].
CAR-T cell recipients share many of the conventional risk factors for invasive fungal infection in patients with hematological malignancies and those undergoing stem cell transplantation. More precisely, a long-lasting and significant cumulative risk is probably influenced by prolonged neutropenia and lymphopenia [167]. Patients undergoing treatment who have a high net state of immunosuppression may be more susceptible to fungal infection overall and have many risk factors present than those who received CAR-T cells soon after cancer diagnosis. Tocilizumab is unlikely to significantly increase the risk of fungal disease when utilized as treatment for CRS [168]. CAR-T cell therapy is a unique treatment option for persons with hematological malignancies. The rate of therapy-related toxicity is high, contributing to patient morbidity. Infectious diseases are very prevalent and might potentially be reduced with preventative measures.
Conclusion
CAR-T cell therapy is a cutting-edge, novel, and successful treatment for a variety of hematological cancers. There are five generations of CAR-T cells available on the market to overcome these challenges. The use of CAR-T cells in the treatment of many forms of blood cancer has grown. Still, its success rate remains unsatisfactory when it comes to solid tumors. Cost-effectiveness, safety, and quality assurance are issues that need to be thoroughly researched in order to apply this therapy for different types of cancer. Several TAAs of solid tumors have been discovered by scientists, and some of these were tested in phase I and II clinical studies. Scientists developed multiple strategies to address the numerous challenges that CAR-T cell therapy has faced when treating solid tumors [169]. Certain lethal toxicities and off-target consequences are linked to several difficulties and drawbacks with CAR-T cell treatment. Given the need for CAR-T cells to concurrently detect the presence of two or more TAAs on tumors, it is essential that future studies broaden multitarget CAR-T cell development to improve their tumor-killing specificity and decrease off-target effects. Furthermore, it is essential to develop a "device switch" to regulate the timing of T-cell activation or a "suicide gene" system to eliminate infused CAR-T cells to regulate their proliferation and reduce their toxicity. CAR-T has significant short- and long-term toxicities, as well as infection risks, for patients who received several prior treatments, often involving hematopoietic cell transplantation. When CAR-T cell therapy is combined with another immunotherapy, its effectiveness can be boosted. Despite the challenges, novel approaches and effective solutions are always developing, which will result in more potent and secure future treatments.
Acknowledgments
This work was supported by the National Cancer Institute (1R37CA251318-01, 1R01CA248111-01A1, R01CA258477-01, R01CA278911, R01CA288403), and CPRIT Scholar Award (RR210067).
Declaration of Interests
The authors declare no competing financial interests.
References
2. Bashor CJ, Hilton IB, Bandukwala H, Smith DM, Veiseh O. Engineering the next generation of cell-based therapeutics. Nature Reviews Drug Discovery. 2022 Sep;21(9):655-75.
3. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proceedings of the National Academy of Sciences. 1993 Jan 15;90(2):720-4.
4. Hu L, Charwudzi A, Li Q, Zhu W, Tao Q, Xiong S, et al. Anti-CD19 CAR-T cell therapy bridge to HSCT decreases the relapse rate and improves the long-term survival of R/R B-ALL patients: a systematic review and meta-analysis. Annals of Hematology. 2021 Apr;100:1003-12.
5. Finney OC, Brakke H, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. The Journal of Clinical Investigation. 2019 May 1;129(5):2123-32.
6. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discovery. 2018 Oct 1;8(10):1219-26.
7. Lesch S, Benmebarek MR, Cadilha BL, Stoiber S, Subklewe M, Endres S, et al. Determinants of response and resistance to CAR T cell therapy. Seminars in Cancer Biology. 2020 Oct 1;65:80-90.
8. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer Journal. 2021 Apr 6;11(4):69.
9. Carmenate T, Ortíz Y, Enamorado M, García-Martínez K, Avellanet J, Moreno E, et al. Blocking IL-2 signal in vivo with an IL-2 antagonist reduces tumor growth through the control of regulatory T cells. The Journal of Immunology. 2018 May 15;200(10):3475-84.
10. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New England Journal of Medicine. 2017 Dec 28;377(26):2531-44.
11. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New England Journal of Medicine. 2018 Feb 1;378(5):439-48.
12. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. New England Journal of Medicine. 2020 Apr 2;382(14):1331-42.
13. Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. 2023 Feb 23;614(7949):635-48.
14. Strati P, Neelapu SS. Chimeric antigen receptor–engineered T cell therapy in lymphoma. Current Oncology Reports. 2019 May;21:38.
15. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. The Journal of Immunology. 2004 Dec 15;173(12):7647-53.
16. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Research. 2015 Sep 1;75(17):3596-607.
17. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Research. 2015 Sep 1;75(17):3505-18.
18. Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Molecular Therapy. 2017 Aug 2;25(8):1946-58.
19. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clinical Cancer Research. 2013 Jun 15;19(12):3153-64.
20. Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS, et al. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia. 2016 Jun;30(6):1355-64.
21. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nature Medicine. 2019 Sep;25(9):1408-14.
22. Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proceedings of the National Academy of Sciences. 2013 Apr 23;110(17):6973-8.
23. Oren R, Hod-Marco M, Haus-Cohen M, Thomas S, Blat D, Duvshani N, et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. The Journal of Immunology. 2014 Dec 1;193(11):5733-43.
24. Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, et al. Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Molecular Therapy. 2017 Aug 2;25(8):1933-45.
25. Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Reports. 2017 Oct 3;21(1):17-26.
26. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017 Mar 2;543(7643):113-7.
27. Hochweller K, Wabnitz GH, Samstag Y, Suffner J, Hämmerling GJ, Garbi N. Dendritic cells control T cell tonic signaling required for responsiveness to foreign antigen. Proceedings of the National Academy of Sciences. 2010 Mar 30;107(13):5931-6.
28. Gauld SB, Dal Porto JM, Cambier JC. B cell antigen receptor signaling: roles in cell development and disease. Science. 2002 May 31;296(5573):1641-2.
29. Calderon H, Mamonkin M, Guedan S. Analysis of CAR-mediated tonic signaling. Chimeric Antigen Receptor T Cells: Development and Production. 2020:223-36.
30. Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. 2016 Dec 1;5(12):e1253656.
31. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature Medicine. 2015 Jun;21(6):581-90.
32. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunology Research. 2015 Apr 1;3(4):356-67.
33. Ajina A, Maher J. Strategies to address chimeric antigen receptor tonic signaling. Molecular Cancer Therapeutics. 2018 Sep 1;17(9):1795-815.
34. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. The Journal of Immunology. 2008 Apr 1;180(7):4901-9.
35. Zhang Z, Jiang D, Yang H, He Z, Liu X, Qin W, et al. Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death & Disease. 2019 Jun 17;10(7):476.
36. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunology Research. 2016 Jun 1;4(6):498-508.
37. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunology Research. 2015 Feb 1;3(2):125-35.
38. Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O'Neill A, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203-11.
39. James SE, Greenberg PD, Jensen MC, Lin Y, Wang J, Till BG, et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J Immunol. 2008;180(10):7028-38.
40. Woodsworth DJ, Dunsing V, Coombs D. Design parameters for granzyme-mediated cytotoxic lymphocyte target-cell killing and specificity. Biophysical Journal. 2015 Aug 4;109(3):477-88.
41. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proceedings of the National Academy of Sciences. 2018 Feb 27;115(9):E2068-76.
42. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nature Chemical Biology. 2018 Mar 1;14(3):317-24.
43. Smith EL, Harrington K, Staehr M, Masakayan R, Jones J, Long TJ, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Science Translational Medicine. 2019 Mar 27;11(485):eaau7746.
44. Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJ, Sievers SA, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Molecular Therapy. 2017 Nov 1;25(11):2452-65.
45. Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proceedings of the National Academy of Sciences. 2016 Jan 26;113(4):E459-68.
46. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and design of chimeric antigen receptors. Molecular Therapy-Methods & Clinical Development. 2019 Mar 15;12:145-56.
47. Zhang C, Liu J, Zhong JF, Zhang X. Engineering car-t cells. Biomarker Research. 2017 Dec;5:22.
48. Elazar A, Chandler NJ, Davey AS, Weinstein JY, Nguyen JV, Trenker R, et al. De novo-designed transmembrane domains tune engineered receptor functions. Elife. 2022 May 4;11:e75660.
49. Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunological Reviews. 2019 Jul;290(1):127-47.
50. Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3ɛ cytoplasmic tyrosine-based motif. Cell. 2008 Nov 14;135(4):702-13.
51. Hartl FA, Beck-Garcìa E, Woessner NM, Flachsmann LJ, Cárdenas RM, Brandl SM, et al. Noncanonical binding of Lck to CD3ε promotes TCR signaling and CAR function. Nature immunology. 2020 Aug;21(8):902-13.
52. Dietrich J, Hou X, Wegener AM, Geisler C. CD3 gamma contains a phosphoserine‐dependent di‐leucine motif involved in down‐regulation of the T cell receptor. The EMBO journal. 1994 May 1;13(9):2156-66.
53. Velasco Cárdenas RM, Brandl SM, Meléndez AV, Schlaak AE, Buschky A, Peters T, et al. Harnessing CD3 diversity to optimize CAR T cells. Nature Immunology. 2023 Dec;24(12):2135-49.
54. Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent advances in CAR-T cell engineering. Journal of Hematology & Oncology. 2020 Dec;13:86.
55. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor‐expressing T cells. Immunological Reviews. 2014 Jan;257(1):107-26.
56. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AH, et al. CAR-T design: Elements and their synergistic function. EBioMedicine. 2020 Aug 1;58:102931.
57. Sun M, Xu P, Wang E, Zhou M, Xu T, Wang J, et al. Novel two-chain structure utilizing KIRS2/DAP12 domain improves the safety and efficacy of CAR-T cells in adults with r/r B-ALL. Molecular Therapy-Oncolytics. 2021 Dec 17;23:96-106.
58. Liang X, Huang Y, Li D, Yang X, Jiang L, Zhou W, et al. Distinct functions of CAR-T cells possessing a dectin-1 intracellular signaling domain. Gene Therapy. 2023 May;30(5):411-20.
59. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nature Medicine. 2018 Mar 1;24(3):352-9.
60. Nair S, Wang JB, Tsao ST, Liu Y, Zhu W, Slayton WB, et al. Functional improvement of chimeric antigen receptor through intrinsic interleukin-15Rα signaling. Current Gene Therapy. 2019 Feb 1;19(1):40-53.
61. Brocker T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood. 2000 Sep 1;96(5):1999-2001.
62. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery. 2013 Apr 1;3(4):388-98.
63. Akhoundi M, Mohammadi M, Sahraei SS, Sheykhhasan M, Fayazi N. CAR T cell therapy as a promising approach in cancer immunotherapy: challenges and opportunities. Cellular Oncology. 2021 Jun;44:495-523.
64. Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Current Opinion in Immunology. 2009 Apr 1;21(2):215-23.
65. Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, et al. Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single-chain variable domain chimeras containing TCR-ζ vs FcεRI-γ. The Journal of Immunology. 2001 Jan 1;166(1):182-7.
66. Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. Journal of Hematology & Oncology. 2018 Dec;11:22.
67. Imai CM, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004 Apr;18(4):676-84.
68. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCRζ chain. The Journal of Immunology. 2004 Jan 1;172(1):104-13.
69. Ramos CA, Rouce R, Robertson CS, Reyna A, Narala N, Vyas G, et al. In vivo fate and activity of second-versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin's lymphomas. Molecular Therapy. 2018 Dec 5;26(12):2727-37.
70. Doherty K. Obe-Cel may mark additional treatment option for R/R B-ALL. Supplements And Featured Publications. 2022.
71. Zhao X, Yang J, Zhang X, Lu XA, Xiong M, Zhang J, et al. Efficacy and safety of CD28-or 4-1BB-based CD19 CAR-T cells in B cell acute lymphoblastic leukemia. Molecular Therapy-Oncolytics. 2020 Sep 25;18:272-81.
72. Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nature reviews Clinical Oncology. 2021 Nov;18(11):715-27.
73. Xiong W, Chen Y, Kang X, Chen Z, Zheng P, Hsu YH, et al. Immunological Synapse Predicts Effectiveness of Chimeric Antigen Receptor Cells. Molecular therapy: the journal of the American Society of Gene Therapy. 2021 Mar 3;29(3):1349-51.
74. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ,et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Science Signaling. 2018 Aug 21;11(544):eaat6753.
