Abstract
Ca2+/ calmodulin (CaM) signaling is important for a wide range of cellular functions. It is not surprised the role of this signaling has been recognized in tumor progressions, such as proliferation, invasion, and migration. However, its role in leukemia has not been well appreciated. The multifunctional Ca2+/CaM-dependent protein kinases (CaMKs) are critical intermediates of this signaling and play key roles in cancer development. The most investigated CaMKs in leukemia, especially myeloid leukemia, are CaMKI, CaMKII, and CaMKIV. The function and mechanism of these kinases in leukemia development are summarized in this study.
Keywords
CaMKII, CaMKI, CaMKIV, Leukemia, ITIM containing receptor, Signaling pathway, Therapeutic target
Introduction
Calcium (Ca2+) is an intracellular universal second messenger that regulates a variety of cellular processes. Many biological processes, including gene transcription, cell cycle, migration, and apoptosis, are affected by changes in intracellular Ca2+ signaling [1,2]. Disruption of normal Ca2+ signaling can cause tumorigenic phenotypes [3].
Ca2+ signaling works by forming a complex with calmodulin (CaM), a 148-amino-acid protein that transduces signals in response to intracellular Ca2+ elevation. Ca2+ binding significantly alters CaM’s conformation and enhances its affinity for a wide range of CaM-binding proteins. Since the discovery of CaM in 1970 as a Ca2+ regulator, there have been over 80 Ca2+/CaM-regulated protein kinases described [4]. However, based on their substrate specificity, not all CaM-regulated kinases are Ca2+/CaMdependent protein kinases (CaMKs). For example, the once called CaMKIII is now termed eukaryotic elongation factor 2 (eEF2) kinase, due to containing a small number of substrates [5].
Structure and Activation
The classic CaMKs include CaMKI, CaMKII, and CaMKIV, each of which has multiple isoforms. They are multifunctional serine/threonine protein kinases that regulate the development and activity of different kinds of cell types through a variety of substrates [6-8]. The structure of CaMKs is critical for their activation and regulation (Figure 1). Similarly, they all have an N-terminal kinase domain, followed by a regulatory domain (consisting of Ca2+/CaM binding domain (CBD) and auto-inhibitory domain (AID)). The ADP/ATP binding site locates between the small and large lobes of CaMKs’ kinase domains. CaMKII, on the other hand, has a special self-association domain at the C-terminus that allows it to form holoenzymes [8-10].
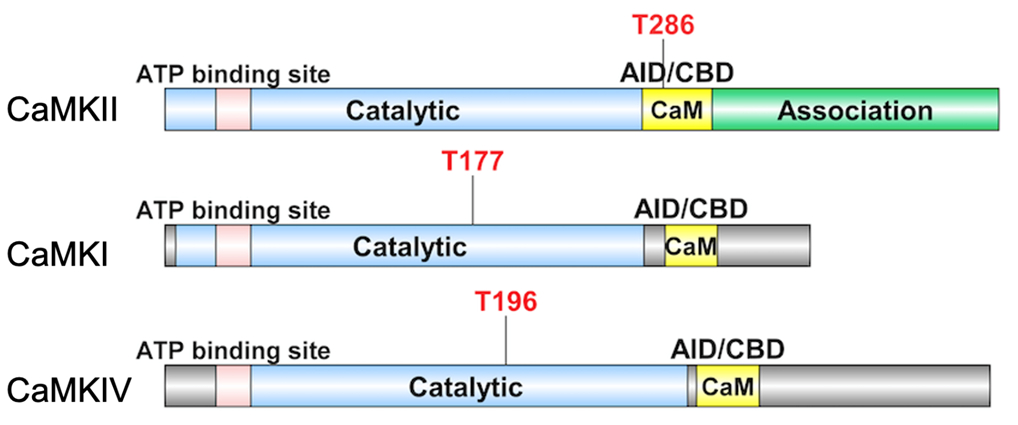
CaMKs have a special phosphorylation-dependent mechanism for the regulation of kinase activity.
