Neuropathology of Alzheimer’s Disease
Alzheimer’s Disease (AD) is the most common form of dementia [1]. Patients diagnosed with AD experience disordered cognition and memory, as well as changes in behaviour and personality [2]. The vast majority of AD is diagnosed in patients aged over 65 years and classified as late onset (LOAD), with the remaining ~1% of cases termed early onset AD (EOAD) [3]. The molecular neuropathology of AD includes the presence of extracellular amyloid beta (Ab) plaques and proteinaceous lesions of aggregated neuronal tau protein across multiple brain regions [4,5]. Implicated regions include brainstem structures as the origin of progressive neurodegeneration [6,7], which then follows a predictable spreading pattern to the cholinergic basal forebrain [8], the (trans)entorhinal cortex, hippocampus, and the neocortex [9]. Sadly, there are currently no specific preventative measures or cures for AD.
Genetics of Alzheimer’s Disease
The heritability of LOAD and EOAD is 60-80% and 90-100% respectively [10], and over 70 genomic loci have been associated with AD to-date [11]. These data support the presence of a strong genetic component in the development of AD (Figure 1). The rarest but most severe AD phenotypes are due to various autosomal dominant gene mutations in APP, PSEN1, and PSEN2; these lead to EOAD [12]. The most important genetic risk factor for developing LOAD is having the allelic variant e4 of APOE (APOE4), whereby homozygous (e4/e4) individuals have a 50-60% lifetime risk of developing LOAD [13].
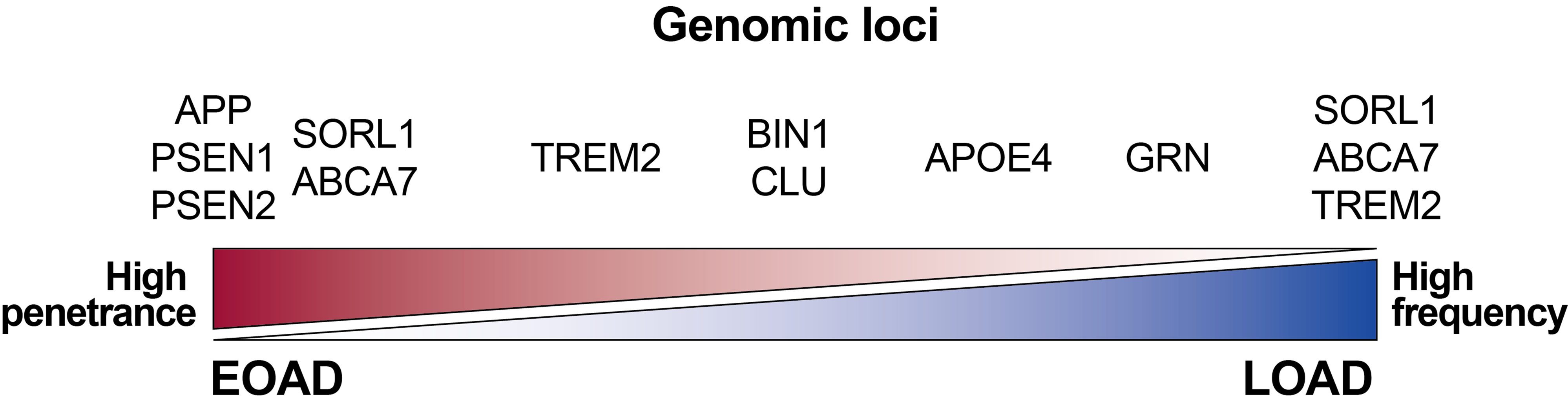
Figure 1. Genes associated with increased risk for developing AD. Gene penetrance and frequency affect AD age at onset. APP & PSEN mutations are 100% penetrant but very rare. Microglial TREM2 mutations can be more common with mixed phenotypic effects. The APOE4 allelic variant is relatively common within the population and significantly increases AD risk. Various other GWAS hits are rare and have small effects. Some genes have multiple loci that are differentially associated with AD, e.g., TREM2, SORL1, ABCA7. Made with reference to three GWAS datasets [11,14,15].
Links between ApoE4 and Amyloid-beta
ApoE was first associated with AD following its discovery in amyloid plaques [16]. Binding of ApoE4 to cell surface receptors induces microglial and astrocytic dysregulation, affecting cholesterol homeostasis and impeding Aβ clearance [17]. In human neurons, ApoE4 stimulates ERK1/2 MAP-kinase signalling through MAP3K12 and MAP2K7 [18]. MAP2K7 then phosphorylates cFos to induce APP expression and subsequent Aβ production and neurodegeneration, but also phosphorylates CREB, paradoxically stimulating synapse formation [18]. Introducing a conditional APP Swedish point mutation in human neurons, Zhou et al., [19] report increased total functional synapses upon expression; an effect that was abolished by inhibiting APP cleaving enzyme BACE1. Restoring Aβ to physiologic levels in neurons not expressing APP was synaptogenic and increased synaptic transmission [19]. These findings support a cautious approach in AD therapeutic strategies involving clearance of Aβ peptides and plaques.