75. Gascoigne NR, Rybakin V, Acuto O, Brzostek J. TCR signal strength and T cell development. Annual Review of Cell and Developmental Biology. 2016 Oct 6;32:327-48.
76. Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends in Biotechnology. 2014 Sep 1;32(9):456-65.
77. Schubert ML, Kunz A, Schmitt A, Neuber B, Wang L, Hückelhoven-Krauss A, et al. Assessment of CAR T Cell Frequencies in Axicabtagene Ciloleucel and Tisagenlecleucel Patients Using Duplex Quantitative PCR. Cancers. 2020 Oct 1;12(10):2820.
78. Salzer B, Schueller CM, Zajc CU, Peters T, Schoeber MA, Kovacic B, et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nature Communications. 2020 Aug 20;11(1):4166.
79. Klampatsa A, Dimou V, Albelda SM. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opinion on Biological Therapy. 2021 Apr 3;21(4):473-86.
80. Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016 Feb 11;164(4):780-91.
81. Liu Y, Liu G, Wang J, Zheng ZY, Jia L, Rui W, et al. Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Science Translational Medicine. 2021 Mar 24;13(586):eabb5191.
82. Wang J, Zhang X, Zhou Z, Liu Y, Yu L, Jia L, et al. A novel adoptive synthetic TCR and antigen receptor (STAR) T‐cell therapy for B‐cell acute lymphoblastic leukemia. American Journal of Hematology. 2022 Aug;97(8):992-1004.
83. Powell Jr DJ, Brennan AL, Zheng Z, Huynh H, Cotte J, Levine BL. Efficient clinical-scale enrichment of lymphocytes for use in adoptive immunotherapy using a modified counterflow centrifugal elutriation program. Cytotherapy. 2009 Jan 1;11(7):923-35.
84. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. 2017 Feb 9;168(4):724-40.
85. Chmielewski M, Abken H. TRUCKS, the fourth-generation CAR T cells: current developments and clinical translation. Advances In Cell And Gene Therapy. 2020 Jul;3(3):e84.
86. Tang L, Pan S, Wei X, Xu X, Wei Q. Arming CAR-T cells with cytokines and more: Innovations in the fourth-generation CAR-T development. Molecular Therapy. 2023 Nov 1;31(11):3146-62.
87. Macian F. NFAT proteins: key regulators of T-cell development and function. Nature Reviews Immunology. 2005 Jun 1;5(6):472-84.
88. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert opinion on biological therapy. 2015 Aug 3;15(8):1145-54.
89. Young RM, Engel NW, Uslu U, Wellhausen N, June CH. Next-generation CAR T-cell therapies. Cancer Discovery. 2022 Jul 6;12(7):1625-33.
90. Tousley AM, Rotiroti MC, Labanieh L, Rysavy LW, Rietberg SP, de la Serna EL, et al. Coopting T cell proximal signaling molecules enables Boolean logic-gated CAR T cell control. BioRxiv. 2022 Jun 17:2022-06.
91. Zhao Z, Sadelain M. CAR T cell design: approaching the elusive AND-gate. Cell Research. 2023 Oct;33(10):739-40.
92. Yee C. The use of endogenous T cells for adoptive transfer. Immunological reviews. 2014 Jan;257(1):250-63.
93. Brudno JN, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for lymphoma. Nature reviews Clinical Oncology. 2018 Jan;15(1):31-46.
94. Mahadeo KM, Khazal SJ, Abdel-Azim H, Fitzgerald JC, Taraseviciute A, Bollard CM, et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nature Reviews Clinical Oncology. 2019 Jan;16(1):45-63.
95. Yu JX, Hubbard-Lucey VM, Tang J. The global pipeline of cell therapies for cancer. Nat Rev Drug Discov. 2019 Oct 1;18(11):821-2.
96. Abbasi S, Totmaj MA, Abbasi M, Hajazimian S, Goleij P, Behroozi J, et al. Chimeric antigen receptor T (CAR-T) cells: Novel cell therapy for hematological malignancies. Cancer Medicine. 2023 Apr;12(7):7844-58.
97. Cullen SP, Martin SJ. Mechanisms of granule-dependent killing. Cell Death & Differentiation. 2008 Feb;15(2):251-62.
98. de Saint Basile G, Ménasché G, Fischer A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nature Reviews Immunology. 2010 Aug;10(8):568-79.
