Introduction
Gliomas are an aggressive class of primary brain tumors with high rates of recurrence and a dismal overall survival [1]. While existing therapeutic strategies provide some benefit, their effects are variable, and no curative modalities exist. The poor prognosis of these patients largely stems from the heterogenous molecular profile of these tumors and their tumor immune microenvironment (TIME) [2,3]. In addition to tumor expression of PD-L1, glioma cells release immunosuppressive cytokines and chemokines, like IL-10, TGF-β, and PGE2 in the intercellular tumor milieu, which have been shown to reduce antigen presenting activity and downregulate the proliferation of natural killer (NK) and CD8+ cytotoxic T-cells [4-7]. These tumor-derived factors facilitate the increased presence of immunosuppressive cell populations like myeloidderived suppressor cells (MDSC), tumor-associated macrophages (TAM), and regulatory T-cells (Tregs), which synergistically contribute to immune evasion and poor response to immunotherapies [4,8,9].
There is a growing body of evidence supporting the cancer immunoediting model as a critical mechanism underlying the immunosuppressive features acquired by cancers, and the ultimate failure of immunotherapies observed in aggressive tumors [10-12]. This process, as previously described in three dynamic phases, suggests that selective pressure from the body’s immune system drives the evolution of cancer cell subclones that can eventually escape immune surveillance [10,13,14]. Elimination is a subclinical phase in which developing cancer cells are recognized and destroyed by immune defenses. Equilibrium is reached when cancer cells remain contained by the immune system, yet are not eliminated, and fail to increase in number. Escape is the final phase in which tumor cells expand to a clinically appreciable disease and are able to evade immune response [13,14]. The reduced immunogenicity observed in “escaped” cancer cell populations has been previously shown to be associated with MHC-I and antigen presentation machinery (APM) defects [14,15]. Progression through these three states can be reversible and likely driven by a cumulative compounding of epigenetic and genetic alterations resulting from a dynamic tumor-immune interaction.
The cancer immunoediting process has been previously described in the context of aggressive tumors like melanoma, lung, and head and neck cancers, and there is a growing body of preclinical and clinical evidence supporting the role of this process in glioma development [14,16,17]. In this sense, the cancer immunoediting phenomena occurring in gliomas is highly relevant since understanding the mechanisms through which the immune system selects immuno-evasive tumor clones could help improve therapeutic strategies for these patients.
Evidence of Cancer Immunoediting in Gliomas
The factors contributing to gliomagenesis are likely multifactorial, and it has recently been suggested that advanced-age related immunosuppression may be associated with glioma onset and lack of response to immunotherapy [18,19]. A state of reduced immunity potentially serves to preclude glioma cell killing as a result of the reduced number of lymphocytes in the elimination phase. Although evidence of elimination is difficult to assess because of its subclinical phenotype, temporal analysis of tumor progression demonstrates a CD4+ and CD8+ gene expression in the TIME in mouse glioma models even before onset of symptoms, followed by progressive decline [11,20]. Similarly, new clinical evidence suggests that the most robust selective pressures take place during the early stages of gliomagenesis, as demonstrated by the retention of the cell clonal architecture in newly diagnosed and recurrent gliomas and lack of significant recurrencespecific gene changes undergoing positive selection [21]. This is also manifested by the absence of a cancer immunoediting process between newly diagnosed and recurrent gliomas [21]. One possibility is that during the early stages of glioma development, CD8+ T-cells start to sculpt tumors by negatively selecting immunogenic cancer cell clones as shown in murine models of breast cancer, lymphoma, and fibrosarcoma [22]. Then, at the time of clinical diagnosis, the acquired immunosuppressive phenotype of the tumor is able to effectively overcome anti-tumor immunity, preventing the further homing and survival of lymphocytes. If cancer immunoediting is occurring during glioma development, it is unclear until which point the lymphocytes exert a selective pressure to the tumor as low numbers of exhausted T-cells are commonly noted in glioma patients [23,24].
