Keywords
Decitabine, Epigenetics, Glioblastoma, Immunotherapy
Commentary
Although adoptive T cell therapies and immune checkpoint blockades have been successfully implemented in various cancers, no immunotherapy agents have yet reached FDA approval for glioblastoma (GBM). GBM harbors a uniquely immunosuppressive tumor microenvironment that presents significant barriers to effective immunotherapy. Key hallmarks of the GBM microenvironment include the exclusion of lymphoid cells from the tumor core and the predominance of immunosuppressive tumor-associated macrophages [1-3]. This limited immune cell infiltration into an inhibitory tumor immune microenvironment combined with low mutational burden results in the added complexity of identifying specific and durably-expressed tumor-associated antigens in a heterogenous tumor cell population [4-7]. These obstacles have resulted in variable response rates of current immunotherapies in GBM that starkly contrast to other cancers where these strategies have demonstrated strong clinical efficacy.
Previous studies have demonstrated the potential for epigenetic engineering to remodel the tumor microenvironment for targeted immunotherapies against solid tumors [8-10]. While histone deacetylase inhibitors (HDACi) and Enhancer of Zeste Homolog 2 inhibitors (EZH2i) have demonstrated similar antigenic upregulation in malignant neoplasms, DNA methyltransferase inhibitors (DNMTi) remain the most well studied small molecule epigenetic agents with strong potential as an adjunct to immunotherapy in solid tumors [11]. Specifically, our group recently published a mechanistic rationale to epigenetically prime human gliomas to immunotherapeutic targeting using clinically achievable doses of the FDA-approved DNMTi, Decitabine (DAC) [12]. Through analysis of tumor-intrinsic epigenetic and transcriptional mechanisms at single cell resolution, we demonstrated that DAC concomitantly induced tumor antigens, reactivated human endogenous retroviruses, and stimulated interferon signaling. Notably, this epigenetic remodeling was correlated with improved T cell functionality and increased cytolytic activity against primary GBM cells in vitro.
In this commentary, we synthesize recent work that supports epigenetic remodeling with DNMTi to sensitize immunogenically cold and heterogenous solid tumors to immunotherapy. We summarize previous efforts using DAC to induce targetable tumor antigens, to enhance immunology through epigenetic reactivation of human endogenous retroviruses (hERV) with downstream stimulation of IFN pathways and apoptosis receptors, and to remodel the GBM tumor microenvironment through epigenetic reprogramming of infiltrating lymphoid and myeloid immune cells. In doing so, we argue that epigenetically priming the tumor microenvironment demonstrates the potential to overcome many of the current obstacles inhibiting successful immune therapies in GBM.
DNMTi Induces Targetable Tumor Antigen
Current approaches utilizing DNMTi epigenetic remodeling have concentrated on the induction of cancer testis antigens (CTA) for adoptive T cell targeting. Endogenously expressed in many cancer types, CTA are a family of proteins that are expressed normally in germinal spermatogonia and downregulated in somatic tissue via de novo DNA methylation and other epigenetic programs [8]. In certain immunogenic tumors, aberrant expression of these proteins has been identified for targeting by engineered T cells, with New York esophageal squamous cell carcinoma 1 (NY-ESO-1) considered to be the most immunogenic [13-18]. Indeed, NY-ESO-1 and other well-studied CTA are currently being assessed in a variety of adoptive T cells and cancer vaccine clinical trials in numerous cancer types [19-25].