99. Nagata S, Tanaka M. Programmed cell death and the immune system. Nature Reviews Immunology. 2017 May;17(5):333-40.
100. Davenport AJ, Jenkins MR, Cross RS, Yong CS, Prince HM, Ritchie DS, et al. CAR-T cells inflict sequential killing of multiple tumor target cells. Cancer Immunology Research. 2015 May 1;3(5):483-94.
101. Makita S, Imaizumi K, Kurosawa S, Tobinai K. Chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma: opportunities and challenges. Drugs in Context. 2019;8:212567.
102. Aghajanian H, Rurik JG, Epstein JA. CAR-based therapies: opportunities for immuno-medicine beyond cancer. Nature Metabolism. 2022 Feb;4(2):163-9.
103. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nature Reviews Clinical Oncology. 2020 Mar;17(3):147-67.
104. Liu S, Zhang X, Dai H, Cui W, Yin J, Li Z, et al. Which one is better for refractory/relapsed acute B-cell lymphoblastic leukemia: Single-target (CD19) or dual-target (tandem or sequential CD19/CD22) CAR T-cell therapy?. Blood Cancer Journal. 2023 Apr 24;13(1):60.
105. Bachy E, Le Gouill S, Di Blasi R, Sesques P, Manson G, Cartron G, et al. A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nature Medicine. 2022 Oct;28(10):2145-54.
106. Meng J, Wu X, Sun Z, Xun R, Liu M, Hu R, et al. Efficacy and safety of CAR-T cell products axicabtagene ciloleucel, tisagenlecleucel, and lisocabtagene maraleucel for the treatment of hematologic malignancies: a systematic review and meta-analysis. Frontiers in Oncology. 2021 Jul 26;11:698607.
107. Kwon M, Iacoboni G, Reguera JL, Corral LL, Morales RH, Ortiz-Maldonado V, et al. Axicabtagene ciloleucel compared to tisagenlecleucel for the treatment of aggressive B-cell lymphoma. Haematologica. 2023 Jan 1;108(1):110-21.
108. Frey NV. Approval of brexucabtagene autoleucel for adults with relapsed and refractory acute lymphocytic leukemia. Blood, The Journal of the American Society of Hematology. 2022 Jul 7;140(1):11-5.
109. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. New England Journal of Medicine. 2016 Dec 29;375(26):2561-9.
110. O'Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clinical Cancer Research. 2019 Feb 15;25(4):1142-6.
111. Prasad V. Tisagenlecleucel—the first approved CAR-T-cell therapy: implications for payers and policy makers. Nature Reviews Clinical Oncology. 2018 Jan;15(1):11-2.
112. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New England Journal of Medicine. 2019 Jan 3;380(1):45-56.
113. Schuster SJ, Tam CS, Borchmann P, Worel N, McGuirk JP, Holte H, et al. Long-term clinical outcomes of tisagenlecleucel in patients with relapsed or refractory aggressive B-cell lymphomas (JULIET): a multicentre, open-label, single-arm, phase 2 study. The Lancet Oncology. 2021 Oct 1;22(10):1403-15.
114. Lemal R, Tournilhac O. State-of-the-art for CAR T-cell therapy for chronic lymphocytic leukemia in 2019. Journal for Immunotherapy of Cancer. 2019 Dec;7:202.
115. Elsawy M, Chavez JC, Avivi I, Larouche JF, Wannesson L, Cwynarski K, et al. Patient-Reported Outcomes (PROs) in Zuma-7, a Phase 3, Randomized, Open-Label Study Evaluating the Efficacy of Axicabtagene Ciloleucel (Axi-Cel) Versus Standard-of-Care (SOC) Therapy in Patients with Relapsed/Refractory Large B-Cell Lymphoma (LBCL). In2022 Tandem Meetings| Transplantation & Cellular Therapy Meetings of ASTCT and CIBMTR 2022 Apr 24. Tandem Meetings.
116. Nastoupil LJ, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, et al. Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US Lymphoma CAR T Consortium. Journal of Clinical Oncology. 2020 Sep 9;38(27):3119-28.