Recently, we interrogated a murine glioma model and directly demonstrated that cancer immunoediting by the CD8+ T-cell population plays a critical role in driving the dynamic and immunosuppressive nature of these tumors and their microenvironment (summarized by the schematic, Figure 1) [11]. These mice exhibit CD4+ and CD8+ T-cell infiltration at early stages of tumor development followed by a decline in the number of lymphocytes at late stages of gliomagenesis, supporting the presence of early selection pressure in promoting the transition to an immunosuppressive phenotype [11]. Furthermore, murine gliomas grown in the absence of CD8+ T-cells exhibited unique genomic and TIME characteristics relative to those grown in immunocompetent mice; similar findings have been observed across both antibody-depleted and transgenic CD8+ T-cell KO models [11,25]. These changes in tumor characteristics can be appreciated at a macro-level by assessing grafting behavior: gliomas developed in CD8+ T-cell-depleted mice failed to grow when intracranially implanted in immunocompetent mice; whereas those from mice with intact CD8+ T-cells progressed after transplant in both immunocompromised and immunocompetent mice [11]. Interestingly, it has been noted that the presence or absence of CD8+ T-cells during gliomagenesis does not influence overall survival on these mouse glioma models [11,25]. While it appears that the immune pressure present during tumor progression contributes to a unique tumor phenotype, it does not promote the emergence of disproportionate pathogenicity/aggressiveness. The delicate and reciprocal cross-talk between tumor progression and immune response, and the magnitude of its effects, may provide insight to potential mechanisms underlying the Hellstrom Paradox of cancer biology; that is, the observation that despite an immunological response against cancer cells, this immune reaction is dysfunctional or attenuated to achieve tumor eradication [26,27].
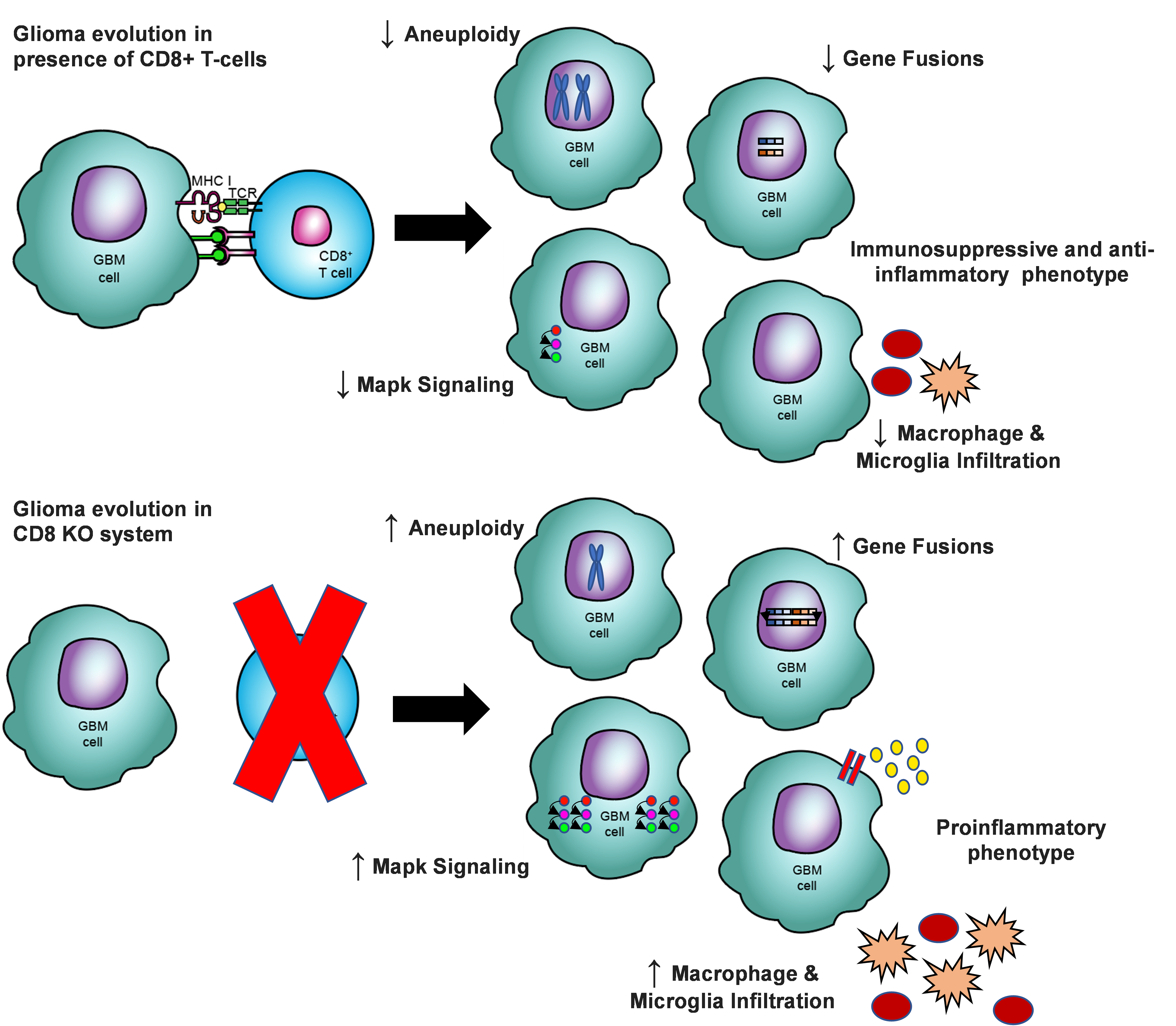
At the molecular level, gliomas developed in CD8+ T-cell-depleted systems have been shown to uniquely exhibit increased immunogenicity. Although the increase in aneuploidy and number of gene fusions observed in these murine tumors are observed less frequently in human gliomas, it has been demonstrated in several other cancers that high aneuploidy is associated with reduced expression of immune-response genes [28]. Interestingly, of the 12 tumor types analyzed by Davoli et al., only aneuploid gliomas (LGG and GBM) failed to exhibit this differential expression signature. These findings lend further support that aneuploidy in gliomas may contribute to immunogenicity and thus drive subsequent loss of this characteristic via negative selection by CD8+ T-cells, as is often observed in the clinical setting.