Despite the potential for tumor specificity and epitope immunogenicity, current adoptive T cell approaches targeting single CTA in GBM have not been successful to date. Single-antigen therapies are susceptible to antigen escape and engineered T cells struggle to overcome the inhibitory tumor-immune microenvironment within some solid tumors [26]. Moreover, in cold tumors resistant to immune therapies like GBM, NY-ESO-1 and other CTA are not frequently endogenously expressed [27]. Indeed, high intratumoral heterogeneity, low mutational burden, and inconsistent antigen expression in glioma patients compound the challenge of targeting GBM-specific tumor antigens. For example, while not classically a CTA, IL13Ra2 expression is restricted to testis in normal tissue and is a well-studied GBM-specific antigen with endogenous expression in the tumor tissue of 75% of glioma patients. However, adoptive chimeric antigen receptor (CAR) T cell therapies targeting IL13Ra2 in GBM have struggled to overcome intratumoral heterogeneity, adapt to antigen escape, and resist immunosuppressive changes in the tumor microenvironment [5,28]. Indeed, tissue analysis from GBM tumors resected after IL13Ra2 CAR T therapy demonstrated that the antigen was eliminated, but tumor growth persisted [29]. Moreover, intraventricular IL13Ra2 CAR T infusion resulted in observable increases in cytokines and immune cells in cerebral spinal fluid, however these enhancements again were unable to overcome the immunosuppressive microenvironment in GBM and prevent relapse in glioma patients receiving therapy [5]. Similar results were observed in CAR T targeting EGFRviii which demonstrated elimination of antigen despite tumor persistence [6]. Therefore, the need to identify, induce, and target multiple antigens that can remodel an immunosuppressive tumor microenvironment in GBM is particularly important.
Cancer testis antigens (CTA) are particularly susceptible to epigenetic induction. Previous work from our group and others have shown that clinically achievable doses of DAC induces durable expression of CTA in GBM tumor tissue but not normal tissue for immunotherapeutic targeting [30-34]. We administered a DAC dose approved by the FDA for myelodysplastic syndrome that was detectable in cerebral spinal and achieved durable expression of NY-ESO-1 upregulation 65-days following initial exposure to DAC [34-36]. Consistent with previous studies demonstrating re-establishment of T cell mediated apoptosis via Fas/Fas Ligand after DAC treatment in glioma, we observed that targeting of these DAC-treated glioma cells with NY-ESO-1 TCR engineered CD8+ cells stimulated polyfunctional antitumor, antiviral, and T helper cytokine repertoire that further enhanced the clearance of NY-ESO-1–expressing tumor cells [30]. Single-cell RNA sequencing of DAC treated serum-free patient-derived gliomaspheres demonstrated that ~70% of tumor cells expressed one or more CTA. Moreover, CTA were upregulated across all clusters despite interpatient and intrapatient heterogeneity marked by mesenchymal, classical, proneural, or cycling cellular clusters.
We speculated that the simultaneous induction of multiple CTA can potentially prime glioma to polyvalent antigen targeting [12]. Preclinical studies of tandem or trivalent CARs targeting multiple antigens IL13Ra2, HER2, and EphA2 in GBM mitigated antigen escape in orthotopic xenograft models of GBM and future work will need to demonstrate the feasibility of this strategy in an immune competent model [37,38]. Moreover, we hypothesize that epigenetic priming with DNMTi can be repeated with DAC being readministered following antigen dropout to re-express targetable antigen. Taken together, the induction of targetable CTA for polyclonal antigen adoptive T cell targeting supports a promising strategy for immunotherapy against GBM.
DNMTi Activates Human Endogenous Retroviruses
Beyond the induction of CTA, recent studies have begun to characterize the pleiotropic epigenetic and transcriptomic effects of DNMTi, with particular focus on how these effects can be leveraged to enhance immunotherapy. Many of these studies have highlighted the reactivation of human endogenous retroviruses (hERV). These transposable elements are fragments of exogenous viruses that have incorporated into the human genome over millions of years. Aberrant activation of these retroviral elements has been implicated in many disease pathologies, including multiple cancer types [39,40]. Until recently, the reactivation of hERV has been difficult to study, and their impact on downstream immune pathways and disease pathologies remain complex.