117. Jacobson CA, Chavez JC, Sehgal A, William BM, Munoz J, Salles GA, et al. Outcomes in ZUMA-5 with axicabtagene ciloleucel (axi-cel) in patients (pts) with relapsed/refractory (R/R) indolent non-Hodgkin lymphoma (iNHL) who had the high-risk feature of progression within 24 months from initiation of first anti-CD20–containing chemoimmunotherapy (POD24). Journal of Clinical Oncology. 2021 May 28;39(15_Suppl):7515.
118. Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. The Lancet Oncology. 2019 Jan 1;20(1):31-42.
119. Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. The Lancet. 2021 Aug 7;398(10299):491-502.
120. Kharfan-Dabaja MA, Yassine F, Moustafa MA, Iqbal M, Murthy H. Lisocabtagene maraleucel in relapsed or refractory diffuse large B cell lymphoma: what is the evidence?. Hematology/Oncology and Stem Cell Therapy. 2022 Dec 23;15(4):168-175.
121. Neelapu SS, Dickinson M, Munoz J, Ulrickson ML, Thieblemont C, Oluwole OO, et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: the phase 2 ZUMA-12 trial. Nature Medicine. 2022 Apr;28(4):735-42.
122. Sehgal A, Hoda D, Riedell PA, Ghosh N, Hamadani M, Hildebrandt GC, et al. Lisocabtagene maraleucel as second-line therapy in adults with relapsed or refractory large B-cell lymphoma who were not intended for haematopoietic stem cell transplantation (PILOT): an open-label, phase 2 study. The Lancet Oncology. 2022 Aug 1;23(8):1066-77.
123. Elmacken M, Peredo-Pinto H, Wang C, Xu Z, Tegenge M, Jaigirdar AA, et al. FDA Approval Summary: Lisocabtagene Maraleucel for Second-Line Treatment of Large B-Cell Lymphoma. Clinical Cancer Research. 2024 Feb 23:OF1-8.
124. Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. The Lancet. 2020 Sep 19;396(10254):839-52.
125. Munshi NC, Anderson Jr LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. New England Journal of Medicine. 2021 Feb 25;384(8):705-16.
126. Sanoyan DA, Seipel K, Bacher U, Kronig MN, Porret N, Wiedemann G, et al. Real-life experiences with CAR T-cell therapy with idecabtagene vicleucel (ide-cel) for triple-class exposed relapsed/refractory multiple myeloma patients. BMC Cancer. 2023 Apr 15;23(1):345.
127. Munshi NC, Anderson LD, Shah N, Jagannath S, Berdeja JG, Lonial S, et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. Journal of Clinical Oncology, 2020. 38(15_suppl): p. 8503-8503.
128. US Food and Drug Administration. FDA approves ciltacabtagene autoleucel for relapsed or refractory multiple myeloma. United States. 2022.
129. Chekol Abebe E, Yibeltal Shiferaw M, Tadele Admasu F, Asmamaw Dejenie T. Ciltacabtagene autoleucel: The second anti-BCMA CAR T-cell therapeutic armamentarium of relapsed or refractory multiple myeloma. Frontiers in Immunology. 2022 Sep 2;13:991092.
130. Jagannath S, Lin Y, Goldschmidt H, Reece D, Nooka A, Senin A, et al. KarMMa-RW: comparison of idecabtagene vicleucel with real-world outcomes in relapsed and refractory multiple myeloma. Blood Cancer Journal. 2021 Jun 18;11(6):116.
131. Martin T, Usmani SZ, Berdeja JG, Agha M, Cohen AD, Hari P, et al. Ciltacabtagene autoleucel, an anti–B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. Journal of Clinical Oncology. 2023 Feb 2;41(6):1265-74.
132. Liu G, Rui W, Zhao X, Lin X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cellular & Molecular Immunology. 2021 May;18(5):1085-95.
133. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nature Reviews Clinical Oncology. 2022 Dec;19(12):775-90.
134. Gerlach C, Van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. Journal of Experimental Medicine. 2010 Jun 7;207(6):1235-46.
135. Sun L, Su Y, Jiao A, Wang X, Zhang B. T cells in health and disease. Signal Transduct Target Ther. 2023 Jun 19;8(1):235.
136. Cosma GL, Eisenlohr LC. Impact of epitope density on CD8+ T cell development and function. Molecular Immunology. 2019 Sep 1;113:120-5.