Additionally, gene fusions are known to generate neoantigens which demonstrate a high affinity for MHC-I [29]. An analysis of IDH wild-type high grade gliomas (HGG) showed that chromosomal instability through whole genome duplication was noted to be an early event in tumorigenesis and was associated with increased gene fusions and tumor progression [30]. Similarly, we found that gliomas that did not undergo CD8+ T-cell pressure develop more gene fusions than tumors developed with this immune pressure [11]. These findings support that negative selection against cancer cells with gene fusions may be occurring early in tumorigenesis. Aneuploid gliomas are also associated with a more aggressive clinical course and shorter overall survival [21]. At the same time, chromosomal instability has been shown to activate the cGAS-STING pathway which subsequently leads to a memory phenotype of CD8+ T-cells, further supporting negative selection of these features by the immune system [31,32]. In a murine model, it has been shown that tumors evolved to possess high chromosomal instability are initially immunogenic through cGAS-STING, contributing to increased interferon and APM activation. The presence of immune pressure, however, leads to eventual suppression of anti-tumor mechanisms and the emergence of immune evasive mechanism, notably by hypermethylation of genes associated to the MHC-I and APM genes [33]. Previously, we performed a comprehensive analysis of the genes that are part of the APM machinery in GBM and LGG from TCGA. In this analysis, we showed that almost a half of GBM patients and a third of LGG patients acquired defects in the genes coding for the MHC-I and APM pathway [14]. Moreover, these findings further support an immune editing-driven mechanism to explain the paradoxically aggressive clinical course of gliomas which exhibit aneuploidy and how tumors that may retain chromosomal instability may evade immune response.
Analysis of the cellular composition and dynamics in the glioma tumor microenvironment provide further evidence of immune system-mediated evolution. Immunosuppressive macrophages infiltrate gliomas and other tumors during progression to suppress the immune system and promote tumor growth [34,35]. Uniquely, gliomas developed in the absence of CD8+ T-cells exhibited a robust macrophage and microglial infiltration; however, their phenotype is changed to one that is proinflammatory, rather than tumor-supportive [11,25]. There is a strong correlation between these proinflammatory cell populations and MAPK signaling, suggesting a potential mechanistic link for activating CD8+ T-cell activity [11,36]. Interestingly, we found that gliomas arising in CD8+ T-cell-depleted mice similarly exhibited phosphorylation/activation of Erk and p38, relative to gliomas in immunocompetent mice, suggesting the attenuation of this cascade is facilitated by selective pressure from CD8+ T-cell-mediated immune response [11,25].