The changes in intracellular Ca2+ concentration trigger Ca2+ binding to the ubiquitously expressed CaM induces a conformational transition, which sparks its binding to CBD of CaMKs. The adjacent AID will then be released, triggering CaMK activation. The difference is, for CaMKII, the Thr-286 residue in the regulatory domain is auto- phosphorylated before kinase activation. While CaMKI/ CaMKIV is phosphorylated by an upstream kinase, CaMKK at Thr-177 and Thr-196 residue, respectively, both located in the kinase domain. Despite the fact that CAMKK activates both CaMKI and CaMKIV, their distinct subcellular distributions after phosphorylation, cytosolic for CaMKI versus nuclear localization for CaMKIV, can allow them to play different roles in distinct cellular settings [9,11].
CaMKs in Cancer Development
Effective cell migration, which is critical for cancer metastasis, requires proper Ca2+ signaling control. Many CaMKs including CaMKI, CaMKII, and CaMKIV play a role in cell-migration-related cytoskeleton dynamics. Thus, the role of CaMKs in tumor cell invasiveness and metastatic potential is well implicated [3,12].
Iwatsubo’s group found that store-operated Ca2+ entry regulates melanoma proliferation and cell migration by activating CaMKII. Further, they demonstrated that CaMKII inhibition suppressed MAPK signaling pathway, which can inhibit human melanoma cell migration and metastasis in the lungs [3,13,14]. Interestingly, the CaMKII/ MAPK signaling axis was also linked to colon cancer, inhibiting CaMKII decreased cancer cell proliferation, migration, and invasion [15]. The CaMKK pathway has been shown to promote cerebellar granule precursor migration and differentiation during normal cerebellar development via CaMKIV [16], while CaMKK/CaMKI cascade regulates basal medulloblastoma cell migration via Rac1. In addition, pharmacological CaMKK inhibition blocks both estrogen-induced Rac1 activation and medulloblastoma migration [17]. These findings indicate that the differential regulation of CaMKs in normal and malignant scenarios is context-dependent. McDonnell’s group found that CaMMKβ is highly expressed in the prostate and is further elevated in prostate cancers. Using cellular models of prostate cancer, they demonstrated that CaMKK/AMPK regulates androgen-dependent migration of prostate cancer cells [18]. All these studies indicate CaMKs may be involved in different signaling pathways to manipulate cancer development.
By using a variety of CaM antagonists or CaMKs specific inhibitors, the researchers have corroborated the roles of CaMKs in a multitude of tumor types, which can prevent cell growth, invasiveness, and /or metastasis [19-24]. For detail reviews on the role of CaMKs in cell migration and cancer metastasis, see [3,25-27].
Relevance to Leukemia
In comparison to a large number of studies on CaMKs in neurology and solid tumors, research on CaMKs’ function in controlling hematopoiesis and leukemia has been scarce. Leukemia diseases are not classified as metastatic cancer. Because they are thought to already be widespread when they are diagnosed. Below, we discuss the role of CaMKs in leukemia development based on work of ours and others. Emphasis is given to CaMK’s function in myeloid leukemia, as well as the signaling pathway they are involved.
Using the TCGA database of AML patients, we conducted an in silico study of the relationship between gene expression and overall survival in AML patients. (https:// tcga-data.nci.nih.gov/tcga/). The expression of most CAMKs analyzed showed a negative correlation between expression and patient survival, which including CAMKI, CAMK2A, CAMK2D, CAMK4, CAMKK2 [28,29]. These results suggest that many CaMKs directly support human leukemia cell growth. Here, we summarize the leukemiarelated roles of individual CaMKs.
CaMKII
CaMKII is the most widely studied CaMKs, which consists of four homologous (CaMKIIa, CaMKIIb, CaMKIIg, and CaMKIId) [1]. CaMKII’s autophosphorylation is one of its most important functional characteristics which means that its activation is self-contained and less influenced by Ca2+ concentration or calmodulin regulation. CaMKII makes up about 1% to 2% of total brain protein, and numerous studies have shown that it plays an important role in controlling neuronal cell growth and function [26].
