Short communication
Glioblastomas (GB) are amongst the most lethal human tumors exhibiting a highly aggressive behavior manifested by tumor cell infiltration into surrounding tissue. Furthermore, GBs are notorious for their high degree of resistance to cytotoxic treatments [1-3]. Despite extensive clinical and experimental research, the prognosis for patients diagnosed with GB is grim; GB has a final mortality rate of nearly 100%, less than a 10% five-year survival rate, and a median overall survival of 15 months [4]. The current standard of care for GB patients consists of surgical resection, radiation, and chemotherapy. However, despite these therapeutic options, the recurrence rate of GB is high [5,6].
One of the main reasons for the difficulty in cancer treatment is that malignant tumors, in general, possess strong capabilities to suppress anti-tumor immune responses [7]. The number of immunosuppressive strategies utilized by tumor cells is expansive. An example of a general mechanism used is the recruitment of tolerance inducing immune cells to the tumor microenvironment (TME), such as tolerogenic dendritic cells, tumor associated macrophages, regulatory T cells (Treg), and in case of GB, microglial cells [8,9]. Additionally, the innate genetic instability of tumor cells leads to alterations of surface proteins and antigenic structures as well as the downregulation of human leukocyte antigens (HLA) molecules, which significantly impairs recognition of tumor cells by the immune system [10]. Malignant tumors also produce a range of soluble factors that aid in the suppression of immune responses, including IL-10 and TGF-β, and proangiogenic factors, such as vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), which promote tumor angiogenesis [11,12]. These mechanisms collectively contribute to the formation of an inhibitory TME, which works to suppress and prevent antitumor immune responses.
Immunotherapies present as a promising alternative to traditional therapeutic approaches, which are often characterized by their severe cytotoxic side effects. Due to the systemic nature of circulating immune cells, tumor cells dispersed throughout the body can be targeted.
However, there is little known about the factors that influence immune cell migration and effective immune responses in the TME of GBs, both in the context of a newly diagnosed tumor and at the stage of post-treatment tumor recurrence. Tumor immune evasion strategies, whether active or general, all contribute to the formation of an inhibitory and immunosuppressive TME [7]. Overcoming this immunosuppressive TME represents one of the greatest challenges to achieve clinically meaningful outcomes of immunotherapies.
Therefore, delineation of regulatory molecules and pathways determining the ability of tumor cells to affect the tolerogenic TME in order to find novel immunotherapeutic targets is a matter of the utmost importance. GARP (glycoprotein A repetition predominant) is a transmembrane protein with an extracellular 21 leucine-rich domain (LRR) and a specific marker of activated Treg [13,14]. GARP is required for the formation, binding, and surface expression of latent TGF-β [13,15]. Production of TGF-β is an immune escape mechanism to induce tolerance in a variety of immune cells. In particular, TGF-β1 has also been associated with poor prognosis of patients with malignant tumors [16]. We have previously demonstrated that GARP contributes significantly to immunological tolerance in humans through the induction of peripheral Treg, regulatory M2 macrophages, and the inhibition of antigen specific T effector cells [17]. Furthermore, we have determined that GARP is also expressed on primary malignant melanoma and melanoma cerebral metastases [18]. In the present study, we evaluated GARP as potential biomarker, factor for immunoregulation, and its relevance as a therapeutic target in GB [19].
To determine in situ GARP expression, its relevance in the TME of primary brain tumors, and its comparative expression levels to astrocytomas grade II and III, 37 patients with histologically confirmed astrocytomas were recruited for this study [19]. Immunohistochemistry was performed on excised tumor tissue and stained for GARP. Notably, all astrocytomas grade II and III and GBs, except for one GB sample, exhibited high levels of GARP expression (Figure 1a, b, d, e, f). Altogether, the prevalent expression of the inhibitory molecule GARP in brain tumor tissue (Figure 1a, b, d, e, f), such as GB and lowgrade glioma, and relatively low abundance in healthy brain tissue (Figure 1c), suggest potential important roles of GARP in the immunosuppressive TME and as a biomarker.