The use of anti-PD-1 therapy against gliomas in a CD8 KO mouse model similarly demonstrates increased infiltration of proinflammatory CD11b+ and Iba1+ monocytes and macrophages which are associated with reduced tumor proliferation [25,37]. In addition, there is an anti-PD-1 mediated reduction of macrophages and microglia from the TIME, suggesting an alternative therapeutic mechanism of action from previously explored CD8+ T-cell activation [25]. Nonetheless, it appears that CD8+ T-cell selection pressure against proinflammatory cellular populations in the TIME may serve to mitigate this phenotype. Moreover, tumor subclones which are able to shift the TIME phenotype to one that is more immunosuppressive, as often observed in clinical stage gliomas, will be able to escape elimination by CD8+ T-cells.
Transcriptomic analysis of glioma cells at different stages of progression in another preclinical model showed LGG were not able to graft in immunocompetent mice with greater abundance of infiltrating lymphocytes and immunostimulatory transcript profile, suggestive of a TIME associate with the elimination stage [38]. Similar to CD8+ T-cell-depleted systems, temporal analysis demonstrated that these genes are downregulated during glioma progression and immune infiltrate shifts to a more pro-tumorigenic phenotype. Gliomas eliciting neurological symptoms at an earlier time period in mice are noted to be less malignant and more immunogenic than those that manifest clinically at a later time point [38]. This is consistent with the cancer immunoediting model, as tumors early in development may have been exposed to milder selection pressures and thus may not have developed as robust of an immune-evasive arsenal.
A recent preclinical study employing GL261 glioma model in Pfp-/- Rag2-/- and wild-type murine hosts showed differences in the diversity of the tumor clonality [39]. By using a multicolor cell tracking system, it was shown that tumors injected into the brains of immunodeficient mice displayed a highly clonal composition compared to tumors grown in immunocompetent mouse hosts. Although some of the findings of this study are different to ours, likely due to the glioma models employed, our results were consistent in showing that the immune system shapes the tumor clonal architecture. These findings also further highlight the complexity of the interaction between the immune system and cancer cells at different time points of gliomagenesis.
Immunotherapies as Drivers of Cancer Immunoediting
The implications of cancer immunoediting in glioma will be important to consider when making treatment decisions for patients and should help further guide the development of therapeutic strategies. Current immunotherapies, which rely on augmenting immunogenicity directly through activation of the immune response and/or indirectly through the TIME, may effectively drive the cancer immunoediting process in eliminating tumor cells initially, and subsequently contribute to the positive selection of resistant cancer clones leading to tumor escape [40]. The failure of many existing immunotherapies for gliomas may in part be attributable to the editing-out of neoantigens that many of these treatments target [10,14]. Previous studies have demonstrated the reduced expression of therapeutic targets at glioma recurrence, notably following EGFRvIII peptide vaccination and CAR-T targeting of IL13Ra2 [41,42].
Although successful for melanoma and non-small cell lung cancer, immunotherapies have shown limited effects in glioma, largely due to the scarcity of CD8+ T-cells and predominance of immunosuppressive macrophages and Tregs in the TIME [43,44]. Our group has previously demonstrated that recurrent GBM treated with anti- PD-1 therapy demonstrate progressive loss of non-silent mutations predicted to be antigenic, and supporting the case for therapy-driven immunoediting [12]. Recurrent GBM tumors that have been shown to be more susceptible to anti-PD-1 immunotherapy share important characteristics with tumors grown in a CD8+ T-celldepleted environment, notably MAPK pathway alterations and a more immunogenic TIME [12]. Thus, this may contribute to greater potential for evolutionary sculpting by the activated immune response and may lead to the development of resistance and recurrence in the future.
Other approaches, like IL-12-based therapies, have attempted to regulate the tumor microenvironment to induce greater differentiation into Th1 cells, effectively promoting IFNγ release and thus indirectly increasing CD8+ T-cell activation [40]. It will be important to further assess the potency and effectiveness of such TIME-augmenting strategies given that they may also indirectly accelerate cancer immunoediting via CD8+ T-cell activation, and subsequently drive tumor escape.