Although aberrant activation of CAMKII has been linked to different hematopoietic malignancies [30,31], most studies focus on one of its isoforms, CaMKIIg, and its role in myeloid leukemia. CaMKIIg is preferentially expressed in myeloid cells [32,33]. Furthermore, the activation of CaMKIIg is greatly increased in leukemia stem/progenitor cells but not in normal hematopoietic cells [34-36]. By phosphorylating and inhibiting the transcriptional function of retinoic acid receptors (RARs), CaMKIIg prevents the differentiation of myeloid leukemia cells. Coordinately, CaMKII inhibitors improve the differentiation of myeloid leukemia cells [32]. This finding was further extended to non-retinoic acid responsive myeloid leukemia cells, which demonstrates that CaMKIIg plays a critical and central role in controlling the proliferation of a broad range of myeloid leukemia cells [33]. The mechanistic studies showed that CaMKII activation induced by different stress contributes to mitogenic signaling and promotes the proliferation of leukemia cells [14,37]. Strikingly, Huang’s group proved that CaMKIIg is a key regulator of leukemia stem cells in chronic myeloid leukemia (CML) [35]. The findings in mouse CML leukemia model and human CML patients indicate that CaMKIIg could be a critical regulator in the progression of CML blast crisis, and they reveal a novel mechanism by which CaMKIIg promotes leukemia stem cell (LSC) self-renewal by inhibiting nuclear p27Kip1 and reawakening dormant LSCs [34,38]. The signaling studies in both AML and CML revealed that CaMKIIg acts as an important regulator of multiple cancer-related signaling pathways including NF-κB, Wnt/b-catenin, p42/p44/ MAPK, and Stat3/5 networks [32,35]. The supportive role of CaMKIIg in the development and progression of myeloid leukemia was verified by the treatment of specific inhibitors or by depletion of CaMKIIg [14,32,33,35-37]. In summary, inhibition of CaMKIIg activity may be beneficial in the treatment of myeloid leukemia.
CaMKI
Similar to CaMKII, CaMKI is multifunctional kinases that have been linked to neuronal plasticity and gene regulation [9]. In human, the CaMKI family consists of four members, each of which is encoded by a different gene: CAMK1, PNCK, CAMK1G and CAMK1D, which generate CaMKIa, CaMKIb, CaMKIg, and CaMKId, respectively [39].
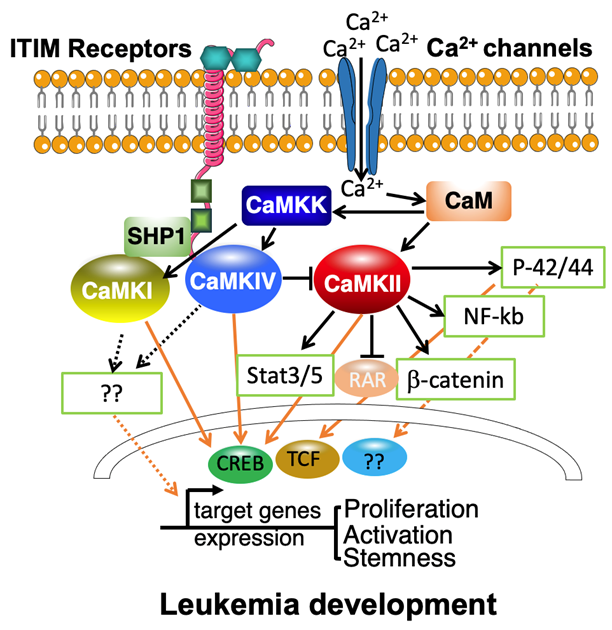
It is reported that the isoforms of CaMKI are ubiquitously expressed at low levels [40], and expressed at high levels in several brain regions [41,42]. Interestingly, we found that CaMKI protein is well detected in both normal hematopoietic cells and AML cells. An in silico analysis of human CAMK1 mRNA expression in 43 human AML samples showed that it is highly expressed in M5 AML cells [28,43]. Moreover, CaMKI is greatly expressed in AML LSCs in mouse MLL-AF9 model. Gain-offunction and loss-of-function analyses of CaMKI in AML cells in vivo proved that CaMKI is essential for the growth of human and mouse AML cells [28,29]. The mechanistic studies indicated CaMKI participates in the Inhibitory leukocyte immunoglobulin-like (ITIM) receptors signaling axis in leukemia development by the recruitment of SH2 domain-containing phosphatase1 (SHP-1). The activated CaMKI can be transported into the nucleus [44] and activate the downstream transcription factor cyclic AMP element-binding protein (CREB) in AML cells [29]. CREB is one of the well-known targets of CaMKs in hematopoietic cells, which can be phosphorylated and activated by CaMKs [45,46]. This novel signaling pathway identified in AML stem cells may represent a target for AML treatment [47].
