Commentary
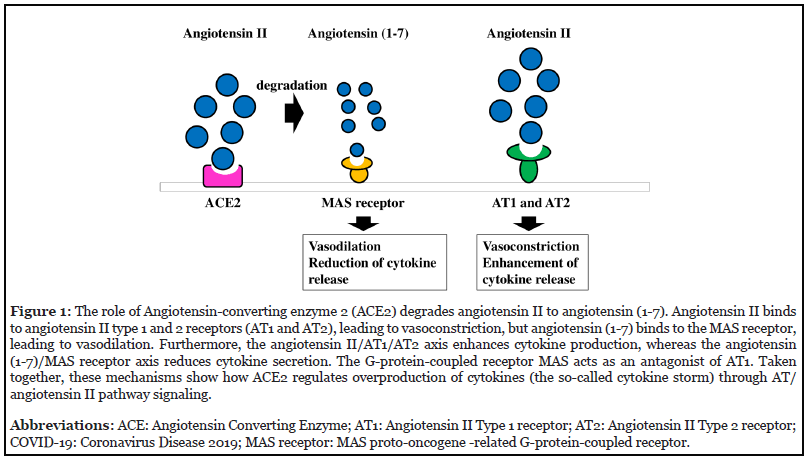
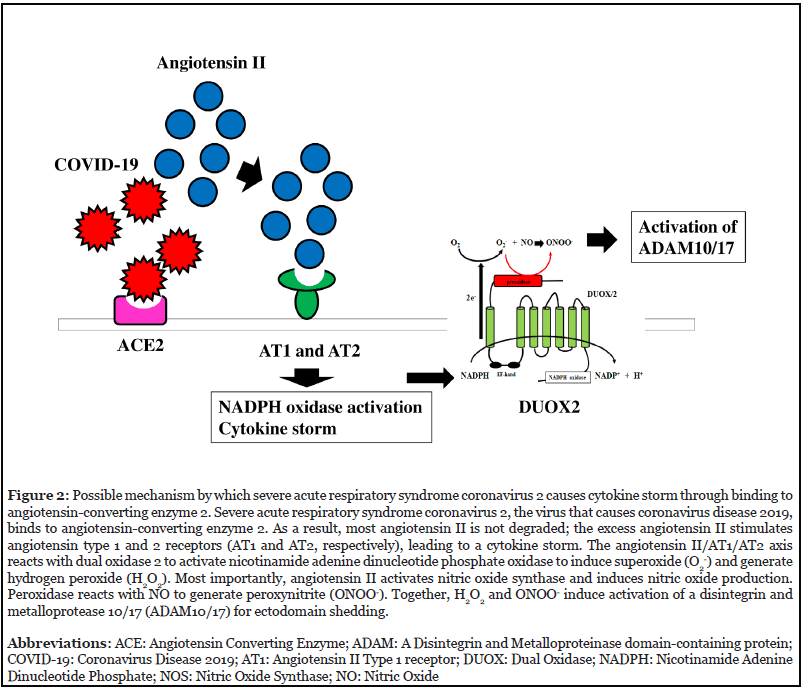
Ectodomain shedding mediated by a disintegrin and metalloprotease 10/17 (ADAM10/17) modulates the function of immune effector cells and may be involved in the novel coronavirus disease COVID-19. Toll-like receptor 7/8 (TLR7/8) recognizes single-strand RNA from viruses such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, the virus that causes COVID-19) during the innate immune response [1], and TLR7/8 agonist activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase to generate reactive oxygen species (ROS) [2]. ADAM10/7 was found to mediate ectodomain shedding to modulate immune responses [3] and to be activated by ROS [4]. These findings suggest that SARS-CoV-2 contributes to and induces ectodomain shedding, which may be associated with disease severity. In patients with COVID-19, studies found a higher blood concentration of the chemokine fractalkine [5]. Cell membrane-bound angiotensin-converting enzyme 2 (ACE2) has been identified as a binding site and entry receptor for the spike protein of SARS-CoV-2. After the virus binds to ACE2, ACE2 is internalized; ACE2 shedding also is mediated and enhanced by ADAM10/17 [6,7]. ACE2 deficiency increases expression of fractalkine [5,8]. ACE2 catalyzes and degrades angiotensin II, leading to the production of angiotensin 1-7, which binds to the angiotensin 1-7 (MAS) receptor and acts as a vasodilator (Figure 1). Indeed, binding of SARS-CoV-2 to ACE2 leads to ACE2 deficiency, which potentiates angiotensin II activity. Excess angiotensin II then activates NF-κB through the angiotensin type 1 receptor (AT1) and type 2 receptor (AT2) signaling pathway to enhance cytokine production (Figure 2). This mechanism explains how COVID-19 induces a cytokine storm [9]. Most importantly, angiotensin II activates NADPH oxidase to generate ROS [10], ie, superoxide (O2-) and hydrogen peroxide (H2O2). In cells stimulated with interleukin-1β (IL-1β), the angiotensin II/AT1 and AT2 axis augments expression of inducible nitric oxide synthase (iNOS) to generate nitric oxide (NO) [11]; NO reacts with the peroxidase domain of the enzyme dual oxidase 2, which has both a peroxidase domain and an NADPH oxidase domain, to produce the strong biological oxidant agent peroxynitrite (ONOO-). Both H2O2 and ONOO- contribute to enhanced activation of ectodomain shedding by ADAM10/17. The fractalkine receptor CX3CR1 is highly expressed by macrophages, and