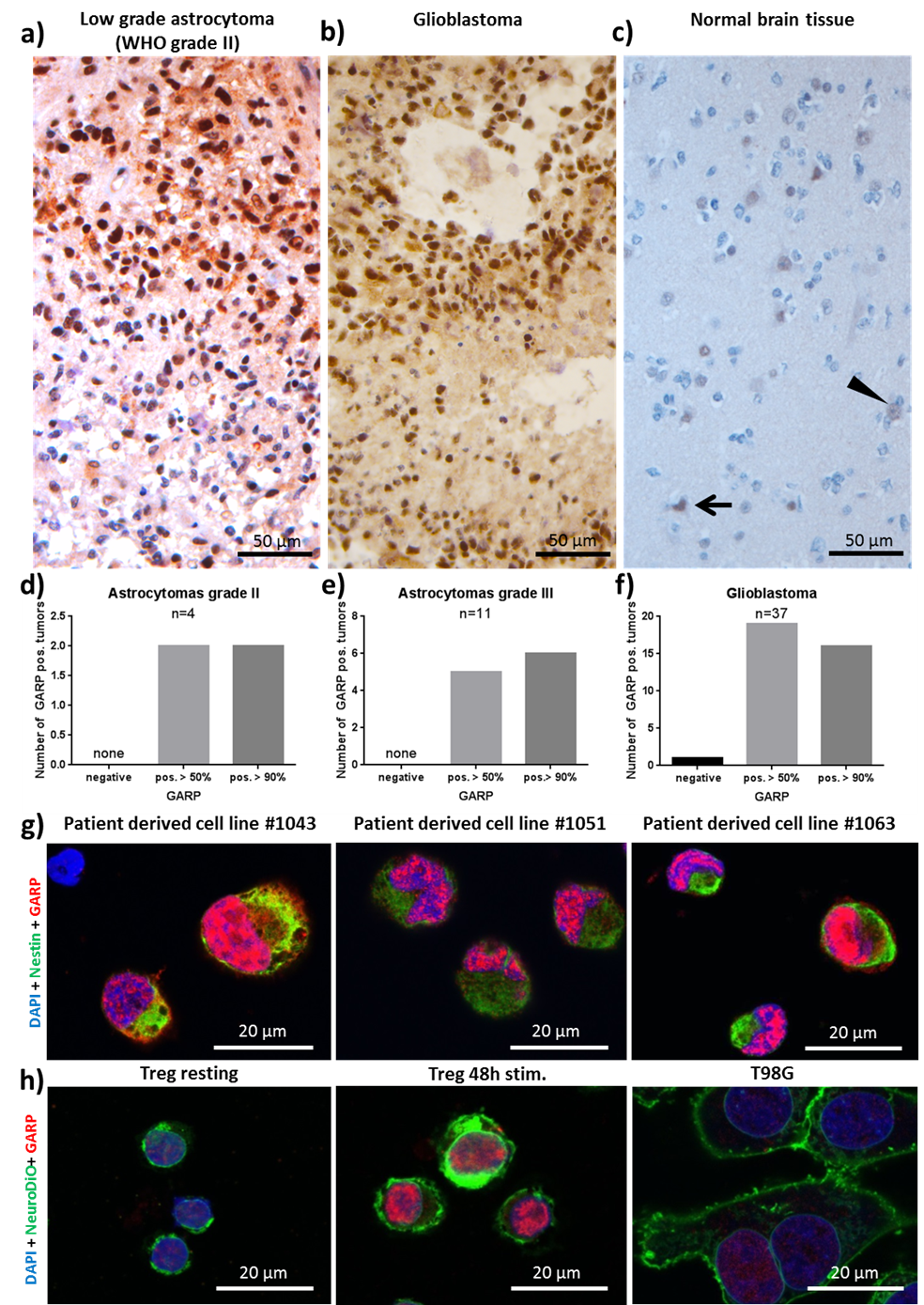
Next, we investigated GARP expression on the surface of GB tumor cell cultures. This was addressed in a commercially available GB cell line (T98G) and three GB cell lines (#1043, #1051, #1063) established in our group.
Resting and activated Treg as well as one melanoma cell line (MaMel-19) served as positive controls for GARP expression. Flow cytometry and confocal microscopy were performed to assess GARP expression. Flow cytometry confirmed that all GB cell lines used in our study express GARP on their surfaces and further supported our earlier in situ results showing GARP expression in brain tumor tissues [19]. Intriguingly, we also observed intracellular and intranuclear localization of GARP in resting and activated Treg, T98G, the three established GB cell lines (#1043, #1051, #1063), and in MaMel-19 cells (Figure 1g, h). This finding is intriguing because intracellular expression of GARP has not been previously reported. Moreover, while intracellular GARP expression was significantly higher than surface GARP expression, the intranuclear levels were even greater (Figure 1g, h). Our data demonstrates for the first time that GARP is expressed intracellularly in Treg and in GB and melanoma cell lines.
Co-culture experiments were then performed to elucidate the GARP-dependent modulatory effect of GB cells on T effector cells. Our results showed GARPdependent inhibition of T effector cell proliferation and cytokine production, in concordance with our previous findings of T effector cell suppression by melanoma tumor cells [17-19]. Collectively, these findings support the hypothesis that GARP has a role in the suppression of T effector cells in GB.