CaMKIV
CaMKIV is encoded by the CAMK4 gene. Alternative processing yields two distinct isoforms (CaMKIVa and CaMKIVb) [48]. The CaMKIV expression pattern is similar to other CaMKs, with primarily being expressed in the brain, however, CaMKIV is also present in hematopoietic cells, testes and ovaries [49-52]. CaMKIV has been implicated in the regulation of homeostatic plasticity, neurite outgrowth, fear memory, immune and inflammatory responses [1,53].
CaMKIV have been shown to play a significant role in hematopoietic physiology and pathology in a few studies. CaMKIV/CREB/BCL-2 signaling is required for hematopoietic stem cell (HSC) activity. CaMKIV is correlated with increased apoptosis and proliferation of HSCs in vivo and in vitro [50]. We also found that, in human cord blood CD34+ cells, Angptl binding to ITIM receptors can trigger phosphorylation of CaMKIV [54]. CaMKIV is also a key player in the imbalance between Treg and Th17 cells. CaMKIV contributes to the reduction of IL-2 and the restriction of Treg cells in patients with SLE [55]. Since IL-2 has been shown to inhibit Th17 differentiation, CaMK4 inhibition possibly promotes Th17 differentiation indirectly [56,57]. Although HSCs in CAMKIV-knockout mice showed only mild defects [50], The loss of function of LILRB2/PirB or CaMKIV, on the other hand, is detrimental to AML growth. Importantly, inhibiting CaMKIV kinase activity or deleting CaMKIV significantly reduced AML stem cell activity. We discovered that phosphorylation of CREB is important downstream of CaMKIV signaling in AML cells using a rescue assay. Our findings showed that CaMKIV signaling promotes AML cell self-renewal and inhibits apoptosis [28]. As a result, inhibiting CaMKIV is likely to be successful in treating leukemia with limited hematopoietic system side effects.
Concluding Remarks and Perspectives
Targeted therapy is an effective cancer treatment technique that has seen a lot of success in clinical trials [58]. Currently, many of the clinically used targeted cancer drugs are tyrosine kinase inhibitors. However, even though these medications have extraordinary effectiveness at first, drug resistance emerges later, limiting their utility [59]. Finding new drug targets and developing new targeted anticancer agents have accordingly become urgent for drug discovery and development. Because the different structure between tyrosine kinases and serine-threonine kinases, drugs that target CaMKs are less likely to modulate conventional kinases and, in theory, do not have or trigger cross-resistance with traditional kinase drugs. The CaMK family members, particularly CaMKII, CaMKI, CaMKIV, are appealing anti-cancer targets because they are overexpressed in myeloid leukemia as compared to normal blood cells and play a critical role in leukemia cell proliferation, differentiation and self-renewal.
In contrast to studies in other forms of tumors, there has been very little research into the role of CaMKs in leukemia. Our review reveals that most of these limited studies focus on myeloid leukemia diseases, which may be due to the relative high expression of CAMKs in myeloid cell lineage. Because inhibition of the expression and/or activity of CaMKs directly blocks leukemia cell growth and activity, and suppresses tumorigenesis and leukemia development, but does not much disturb normal development, CaMKs may represent ideal targets for treating leukemia. Since there are many different CaMKs, which can express differently in diverse leukemia types or subtypes. It is fascinated to see how these CaMKs interact individually and in combination in cancer cells. For example, in leukemia cell lines (U937), inhibiting CaMKII activity causes an upregulation of CaMKIV mRNA and protein. CaMKIV expression, on the other hand, inhibits CaMKII autophosphorylation and activation, as well as G0/G1 cell cycle arrest, which inhibits cell proliferation [38]. This data indicates that CaMKII suppresses CaMKIV expression to promote leukemia cell proliferation.