Previous studies have identified a role for DNA demethylation in the regulation of hERV expression which subsequently activates innate immune pattern recognition receptors (PRR) for downstream inflammatory signaling. Administration of DNMTi in solid tumors has been shown to reactivate hERVs which triggers antiviral interferon responses via Toll-Like Receptor 3 (TLR3) pathways and enhances the tumor's sensitivity to immune checkpoint therapies [41]. In primary GBM cells specifically, we identified significantly upregulated hERV after DAC treatment that positively correlated with downstream type I interferon (IFN) signaling. Consistent with previous studies in glioma, we observed enhanced MHC-I expression after DAC treatment that correlated with IFN signaling instead of direct MHC-I demethylation [30]. Similar findings have been observed in clear cell renal cell carcinoma where increased hERV signatures and associated type I IFN signaling via RIG-I have been predictive of immunotherapy [42]. In small-cell lung cancer, interferon inducible endogenous retroviruses engaged the PRRs Mitochondrial Antiviral Protein (MAVS) and Stimulator of Interferon Genes (STING), resulting in feed-forward innate immune signaling and resultant T cell infiltration [43]. We speculate that similar enhancement of IFN signaling may occur in glioma to alter the cellular composition of the tumor immune microenvironment and overcome the epigenetic mechanisms of T cell exhaustion in glioma [3,44,45].
Interestingly, recent studies have questioned the role of interferon signaling as an inhibitor rather than an enhancer of immunotherapeutic efficacy. We previously reported that TLR3 agonist, Poly-ICLC, in conjunction with dendritic cell vaccination induced enhanced interferon signaling that was associated with prolonged survival and delayed tumor progression in a randomized phase II clinical trial for the treatment of recurrent GBM [44]. These findings underscore the essential role of interferon in initiating robust anti-tumor immunity by enhancing antigen presentation, promoting T cell recruitment, and increasing MHC class I expression.
However, chronic inflammation secondary to type I IFN signaling has been associated with T cell exhaustion and resistance to immune checkpoint blockade (ICB) therapy in multiple solid tumors. Indeed, recent studies characterizing the use of a Janus kinase inhibitor (JAKi), Ruxolitinib, to inhibit downstream IFN signaling in combination with ICB has demonstrated enhanced efficacy in Hodgkin lymphoma and non-small cell lung cancer patients who had previously failed ICB [46,47]. Immunologically cold tumors need some IFN signaling to mount an effective immune response, however the role of IFN is less defined than previously thought [48]. In the context of DNMTi therapy and hERV activation in GBM, it will be crucial to carefully balance the activation of IFN signaling to avoid tipping the immune response toward exhaustion. Strategic dosing regimens or combinatorial approaches will need to be carefully considered to briefly pulse IFN and avoid chronic activation. Ongoing studies are being performed to better characterize the immunologic consequences of IFN signaling downstream of DNMTi and hERV activation in GBM.
Despite the contrasting effect of interferon stimulation in immunotherapeutic response, modulation of the glioma tumor immune microenvironment through epigenetic reactivation of hERV joins established viral strategies for the treatment of GBM. Oncolytic viruses have long been studied for their ability to stimulate host immune response in GBM and cytomegalovirus-specific CD8+ T cell responses have been induced in GBM patients treated after autologous tumor-lysate pulsed dendritic cell vaccination [49,50]. Future work will need to mechanistically establish the specific hERV upregulation and downstream interferon activation in glioma.
DNMTi Reprograms Immune Cells
Although most studies on DNMTi have focused on tumor-intrinsic epigenetic and transcriptional alterations, emerging research highlights the impact of DNMTi on immune cell function. In tumor-reactive effector cells, de novo hypermethylation programs have been associated with exhausted T-cell phenotypes that limit the efficacy of ICB [51]. Encouragingly, administration of DAC prior to ICB in a chronic LCMV in vivo model overcame de novo exhaustion-associated hypermethylated regions within IFN-g and Myc loci that rejuvenated exhausted CD8 T cells [52]. Moreover, low dose DAC priming prior to adoptive transfer of effector T cells into in vivo tumor models demonstrated enhanced anti-tumor properties, suggesting epigenetic protection from an exhausted T cell phenotype [53,54]. Specifically in GBM, a recent study demonstrated that hypermethylation of a regulatory transcription factor AP-2a is associated with increased expression of PD-L1 [55]. Furthermore, DAC re-expressed AP-2a in the tumor parenchyma to sensitize glioma to anti-PD-1 ICB in vivo [55].