As described earlier, GARP is a transmembrane protein characterized by its extracellular 21 LRR domain [13,14]. A variety of proteins have been discovered containing LRRs with many different functions, including signal transduction, cell differentiation, and migration. Such proteins are frequently membrane bound, but they also can be secreted or localized to the cytoplasm or nucleus [20]. Examples of molecules containing LRRs are adhesion molecules, enzymes, and tyrosine kinase receptors (RTK). Although RTKs are usually localized to the plasma membrane, they have also been observed in the nucleus where they can interact with transcription factors which regulate cell proliferation, survival, and migration [21]. In the light of our findings of intranuclear GARP in Treg and in tumor cells and the structural similarities between GARP and other LRRs-containing proteins, it is tempting to speculate that GARP may have some previously unrecognized functions that may or may not be related to the canonical TGF-β dependent mechanism of tumor immunosuppression mediated by GARP. However, further studies are needed to confirm or rule out this assumption.
Our findings may also further enhance the understanding of tumor immune escape mechanisms used by GB, especially in the context of GB progression and therapy resistance. Specifically, GARP is known to suppress antitumor responses via the TGF-β pathway, which is involved in the progression of GB and in the maintenance of self-renewal in glioma stem cells [17,22]. Targeting the factors involved in the formation of an immunosuppressive TME, like GARP, has become increasingly recognized as a promising strategy to improve the efficacy of immunotherapies for GB [23,24]. However, the unequivocal evidence to support this line of thought is still missing and further studies involving larger groups of patients must be conducted to confirm this hypothesis.
The need for individualized targeted therapies and immunotherapies for the treatment of malignant tumors has become widely recognized. Individual differences in response rates, side effects, and outcomes from different therapies can be attributed in part to differences in patients’ unique TMEs. Characterization of TMEs through the use of biomarkers is a promising approach to individually describe patients’ TMEs and to better predict possible prognoses, therapy options, side effects, response rates, and costs for each patient. The discovery of new biomarkers is a prerequisite to the development of novel immunotherapeutic approaches [25-27]. This study reveals for the first time, the expression of GARP inside glioma cells suggesting that the impact of this immunoregulatory molecule in GBs may extend beyond the canonical role of GARP in contributing to the immunosuppressive TME by modulating immune cells activities. We found a correlation between GARP expression in GB cells and decreased T effector cell function, similar to the relationship between GARP expression in melanoma cells and T effector cell suppression reported in our recent study [18,19]. It is tempting to speculate that GARP may have a conserved role in different types of cancer cells. The expression of GARP on GB cells in conjunction with its relatively low abundance in non-neoplastic brain underscores the potential diagnostic merit of GARP as a tumor cell biomarker and a prognostic marker for GB.
In summary, our present findings have described for the first time that GARP is expressed in the TME of primary brain tumors, including GB and astrocytomas grade II and III [19]. The presence of GARP on the surface of primary brain tumors, malignant melanoma, and activated Treg, is all suggestive of GARP being an interesting novel target for antibody based immunotherapeutic targeting. The results from this study provide novel insights that may have an impact on the development of new immunotherapeutic strategies for GB diagnostics and treatment.
Author Contributions
Conceptualization, A.T., E.K., J.T., and C.S.; methodology, N.Z., J.S., P.L., F.K., and B.S.; validation, N.Z. and B.S.; formal analysis, F.K.; resources, A.T.; writing—original draft preparation, A.T., N.Z., and E.K.; writing—commentary preparation, E.T., N.Z., A.T.; writing—review and editing of original draft, C.S., J.T., and F.R.; writing—review and editing of commentary, E.K., J.T.; draft supervision, A.T.; project administration, N.Z.; funding acquisition, A.T.
References
2. Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad—tumor-associated microglia/ macrophages in glioma. International journal of molecular sciences. 2018 Feb;19(2):436.
3. See AP, Parker JJ, Waziri A. The role of regulatory T cells and microglia in glioblastoma-associated immunosuppression. Journal of neuro-oncology. 2015 Jul 1;123(3):405-12.
4. Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine. 2005 Mar 10;352(10):987-96.
5. Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma—are we there yet? Neuro-oncology. 2012 Nov 7;15(1):4-27.
6. Seystahl K, Wick W, Weller M. Therapeutic options in recurrent glioblastoma—an update. Critical reviews in oncology/hematology. 2016 Mar 1;99:389-408.
7. Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral oncology. 2016 Jul 1;58:59-70.
8. Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, Franciszkiewicz K, Chouaib S, Kaminska B. Microglia-derived TGF-β as an important regulator of glioblastoma invasion— an inhibition of TGF-β-dependent effects by shRNA against human TGF-β type II receptor. Oncogene. 2008 Feb;27(7):918.
9. Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G. Immune evasion strategies of glioblastoma. Frontiers in surgery. 2016 Mar 2;3:11.
10. Perea F, Bernal M, Sánchez-Palencia A, Carretero J, Torres C, Bayarri C, Gómez-Morales M, Garrido F, Ruiz- Cabello F. The absence of HLA class I expression in nonsmall cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration. International journal of cancer. 2017 Feb 15;140(4):888-99.
11. Gajewski TF. Identifying and overcoming immune resistance mechanisms in the melanoma tumor microenvironment. Clinical Cancer Research. 2006 Apr 1;12(7):2326s-30s.
12. Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro-oncology. 2015, 17 Suppl 7, vii9-vii14, doi:10.1093/ neuonc/nov151. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro-oncology. 2015 Oct 29;17(suppl_7):vii9-14.
13. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proceedings of the National Academy of Sciences. 2009 Aug 11;106(32):13445-50.
14. Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PloS one. 2008 Jul 16;3(7):e2705.
15. Vermeersch E, Denorme F, Maes W, De Meyer SF, Vanhoorelbeke K, Edwards J, Shevach EM, Unutmaz D, Fujii H, Deckmyn H, Tersteeg C. The role of platelet and endothelial GARP in thrombosis and hemostasis. PloS one. 2017 Mar 9;12(3):e0173329.
16. Guo SK, Shen MF, Yao HW, Liu YS. Enhanced Expression of TGFBI Promotes the Proliferation and Migration of Glioma Cells. Cellular Physiology and Biochemistry. 2018;49(3):1138-50.
17. Hahn SA, Stahl HF, Becker C, Correll A, Schneider FJ, Tuettenberg A, Jonuleit H. Soluble GARP has potent antiinflammatory and immunomodulatory impact on human CD4+ T cells. Blood. 2013 Aug 15;122(7):1182-91.
18. Hahn SA, Neuhoff A, Landsberg J, Schupp J, Eberts D, Leukel P, Bros M, Weilbaecher M, Schuppan D, Grabbe S, Tueting T. A key role of GARP in the immune suppressive tumor microenvironment. Oncotarget. 2016 Jul 12;7(28):42996.
19. Zimmer N, Kim E, Schupp J, Sprang B, Leukel P, Khafaji F, Ringel F, Sommer C, Tuettenberg J, Tuettenberg A. GARP as an Immune Regulatory Molecule in the Tumor Microenvironment of Glioblastoma Multiforme. International journal of molecular sciences. 2019 Jan;20(15):3676.
20. Ollendorff V, Noguchi T, Delapeyriere O, Birnbaum D. The GARP gene encodes a new member of the family of leucine-rich repeat-containing proteins. Cell Growth and Differentiation-Publication American Association for Cancer Research. 1994 Feb 1;5(2):213-20.
21. Bencheikh L, Rivière J, Imanci A, Pierron G, Souquere S, Naimo A, Morabito M, Dussiot M, De Leeuw F, Lobry C, Solary E. Dynamic gene regulation by nuclear colony-stimulating factor 1 receptor in human monocytes and macrophages. Nature communications. 2019 Apr 26;10(1):1935.
22. Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-β signaling and its targeting for glioma treatment. American journal of cancer research. 2015;5(3):945.
23. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nature reviews Clinical oncology. 2018 Jul;15(7):422-42.
24. Xue S, Hu M, Iyer V, Yu J. Blocking the PD-1/PD-L1 pathway in glioma: a potential new treatment strategy. Journal of hematology & oncology. 2017 Dec;10(1):81.
25. de Groot JF, Piao Y, Tran H, Gilbert M, Wu HK, Liu J, Bekele BN, Cloughesy T, Mehta M, Robins HI, Lassman A. Myeloid biomarkers associated with glioblastoma response to anti-VEGF therapy with aflibercept. Clinical Cancer Research. 2011 Jul 15;17(14):4872-81.
26. Ilhan-Mutlu A, Wagner L, Woehrer A, Jungwirth S, Marosi C, Fischer P, Preusser M. Blood alterations preceding clinical manifestation of glioblastoma. Cancer investigation. 2012 Oct 25;30(9):625-9.
27. Martens A, Wistuba-Hamprecht K, Foppen MG, Yuan J, Postow MA, Wong P, Romano E, Khammari A, Dreno B, Capone M, Ascierto PA. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clinical Cancer Research. 2016 Jun 15;22(12):2908-18.