Blocking CaMKs signaling in conjunction with standard therapies may be an effective method for destroying leukemia cells. In light of the tremendous importance of CaMKs signaling in leukemia development, it will be critically important to identify more components involved in this signaling (Figure 2). Besides ITIM containing receptors, whether there are any other upstream signaling involved? Our studies have identified CREB as the major downstream signaling that are activated by CaMKI/IV. If there is another transcriptional factor that is crucial for leukemia development and controlled by CaMKI and/or CaMKIV need to be further investigated. For CaMKIIg, how consequential phosphorylation of several target substrates dynamically cooperates to modulate leukemia cell proliferation or activity. These future directions may help further to clarify the role of CaMKs in leukemia development.
Acknowledgements
We would like to thank the NIH (R37CA241603) for generous support. We regret that we have been unable to cite many relevant primary references due to space limitations.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
2. Junho CVC, Caio-Silva W, Trentin-Sonoda M, Carneiro-Ramos MS. An Overview of the Role of Calcium/ Calmodulin-Dependent Protein Kinase in Cardiorenal Syndrome. Front Physiol. 2020;11:735.
3. Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7(1):3-17.
4. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912-34.
5. Zhang B, Zou J, Zhang Q, Wang Z, Wang N, He S, et al. Progress in the Development of Eukaryotic Elongation Factor 2 Kinase (eEF2K) Natural Product and Synthetic Small Molecule Inhibitors for Cancer Chemotherapy. Int J Mol Sci. 2021;22(5).
6. Colbran RJ, Schworer CM, Hashimoto Y, Fong YL, Rich DP, Smith MK, et al. Calcium/calmodulin-dependent protein kinase II. Biochem J. 1989;258(2):313-25.
7. Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8(21):2527-39.
8. Fujisawa H. Regulation of the activities of multifunctional Ca2+/calmodulin-dependent protein kinases. J Biochem. 2001;129(2):193-9.
9. Wayman GA, Tokumitsu H, Davare MA, Soderling TR. Analysis of CaM-kinase signaling in cells. Cell Calcium. 2011;50(1):1-8.
10. Swulius MT, Waxham MN. Ca(2+)/calmodulindependent protein kinases. Cellular and Molecular Life Sciences : CMLS. 2008;65(17):2637-57.
11. Bayer KU, Schulman H. CaM Kinase: Still Inspiring at 40. Neuron. 2019;103(3):380-94.
12. Brzozowski JS, Skelding KA. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals (Basel). 2019;12(1).
13. Umemura M, Baljinnyam E, Feske S, De Lorenzo MS, Xie LH, Feng X, et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS One. 2014;9(2):e89292.
14. Heise N, Palme D, Misovic M, Koka S, Rudner J, Lang F, et al. Non-selective cation channel-mediated Ca2+- entry and activation of Ca2+/calmodulin-dependent kinase II contribute to G2/M cell cycle arrest and survival of irradiated leukemia cells. Cell Physiol Biochem. 2010;26(4-5):597-608.
15. Chen W, An P, Quan XJ, Zhang J, Zhou ZY, Zou LP, et al. Ca(2+)/calmodulin-dependent protein kinase II regulates colon cancer proliferation and migration via ERK1/2 and p38 pathways. World J Gastroenterol. 2017;23(33):6111-8.
16. Neal AP, Molina-Campos E, Marrero-Rosado B, Bradford AB, Fox SM, Kovalova N, et al. CaMKKCaMKI signaling pathways differentially control axon and dendrite elongation in cortical neurons. J Neurosci. 2010;30(8):2807-9.
17. Davare MA, Saneyoshi T, Soderling TR. Calmodulinkinases regulate basal and estrogen stimulated medulloblastoma migration via Rac1. J Neurooncol. 2011;104(1):65-82.
18. Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, et al. CaM kinase kinase betamediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011;71(2):528-37.
19. Gocher AM, Azabdaftari G, Euscher LM, Dai S, Karacosta LG, Franke TF, et al. Akt activation by Ca(2+)/ calmodulin-dependent protein kinase kinase 2 (CaMKK2) in ovarian cancer cells. J Biol Chem. 2017;292(34):14188-204.
20. Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO journal. 2011;30(13):2719-33.
21. Takai N, Ueda T, Nasu K, Yamashita S, Toyofuku M, Narahara H. Targeting calcium/calmodulin-dependence kinase I and II as a potential anti-proliferation remedy for endometrial carcinomas. Cancer Letters. 2009;277(2):235- 43.