137. Wherry EJ. T cell exhaustion. Nat Immunol. 2011 Jun;12(6):492-9.
138. Stewart AG, Henden AS. Infectious complications of CAR T-cell therapy: a clinical update. Therapeutic Advances in Infectious Disease. 2021 Aug;8:20499361211036773.
139. Wudhikarn K, Palomba ML, Pennisi M, Garcia-Recio M, Flynn JR, Devlin SM, et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer Journal. 2020 Aug 5;10(8):79.
140. Logue JM, Zucchetti E, Bachmeier CA, Krivenko GS, Larson V, Ninh D, et al. Immune reconstitution and associated infections following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma. Haematologica. 2021 Apr 4;106(4):978-86.
141. Wittmann Dayagi T, Sherman G, Bielorai B, Adam E, Besser MJ, Shimoni A, et al. Characteristics and risk factors of infections following CD28-based CD19 CAR-T cells. Leukemia & Lymphoma. 2021 Jun 7;62(7):1692-701.
142. Baird JH, Epstein DJ, Tamaresis JS, Ehlinger Z, Spiegel JY, et al. Immune reconstitution and infectious complications following axicabtagene ciloleucel therapy for large B-cell lymphoma. Blood dvances. 2021 Jan 12;5(1):143-55.
143. Hill JA, Li D, Hay KA, Green ML, Cherian S, Chen X, et al. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood, The Journal of the American Society of Hematology. 2018 Jan 4;131(1):121-30.
144. Park JH, Romero FA, Taur Y, Sadelain M, Brentjens RJ, Hohl TM, et al. Cytokine release syndrome grade as a predictive marker for infections in patients with relapsed or refractory B-cell acute lymphoblastic leukemia treated with chimeric antigen receptor T cells. Clinical Infectious Diseases. 2018 Aug 1;67(4):533-40.
145. Abbasi A, Peeke S, Shah N, Mustafa J, Khatun F, Lombardo A, et al. Axicabtagene ciloleucel CD19 CAR-T cell therapy results in high rates of systemic and neurologic remissions in ten patients with refractory large B cell lymphoma including two with HIV and viral hepatitis. Journal of Hematology & Oncology. 2020 Dec;13(1):1.
146. Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Experimental Hematology & Oncology. 2012 Dec;1:36.
147. Tedder TF. CD19: a promising B cell target for rheumatoid arthritis. Nature Reviews Rheumatology. 2009 Oct;5(10):572-7.
148. Van Zelm MC, Reisli I, Van Der Burg M, Castaño D, Van Noesel CJ, Van Tol MJ, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. New England Journal of Medicine. 2006 May 4;354(18):1901-12.
149. Bu DX, Singh R, Choi EE, Ruella M, Nunez-Cruz S, Mansfield KG, et al. Pre-clinical validation of B cell maturation antigen (BCMA) as a target for T cell immunotherapy of multiple myeloma. Oncotarget. 2018 May 5;9(40):25764-80.
150. Osório C, Chacón PJ, White M, Kisiswa L, Wyatt S, Rodríguez-Tébar A, et al. Selective regulation of axonal growth from developing hippocampal neurons by tumor necrosis factor superfamily member APRIL. Molecular and Cellular Neuroscience. 2014 Mar 1;59:24-36.
151. Wang Y, Liu Y, Tan X, Pan B, Ge J, Qi K, et al. Safety and efficacy of chimeric antigen receptor (CAR)-T-cell therapy in persons with advanced B-cell cancers and hepatitis B virus-infection. Leukemia. 2020 Oct;34(10):2704-7.
152. Mian A, Andrapalliyal N, Weathers AL, Pohlman B, Hill BT. Late occurrence of progressive multifocal leukoencephalopathy after anti‐CD19 chimeric antigen receptor T‐cell therapy. European Journal of Haematology. 2021 Apr;106(4):584-8.
153. Hill JA, Krantz EM, Hay KA, Dasgupta S, Stevens-Ayers T, Bender Ignacio RA, et al. Durable preservation of antiviral antibodies after CD19-directed chimeric antigen receptor T-cell immunotherapy. Blood Advances. 2019 Nov 26;3(22):3590-601.