The impact of DNMTi on tumor-infiltrating myeloid cells, which constitute most of the infiltrating immune cells in the GBM parenchyma, is less well-defined. Indeed, in vivo studies using a murine ovarian cancer model have identified that, secondary to type I IFN enhancement, DNMTi decreased the proportion of tumor-promoting M2 macrophages characterized by expression of scavenger receptors and indoleamine 2,3-dioxygenase activity and increased anti-tumor M1 characterized by production of TNF, IL-1, and IL-2 macrophages [56-58]. However direct in vitro DNMTi treatment of macrophages promoted M2 markers CD206 and ALOX15 while decreasing IL-1B, TNF-a, IFNg, and IL-10 production in a granuloma-like Mycobacterium tuberculosis model [59]. While the study of DNMTi on tumor-associated lymphocytes and myeloid demonstrates promise, future study of the direct DNMTi effect on the lymphoid and myeloid compartments in the GBM microenvironment remains critical.
Advances in our understanding of the epigenetic contribution to the immunologic milieu in GBM and other solid tumors continue to highlight the necessity for effective immunotherapeutic enhancement. We theorize that minimizing cell cycle inhibition and cytotoxic effects with lower doses and longer administration of DNMTi would enhance immunotherapies in GBM and there are now depot injection forms of drug as well as oral forms that might make this immune adjunct strategy more feasible [60]. The capacity of DNMTi to induce targetable antigens in solid tumors is well-established and this manuscript has reviewed novel strategies that include enhancing tumor-intrinsic interferon signaling and epigenetically reprogramming effector cells, which collectively may overcome the immunosuppressive microenvironment characteristic of GBM. While future preclinical studies will need to directly characterize DAC-mediated effects in the GBM tumor-immune microenvironment within the context of established immunotherapies, we are encouraged by this exciting progress in the study of DNMTi as a promising strategy to induce epigenetic reprogramming capable of enhancing immune therapy for GBM.
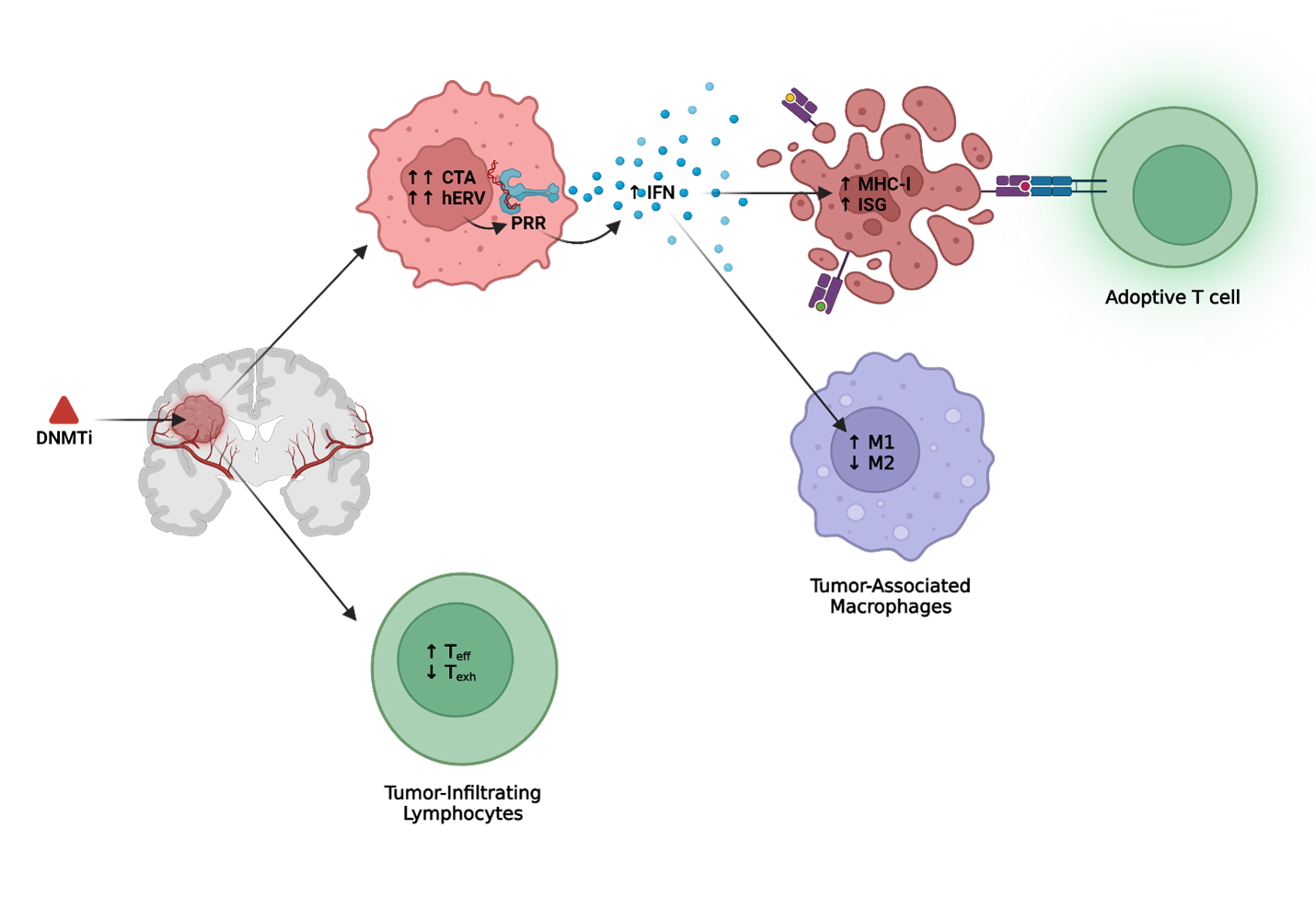
Figure 1: DNMTi enhancement of adoptive T cell therapy and microenvironment in GBM. Created with Biorender.com.
References
2. Fu W, Wang W, Li H, Jiao Y, Huo R, Yan Z, et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front Immunol. 2020 May 7;11:835.