22. Gardner HP, Ha SI, Reynolds C, Chodosh LA. The caM kinase, Pnck, is spatially and temporally regulated during murine mammary gland development and may identify an epithelial cell subtype involved in breast cancer. Cancer Res. 2000;60(19):5571-7.
23. Wu S, Lv Z, Wang Y, Sun L, Jiang Z, Xu C, et al. Increased expression of pregnancy up-regulated nonubiquitous calmodulin kinase is associated with poor prognosis in clear cell renal cell carcinoma. PLoS One. 2013;8(4):e59936.
24. Daft PG, Yuan K, Warram JM, Klein MJ, Siegal GP, Zayzafoon M. Alpha-CaMKII plays a critical role in determining the aggressive behavior of human osteosarcoma. Mol Cancer Res. 2013;11(4):349-59.
25. Tsai FC, Kuo GH, Chang SW, Tsai PJ. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res Int. 2015;2015:409245.
26. Wang YY, Zhao R, Zhe H. The emerging role of CaMKII in cancer. Oncotarget. 2015;6(14):11725-34.
27. Villalobo A, Berchtold MW. The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis. Int J Mol Sci. 2020;21(3).
28. Kang X, Cui C, Wang C, Wu G, Chen H, Lu Z, et al. CAMKs support development of acute myeloid leukemia. J Hematol Oncol. 2018;11(1):30.
29. Kang X, Lu Z, Cui C, Deng M, Fan Y, Dong B, et al. The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nature Cell Biology. 2015;17(5):665-77.
30. Genot EM, Meier KE, Licciardi KA, Ahn NG, Uittenbogaart CH, Wietzerbin J, et al. Phosphorylation of CD20 in cells from a hairy cell leukemia cell line. Evidence for involvement of calcium/calmodulin-dependent protein kinase II. Journal of immunology. 1993;151(1):71-82.
31. Gu Y, Zhang J, Ma X, Kim BW, Wang H, Li J, et al. Stabilization of the c-Myc Protein by CAMKIIγ Promotes T Cell Lymphoma. Cancer cell. 2017;32(1):115-28.e7.
32. Si J, Mueller L, Collins SJ. CaMKII regulates retinoic acid receptor transcriptional activity and the differentiation of myeloid leukemia cells. J Clin Invest. 2007;117(5):1412- 21.
33. Si J, Collins SJ. Activated Ca2+/calmodulindependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res. 2008;68(10):3733-42.
34. Gu Y, Zheng W, Zhang J, Gan X, Ma X, Meng Z, et al. Aberrant activation of CaMKIIγ accelerates chronic myeloid leukemia blast crisis. Leukemia. 2016;30(6):1282- 9.
35. Gu Y, Chen T, Meng Z, Gan Y, Xu X, Lou G, et al. CaMKII γ, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood. 2012;120(24):4829-39.
36. Gonzalez M, De Brasi C, Ferri C, Bengió R, Bianchini M, Larripa I. CAMKIIγ, HSP70 and HSP90 transcripts are differentially expressed in chronic myeloid leukemia cells from patients with resistant mutated disease. Leuk Lymphoma. 2014;55(9):2101-8.
37. Muthalif MM, Ljuca F, Roaten JB, Pentapaty N, Uddin MR, Malik KU. Ca2+/calmodulin-dependent protein kinase II and cytosolic phospholipase A2 contribute to mitogenic signaling in myeloblastic leukemia U-937 cells. J Pharmacol Exp Ther. 2001;298(1):272-8.
38. Monaco S, Rusciano MR, Maione AS, Soprano M, Gomathinayagam R, Todd LR, et al. A novel crosstalk between calcium/calmodulin kinases II and IV regulates cell proliferation in myeloid leukemia cells. Cellular signalling. 2015;27(2):204-14.
39. Løseth OP, de Lecea L, Calbet M, Danielson PE, Gautvik V, Høvring PI, et al. Developmental regulation of two isoforms of Ca(2+)/calmodulin-dependent protein kinase I beta in rat brain. Brain Res. 2000;869(1-2):137- 45.
40. Picciotto MR, Zoli M, Bertuzzi G, Nairn AC. Immunochemical localization of calcium/calmodulindependent protein kinase I. Synapse. 1995;20(1):75-84.