154. Hensley MK, Bain WG, Jacobs J, Nambulli S, Parikh U, Cillo A, et al. Intractable coronavirus disease 2019 (COVID-19) and prolonged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) replication in a chimeric antigen receptor-modified T-cell therapy recipient: a case study. Clinical Infectious Diseases. 2021 Aug 1;73(3):e815-21.
155. Abbasi J. Prolonged SARS-CoV-2 infection in a CAR T-cell therapy recipient. JAMA. 2021 Mar 9;325(10):924.
156. Liang W, Guan W, Chen R, Wang W, Li J, Xu K, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. The Lancet Oncology. 2020 Mar 1;21(3):335-7.
157. Mehta V, Goel S, Kabarriti R, Cole D, Goldfinger M, Acuna-Villaorduna A, et al. Case fatality rate of cancer patients with COVID-19 in a New York hospital system. Cancer Discovery. 2020 Jul 1;10(7):935-41.
158. Passamonti F, Cattaneo C, Arcaini L, Bruna R, Cavo M, Merli F, et al. Clinical characteristics and risk factors associated with COVID-19 severity in patients with haematological malignancies in Italy: a retrospective, multicentre, cohort study. The Lancet Haematology. 2020 Oct 1;7(10):e737-45.
159. Shah GL, DeWolf S, Lee YJ, Tamari R, Dahi PB, Lavery JA, et al. Favorable outcomes of COVID-19 in recipients of hematopoietic cell transplantation. The Journal of Clinical Investigation. 2020 Dec 1;130(12):6656-67.
160. Aydillo T, Gonzalez-Reiche AS, Aslam S, van de Guchte A, Khan Z, Obla A, et al. Shedding of viable SARS-CoV-2 after immunosuppressive therapy for cancer. New England Journal of Medicine. 2020 Dec 24;383(26):2586-8.
161. Maurer K, Saucier A, Kim HT, Acharya U, Mo CC, Porter J, et al. COVID-19 and hematopoietic stem cell transplantation and immune effector cell therapy: a US cancer center experience. Blood Advances. 2021 Feb 9;5(3):861-71.
162. Meir J, Abid MA, Abid MB. State of the CAR-T: risk of infections with chimeric antigen receptor T-cell therapy and determinants of SARS-CoV-2 vaccine responses. Transplantation and Cellular Therapy. 2021 Dec 1;27(12):973-87.
163. Haidar G, Garner W, Hill JA. Infections after anti-CD19 chimeric antigen receptor T-cell therapy for hematologic malignancies: timeline, prevention, and uncertainties. Current Opinion in Infectious Diseases. 2020 Dec 1;33(6):449-57.
164. Garner W, Samanta P, Haidar G. Invasive fungal infections after anti-CD19 chimeric antigen receptor-modified T-cell therapy: state of the evidence and future directions. Journal of Fungi. 2021 Feb 23;7(2):156.
165. Cordeiro A, Bezerra ED, Hirayama AV, Hill JA, Wu QV, Voutsinas J, et al. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biology of Blood and Marrow Transplantation. 2020 Jan 1;26(1):26-33.
166. Rejeski K, Kunz WG, Rudelius M, Bücklein V, Blumenberg V, Schmidt C, et al. Severe Candida glabrata pancolitis and fatal Aspergillus fumigatus pulmonary infection in the setting of bone marrow aplasia after CD19-directed CAR T-cell therapy–a case report. BMC Infectious Diseases. 2021 Dec;21(1):121.
167. Greenhalgh S, Asslan M, Dignan F, Iqbal S, Norman J, Bokhary M, et al. P1413: DOES TOCILIZUMAB INCREASE THE RISK OF INFECTIONS IN PATIENTS UNDERGOING CAR-T THERAPY?. HemaSphere. 2023 Aug 1;7(S3):e8435845.
168. Frigault MJ, Nikiforow S, Mansour MK, Hu ZH, Horowitz MM, Riches ML, et al. Tocilizumab not associated with increased infection risk after CAR T-cell therapy: implications for COVID-19?.Blood, The Journal of the American Society of Hematology. 2020 Jul 2;136(1):137-9.
169. Yan T, Zhu L, Chen J. Current advances and challenges in CAR T-Cell therapy for solid tumors: tumor-associated antigens and the tumor microenvironment. Experimental Hematology & Oncology. 2023 Jan 27;12(1):14.