3. Lee AH, Sun L, Mochizuki AY, Reynoso JG, Orpilla J, Chow F, et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat Commun. 2021 Nov 26;12(1):6938.
4. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019 Mar;25(3):477-86.
5. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016 Dec 29;375(26):2561-9.
6. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017 Jul 19;9(399):eaaa0984.
7. Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023 Jan 1;9(1):112-21.
8. Gonda TA, Fang J, Salas M, Do C, Hsu E, Zhukovskaya A, et al. A DNA Hypomethylating Drug Alters the Tumor Microenvironment and Improves the Effectiveness of Immune Checkpoint Inhibitors in a Mouse Model of Pancreatic Cancer. Cancer Res. 2020 Nov 1;80(21):4754-67.
9. Yu G, Wu Y, Wang W, Xu J, Lv X, Cao X, et al. Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol. 2019 Apr;16(4):401-09.
10. Wolff F, Leisch M, Greil R, Risch A, Pleyer L. The double-edged sword of (re)expression of genes by hypomethylating agents: from viral mimicry to exploitation as priming agents for targeted immune checkpoint modulation. Cell Commun Signal. 2017 Mar 31;15(1):13.
11. Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. 2019 Mar;19(3):151-61.
12. Lai TJ, Sun L, Li K, Prins TJ, Treger J, Li T, et al. Epigenetic Induction of Cancer-Testis Antigens and Endogenous Retroviruses at Single-Cell Level Enhances Immune Recognition and Response in Glioma. Cancer Res Commun. 2024 Jul 1;4(7):1834-49.
13. Nin DS, Deng LW. Biology of Cancer-Testis Antigens and Their Therapeutic Implications in Cancer. Cells. 2023 Mar 17;12(6):926.
14. Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, et al. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front Immunol. 2018 May 1;9:947.
15. Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009 Nov;100(11):2014-21.
16. Yang P, Meng M, Zhou Q. Oncogenic cancer/testis antigens are a hallmarker of cancer and a sensible target for cancer immunotherapy. Biochim Biophys Acta Rev Cancer. 2021 Aug;1876(1):188558.
17. Fratta E, Coral S, Covre A, Parisi G, Colizzi F, Danielli R, et al. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol. 2011 Apr;5(2):164-82.
18. Li XF, Ren P, Shen WZ, Jin X, Zhang J. The expression, modulation and use of cancer-testis antigens as potential biomarkers for cancer immunotherapy. Am J Transl Res. 2020 Nov 15;12(11):7002-19.