soluble fractalkine shed from cells because of cleavage by ADAM10/17 activates macrophages to cause a hyperinflammatory response. SARS-CoV-2 also induces cytotoxic CD8+ T cells to produce perforin and granzyme B, which show aberrant hyperactivation and target cell killing [12]. CX3CR1 is expressed by these cytotoxic CD8+ T cells, and fractalkine acts as a chemoattractant for them. Taken together, these findings indicate that ectodomain shedding may be closely associated with severity of COVID-19.
References
2. Makni-Maalej K, Boussetta T, Hurtado-Nedelec M, Belambri SA, Gougerot-Pocidalo M-A, El-Benna J. The TLR7/8 Agonist CL097 Primes N-Formyl-Methionyl- Leucyl-Phenylalanine–Stimulated NADPH Oxidase Activation in Human Neutrophils: Critical Role of p47phox Phosphorylation and the Proline Isomerase Pin1. J Immunol. 2012;189:4657-4665.
3. Wetzel, S, Seipold, L, Saftig, P. The metalloproteinase ADAM10: a useful therapeutic target? Biochim. Biophys. Acta. 2017;1864:2071-2081.
4. Myers TJ, Brennaman LH, Stevenson M, Higashiyama S, Russell WE, Lee DC, et al. Mitochondrial reactive oxygen species mediate GPCR-induced TACE/ADAM17- dependent transforming growth factor-alpha shedding. Mol Biol Cell. 2009;20:5236-5249.
5. Rivas-Fuentes S, Valdés VJ, Espinosa B, Gorocica- Rosete P, Salgado-Aguayo A. Could SARS-CoV-2 blocking of ACE2 in endothelial cells result in upregulation of CX3CL1, promoting thrombosis in COVID-19 patients? Med Hypotheses. 2021;151:110570.
6. Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297:L84-96.
7. Sharma RK, Li J, Krishnan S, Richards EM, Raizada MK, Mohandas R. Angiotensin-converting enzyme 2 and COVID-19 in cardiorenal diseases. Clin Sci (Lond). 2021;135:1-17.
8. Song B, Zhang Z-Z, Zhong J-C, Yu X-Y, Oudit GY, Hai- Yan Jin, et al. Loss of angiotensin-converting enzyme 2 exacerbates myocardial injury via activation of the CTGFfractalkine signaling pathway. Circ J. 2013;77:2997-3006.
9. Randy Q Cron. COVID-19 cytokine storm: targeting the appropriate cytokine. Lancet Rheumatol. 2021;3:e236-e237.
10. Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal. 2013;19:1110- 20.
11. Ikeda U, Maeda Y, Kawahara Y, Yokoyama M, Shimada K. Angiotensin II Augments Cytokine-Stimulated Nitric Oxide Synthesis in Rat Cardiac Myocytes. Circulation. 1995;92:2683-2689.
12. Kang CK, Han GC, Kim M, Kim G, Shin HM, Song KH, et al. Aberrant hyperactivation of cytotoxic T-cell as a potential determinant of COVID-19 severity. Int J Infect Dis. 2020;97:313-321.