Moving forward, therapies may aim to target other cellular populations (e.g. NK cells, MDSC, B cells, etc.), for direct or indirect manipulation [45,46]. It has recently been shown that anti-PD-1 therapy may also mediate its effects through an innate immune response, in addition to CD8+ T-cell activation, providing an alternative mechanism to augment therapy [25]. The impact of other cell populations on cancer immunoediting will need to be assessed in the future to better understand the extent of selective pressure they may exert on glioma evolution.
Treatment strategies will also benefit from combination therapies based on the unique profile of an individual’s tumor to effectively limit tumor immune escape. By determining the optimal therapeutic regimen at diagnosis, it may be possible to overcome the immunoevasive mechanisms of tumor cells. These findings also highlight the importance of exploring alternative immunotherapeutic innovations to effectively mitigate escape and avoid priming tumors for recurrence with reduced immunogenicity. A better understanding of the cancer immunoediting dynamics may enable future therapies to manipulate tumor evolution towards a more immunogenic path or surpass CD8+ T-cell-mediated immune evasion. Furthermore, because it appears that much of cancer immunoediting is subclinical, it will also be important to investigate potential strategies for early detection of gliomas early in their progression (i.e. while they are still in the elimination or equilibriums stages). In doing so, it may be possible to treat glioma patients with immunotherapies while tumor cells remain in these immunogenic states.
Overall, it is evident that the increase in understanding of glioma biology, its immune system interactions, and the tumor microenvironment support the critical role of cancer immunoediting in sculpting these tumors. The heterogenous and immunosuppressive manifestations of clinical gliomas which contribute to their resistance to immunotherapy appear to be a direct result of this process. Moving forward, it will also be critical to further interrogate the dynamics between different immune system components and glioma tumorigenesis to better understand tumor progression during elimination, equilibrium, and escape and potential avenues for intervention. Notably, the effects of immunotherapies in driving cancer immunoediting, and subsequently contributing to glioma evasion and recurrence, must also be considered as use of this class of therapeutics increases in the clinical setting. In taking to account the mechanisms and implications of cancer immunoediting in gliomas, it may be possible to develop new therapeutic platforms for more effective and absolute treatment for this group of cancers.
Acknowledgments
This work was supported by the NIH grant 1R01NS110703- 01A1 (AMS), 5DP5OD021356-05 (AMS), P50CA221747 SPORE for Translational Approaches to Brain Cancer (AMS), developmental funds from the Robert H. Lurie Cancer Center Support Grant #P30CA060553 (AMS).
References
2. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell. 2019;178(4):835-49 e21.
3. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell. 2017;32(1):42-56 e6.
4. Kamran N, Alghamri MS, Nunez FJ, Shah D, Asad AS, Candolfi M, et al. Current state and future prospects of immunotherapy for glioma. Immunotherapy. 2018;10(4):317-39.
5. Perng P, Lim M. Immunosuppressive Mechanisms of Malignant Gliomas: Parallels at Non-CNS Sites. Front Oncol. 2015;5:153.
6. Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. 2013;19(12):3165-75.
7. Colton CA. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol. 2009;4(4):399-418.
8. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloidderived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013;138(2):105-15.
9. Mirghorbani M, Van Gool S, Rezaei N. Myeloidderived suppressor cells in glioma. Expert Rev Neurother. 2013;13(12):1395-406.
10. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151-67.
11. Kane JR, Zhao J, Tsujiuchi T, Laffleur B, Arrieta VA, Mahajan A, et al. CD8(+) T-cell-mediated immunoediting influences genomic evolution and immune evasion in murine gliomas. Clin Cancer Res. 2020.
12. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462-9.
13. Dunn GP, Fecci PE, Curry WT. Cancer immunoediting in malignant glioma. Neurosurgery. 2012;71(2):201-22; discussion 22-3.
14. Arrieta VA, Cacho-Diaz B, Zhao J, Rabadan R, Chen L, Sonabend AM. The possibility of cancer immune editing in gliomas. A critical review. Oncoimmunology. 2018;7(7):e1445458.
15. Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999-1005.
16. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565- 70.