41. Kamata A, Sakagami H, Tokumitsu H, Owada Y, Fukunaga K, Kondo H. Spatiotemporal expression of four isoforms of Ca2+/calmodulin-dependent protein kinase I in brain and its possible roles in hippocampal dendritic growth. Neurosci Res. 2007;57(1):86-97.
42. Nairn AC, Greengard P. Purification and characterization of Ca2+/calmodulin-dependent protein kinase I from bovine brain. J Biol Chem. 1987;262(15):7273- 81.
43. Lukk M, Kapushesky M, Nikkilä J, Parkinson H, Goncalves A, Huber W, et al. A global map of human gene expression. Nature Biotechnology. 2010;28(4):322-4.
44. Stedman DR, Uboha NV, Stedman TT, Nairn AC, Picciotto MR. Cytoplasmic localization of calcium/ calmodulin-dependent protein kinase I-alpha depends on a nuclear export signal in its regulatory domain. FEBS Letters. 2004;566(1-3):275-80.
45. Cheng JC, Kinjo K, Judelson DR, Chang J, Wu WS, Schmid I, et al. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood. 2008;111(3):1182-92.
46. Esparza SD, Chang J, Shankar DB, Zhang B, Nelson SF, Sakamoto KM. CREB regulates Meis1 expression in normal and malignant hematopoietic cells. Leukemia. 2008;22(3):665-7.
47. Kang X, Kim J, Deng M, John S, Chen H, Wu G, et al. Inhibitory leukocyte immunoglobulin-like receptors: Immune checkpoint proteins and tumor sustaining factors. Cell Cycle. 2016;15(1):25-40.
48. Sakagami H, Kondo H. Cloning and sequencing of a gene encoding the beta polypeptide of Ca2+/calmodulindependent protein kinase IV and its expression confined to the mature cerebellar granule cells. Brain Res Mol Brain Res. 1993;19(3):215-8.
49. Ohmstede CA, Jensen KF, Sahyoun NE. Ca2+/ calmodulin-dependent protein kinase enriched in cerebellar granule cells. Identification of a novel neuronal calmodulin-dependent protein kinase. J Biol Chem. 1989;264(10):5866-75.
50. Kitsos CM, Sankar U, Illario M, Colomer-Font JM, Duncan AW, Ribar TJ, et al. Calmodulin-dependent protein kinase IV regulates hematopoietic stem cell maintenance. J Biol Chem. 2005;280(39):33101-8.
51. Wu JY, Gonzalez-Robayna IJ, Richards JS, Means AR. Female fertility is reduced in mice lacking Ca2+/ calmodulin-dependent protein kinase IV. Endocrinology. 2000;141(12):4777-83.
52. Wu JY, Means AR. Ca(2+)/calmodulin-dependent protein kinase IV is expressed in spermatids and targeted to chromatin and the nuclear matrix. J Biol Chem. 2000;275(11):7994-9.
53. Ferretti AP, Bhargava R, Dahan S, Tsokos MG, Tsokos GC. Calcium/Calmodulin Kinase IV Controls the Function of Both T Cells and Kidney Resident Cells. Front Immunol. 2018;9:2113.
54. Zheng J, Umikawa M, Cui C, Li J, Chen X, Zhang C, et al. Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature. 2012;485(7400):656-60.
55. Juang YT, Wang Y, Solomou EE, Li Y, Mawrin C, Tenbrock K, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest. 2005;115(4):996-1005.
56. Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/ mTOR and CREM-α underlies autoimmunity-associated Th17 imbalance. J Clin Invest. 2014;124(5):2234-45.
57. Otomo K, Koga T, Mizui M, Yoshida N, Kriegel C, Bickerton S, et al. Cutting Edge: Nanogel-Based Delivery of an Inhibitor of CaMK4 to CD4+ T Cells Suppresses Experimental Autoimmune Encephalomyelitis and Lupus-like Disease in Mice. Journal of Immunology. 2015;195(12):5533-7.
58. Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: Treating cancer with specificity. Eur J Pharmacol. 2018;834:188-96.
59. Rosenzweig SA. Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv Cancer Res. 2018;138:71-98.