19. Zhang J, Wang L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol Cancer Res Treat. 2019 Jan 1;18:1533033819831068.
20. Pan Q, Weng D, Liu J, Han Z, Ou Y, Xu B, et al. Phase 1 clinical trial to assess safety and efficacy of NY-ESO-1-specific TCR T cells in HLA-A∗02:01 patients with advanced soft tissue sarcoma. Cell Rep Med. 2023 Aug 15;4(8):101133.
21. Kawai A, Ishihara M, Nakamura T, Kitano S, Iwata S, Takada K, et al. Safety and Efficacy of NY-ESO-1 Antigen-Specific T-Cell Receptor Gene-Transduced T Lymphocytes in Patients with Synovial Sarcoma: A Phase I/II Clinical Trial. Clin Cancer Res. 2023 Dec 15;29(24):5069-78.
22. Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015 Mar 1;21(5):1019-27.
23. Ebert LM, MacRaild SE, Zanker D, Davis ID, Cebon J, Chen W. A cancer vaccine induces expansion of NY-ESO-1-specific regulatory T cells in patients with advanced melanoma. PLoS One. 2012;7(10):e48424.
24. Ishihara M, Nishida Y, Kitano S, Kawai A, Muraoka D, Momose F, et al. A phase 1 trial of NY-ESO-1-specific TCR-engineered T-cell therapy combined with a lymph node-targeting nanoparticulate peptide vaccine for the treatment of advanced soft tissue sarcoma. Int J Cancer. 2023 Jun 15;152(12):2554-66.
25. Xia Y, Tian X, Wang J, Qiao D, Liu X, Xiao L, et al. Treatment of metastatic non-small cell lung cancer with NY-ESO-1 specific TCR engineered-T cells in a phase I clinical trial: A case report. Oncol Lett. 2018 Dec;16(6):6998-7007.
26. Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018 Oct;8(10):1219-26.
27. Freitas M, Malheiros S, Stávale JN, Biassi TP, Zamunér FT, de Souza Begnami M, et al. Expression of cancer/testis antigens is correlated with improved survival in glioblastoma. Oncotarget. 2013 Apr;4(4):636-46.
28. Kringel R, Lamszus K, Mohme M. Chimeric Antigen Receptor T Cells in Glioblastoma-Current Concepts and Promising Future. Cells. 2023 Jul 3;12(13):1770.
29. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res. 2015 Sep 15;21(18):4062-72.
30. Konkankit VV, Kim W, Koya RC, Eskin A, Dam MA, Nelson S, et al. Decitabine immunosensitizes human gliomas to NY-ESO-1 specific T lymphocyte targeting through the Fas/Fas ligand pathway. J Transl Med. 2011 Nov 7;9:192.
31. Ma R, Rei M, Woodhouse I, Ferris K, Kirschner S, Chandran A, et al. Decitabine increases neoantigen and cancer testis antigen expression to enhance T-cell-mediated toxicity against glioblastoma. Neuro Oncol. 2022 Dec 1;24(12):2093-2106.
32. Natsume A, Wakabayashi T, Tsujimura K, Shimato S, Ito M, Kuzushima K, et al. The DNA demethylating agent 5-aza-2'-deoxycytidine activates NY-ESO-1 antigenicity in orthotopic human glioma. Int J Cancer. 2008 Jun 1;122(11):2542-53.
33. Riccadonna C, Yacoub Maroun C, Vuillefroy de Silly R, Boehler M, Calvo Tardón M, Jueliger S, et al. Decitabine Treatment of Glioma-Initiating Cells Enhances Immune Recognition and Killing. PLoS One. 2016 Aug 31;11(8):e0162105
34. Everson RG, Antonios JP, Lisiero DN, Soto H, Scharnweber R, Garrett MC, et al. Efficacy of systemic adoptive transfer immunotherapy targeting NY-ESO-1 for glioblastoma. Neuro Oncol. 2016 Mar;18(3):368-78.
35. Chabot GG, Rivard GE, Momparler RL. Plasma and cerebrospinal fluid pharmacokinetics of 5-Aza-2'-deoxycytidine in rabbits and dogs. Cancer Res. 1983 Feb;43(2):592-7.
36. Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer. 2008 Jun;112(11):2341-51.
37. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018 Mar 27;20(4):506-518.
38. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016 Aug 1;126(8):3036-52.
39. Kitsou K, Lagiou P, Magiorkinis G. Human endogenous retroviruses in cancer: Oncogenesis mechanisms and clinical implications. J Med Virol. 2023 Jan;95(1):e28350.
40. Dopkins N, Nixon DF. Activation of human endogenous retroviruses and its physiological consequences. Nat Rev Mol Cell Biol. 2024 Mar;25(3):212-22.
41. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015 Aug 27;162(5):974-86.
42. Smith CC, Beckermann KE, Bortone DS, De Cubas AA, Bixby LM, Lee SJ, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest. 2018 Nov 1;128(11):4804-20.
43. Cañadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. 2018 Aug;24(8):1143-50.
44. Everson RG, Hugo W, Sun L, Antonios J, Lee A, Ding L, et al. TLR agonists polarize interferon responses in conjunction with dendritic cell vaccination in malignant glioma: a randomized phase II Trial. Nat Commun. 2024 May 8;15(1):3882.
45. Sun L, Kienzler JC, Reynoso JG, Lee A, Shiuan E, Li S, et al. Immune checkpoint blockade induces distinct alterations in the microenvironments of primary and metastatic brain tumors. J Clin Invest. 2023 Sep 1;133(17):e169314.
46. Zak J, Pratumchai I, Marro BS, Marquardt KL, Zavareh RB, Lairson LL, et al. JAK inhibition enhances checkpoint blockade immunotherapy in patients with Hodgkin lymphoma. Science. 2024 Jun 21;384(6702):eade8520.
47. Mathew D, Marmarelis ME, Foley C, Bauml JM, Ye D, Ghinnagow R, et al. Combined JAK inhibition and PD-1 immunotherapy for non-small cell lung cancer patients. Science. 2024 Jun 21;384(6702):eadf1329.
48. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016 Dec 1;167(6):1540-54.e12.
49. Everson RG, Gromeier M, Sampson JH. Viruses in the treatment of malignant glioma. Expert Rev Neurother. 2007 Apr;7(4):321-4.
50. Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Engl J Med. 2008 Jul 31;359(5):539-41.
51. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016 Dec 2;354(6316):1160-65.
52. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017 Jun 29;170(1):142-57.e19.
53. Li X, Li Y, Dong L, Chang Y, Zhang X, Wang C, et al. Decitabine priming increases anti-PD-1 antitumor efficacy by promoting CD8+ progenitor exhausted T cell expansion in tumor models. J Clin Invest. 2023 Apr 3;133(7):e165673.
54. Wang Y, Tong C, Dai H, Wu Z, Han X, Guo Y, et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat Commun. 2021 Jan 18;12(1):409.
55. Long S, Huang G, Ouyang M, Xiao K, Zhou H, Hou A, et al. Epigenetically modified AP-2α by DNA methyltransferase facilitates glioma immune evasion by upregulating PD-L1 expression. Cell Death Dis. 2023 Jun 17;14(6):365.
56. Niu Y, Chen J, Qiao Y. Epigenetic Modifications in Tumor-Associated Macrophages: A New Perspective for an Old Foe. Front Immunol. 2022 Jan 24;13:836223.
57. Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A. 2017 Dec 19;114(51):E10981-90.
58. Travers M, Brown SM, Dunworth M, Holbert CE, Wiehagen KR, Bachman KE, et al. DFMO and 5-Azacytidine Increase M1 Macrophages in the Tumor Microenvironment of Murine Ovarian Cancer. Cancer Res. 2019 Jul 1;79(13):3445-54.
59. Zambuzi FA, Cardoso-Silva PM, Castro RC, Fontanari C, Emery F da S, et al. Decitabine Promotes Modulation in Phenotype and Function of Monocytes and Macrophages That Drive Immune Response Regulation. Cells. 2021;10(4):868.
60. Dhillon S. Decitabine/Cedazuridine: First Approval. Drugs. 2020 Sep;80(13):1373-78.