17. Lee MY, Allen CT. Mechanisms of resistance to T cellbased immunotherapy in head and neck cancer. Head Neck. 2020.
18. Ladomersky E, Scholtens DM, Kocherginsky M, Hibler EA, Bartom ET, Otto-Meyer S, et al. The Coincidence Between Increasing Age, Immunosuppression, and the Incidence of Patients With Glioblastoma. Front Pharmacol. 2019;10:200.
19. Ladomersky E, Zhai L, Lauing KL, Bell A, Xu J, Kocherginsky M, et al. Advanced Age Increases Immunosuppression in the Brain and Decreases Immunotherapeutic Efficacy in Subjects with Glioblastoma. Clin Cancer Res. 2020.
20. Tran Thang NN, Derouazi M, Philippin G, Arcidiaco S, Di Berardino-Besson W, Masson F, et al. Immune infiltration of spontaneous mouse astrocytomas is dominated by immunosuppressive cells from early stages of tumor development. Cancer Res. 2010;70(12):4829-39.
21. Barthel FP, Johnson KC, Varn FS, Moskalik AD, Tanner G, Kocakavuk E, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576(7785):112- 20.
22. Takeda K, Nakayama M, Hayakawa Y, Kojima Y, Ikeda H, Imai N, et al. IFN-gamma is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat Commun. 2017;8:14607.
23. Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res. 2018;24(17):4175-86.
24. Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459-68.
25. Rao G, Latha K, Ott M, Sabbagh A, Marisetty A, Ling X, et al. Anti-PD-1 induces M1 polarization in the glioma microenvironment and exerts therapeutic efficacy in the absence of CD8 cytotoxic T cells. Clin Cancer Res. 2020.
26. Hellstrom I, Hellstrom KE, Pierce GE, Yang JP. Cellular and humoral immunity to different types of human neoplasms. Nature. 1968;220(5174):1352-4.
27. Hellstrom KE, Hellstrom I. From the Hellstrom paradox toward cancer cure. Prog Mol Biol Transl Sci. 2019;164:1-24.
28. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355(6322).
29. Yang W, Lee KW, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. 2019;25(5):767-75.
30. Boisselier B, Dugay F, Belaud-Rotureau MA, Coutolleau A, Garcion E, Menei P, et al. Whole genome duplication is an early event leading to aneuploidy in IDHwild type glioblastoma. Oncotarget. 2018;9(89):36017-28.
31. Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5(214):ra20.
32. Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGASSTING- mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci Transl Med. 2020;12(549).
33. Tripathi R, Modur V, Senovilla L, Kroemer G, Komurov K. Suppression of tumor antigen presentation during aneuploid tumor evolution contributes to immune evasion. Oncoimmunology. 2019;8(11):1657374.
34. Gouveia-Fernandes S. Monocytes and Macrophages in Cancer: Unsuspected Roles. Adv Exp Med Biol. 2020;1219:161-85.
35. Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20-7.
36. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679- 92.
37. Wei J, Chen P, Gupta P, Ott M, Zamler D, Kassab C, et al. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol. 2020;22(2):180-94.
38. Appolloni I, Alessandrini F, Ceresa D, Marubbi D, Gambini E, Reverberi D, et al. Progression from low- to high-grade in a glioblastoma model reveals the pivotal role of immunoediting. Cancer Lett. 2019;442:213-21.
39. Maire CL, Mohme M, Bockmayr M, Fita KD, Riecken K, Bornigen D, et al. Glioma escape signature and clonal development under immune pressure. J Clin Invest. 2020.
40. Chiocca EA, Yu JS, Lukas RV, Solomon IH, Ligon KL, Nakashima H, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci Transl Med. 2019;11(505).
41. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722-9.
42. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375(26):2561-9.
43. de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei BL, O’Brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immunesuppressive macrophages. Neuro Oncol. 2020;22(4):539- 49.
44. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020.
45. Ogbomo H, Cinatl J, Jr., Mody CH, Forsyth PA. Immunotherapy in gliomas: limitations and potential of natural killer (NK) cell therapy. Trends Mol Med. 2011;17(8):433-41.
46. Ding AS, Routkevitch D, Jackson C, Lim M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front Immunol. 2019;10:1715.