Links between ApoE4 and Tau Protein
In the noradrenergic (NA) neurons of the locus coeruleus (LC), the presence of ApoE4 increases tau neurotoxicity by selectively binding vesicular monoamine transporter 2, inhibiting vesicular uptake of neurotransmitter NA, leading to NA oxidation to DOPEGAL [20]. This toxic metabolite activates enzymatic cleavage of tau at site N368, triggering LC neurodegeneration, reducing hippocampal volume, and inducing cognitive dysfunction [20]. In a transgenic mouse model of tauopathy (P301S), human APOE4 knock-in (KI) mice (P301S/E4) show significantly exacerbated tau-mediated brain-wide neurodegeneration relative to E2/E3 KI and APOE knock-out (EKO) mice [21].
Wang et al. [22], showed that depletion of astrocytic ApoE4 rescues loss of synapses, reduces tau-mediated neurodegeneration and levels of hyperphosphorylated tau (pTau) in P301S mouse brain. Consistent with these findings, a binding partner of ApoE4, the astrocyte-secreted protein glypican-4 (Gpc4), drives tau hyperphosphorylation and propagation in vitro and in vivo [23]. Glpc4 enhances ApoE4-dependent trafficking of surface ApoE receptor LRP1 and tau propagation by internalising LRP1-ApoE4-Gpc4-pTau complexes [23].
The Re-evaluated Amyloid Cascade Hypothesis
The proteolytic cleavage of APP by β-/g-secretases leads to the generation of 40- and 42-amino acid-long Aβ peptides (Aβ40, Aβ42), of which Aβ42 is less common but more toxic than Aβ40 [24]. According to the amyloid cascade hypothesis the cause of AD is excessive levels of extracellular Aβ42 [25]. Factors contributing to this include missense mutations in APP and PSEN1/2 genes which increase production of Aβ42, and/or APOE4 inheritance and neuroinflammation leading to failures of Aβ42 clearance [24]. These Aβ42-driven effects purportedly culminate in dysregulated kinase-phosphatase signalling, inducing tau hyperphosphorylation, propagation, and deposition of aggregated tau, leading to neuronal cell death [26,27].
Biomarkers for Diagnosing, Treating, and Preventing Alzheimer’s Disease
Protein changes in AD include metabolism/processing of Aβ and pTau, and accumulation of plaques and tangles respectively. These changes can be targeted and used as biomarkers, detected by combining imaging techniques (magnetic resonance imaging, MRI; positron emission tomography, PET) and fluid (blood; cerebrospinal fluid, CSF) analysis techniques. For instance, using a radioligand with affinity for neurofibrillary tangle tau and the spatial topography information conferred by PET, Therriault et al. [28], were able to apply Braak staging to living humans. Late tau-PET-based Braak stages (IV-VI) correlated with higher Aβ42/Aβ40 ratios in CSF. Elevated concentrations of CSF pTau181, pTau217, pTau231, pTau235, and plasma pTau231 were detectable from PET-based Braak stage II compared to stage 0 [28].
Blood-based biomarkers, including soluble Aβ, total/phosphorylated tau, and inflammatory factors, have attracted research interest, as blood collection and biomarker detection are relatively non-invasive and economical. Despite this gain in popularity, blood-based testing has limitations. Minute biomarker concentrations with indistinguishable contributions from peripheral and central nervous systems, coupled to an inherently complex matrix of proteins in human blood can interfere with reliable protein identification and quantification.
Regarding treatment of AD, as of 26/12/2022 (index date for this editorial) there are 235 interventional clinical trials for AD, whereby the vast majority (197; 83.8%) involve drug therapy (Table 1).
|
Interventions |
Study n |
% Studies |
Participants |
% Participants |
|---|---|---|---|---|
|
Drug (small molecule) |
197 |
83.83 |
69030 |
80.21 |
|
Behavioral (cognitive/behavioural training, biofeedback, social aid, memory aid) |
5 |
2.13 |
8025 |
9.33 |
|
Biological (antibodies, gene therapy, bacterial, vaccine) |
12 |
5.11 |
5550 |
6.45 |
|
Other (diet, brain apps, oxygen therapy) |
5 |
2.13 |
1621 |
1.88 |
|
Device (TMS, photobiomodulation) |
7 |
2.98 |
828 |
0.96 |
|
Combination product (cognitive behavioural therapy, PET scans, sleep hygiene) |
5 |
2.13 |
817 |
0.95 |
|
Dietary Supplements |
2 |
0.85 |
96 |
0.11 |
|
Radiation (low-dose whole brain irradiation) |
1 |
0.43 |
60 |
0.07 |
|
Diagnostic test (PET/MRI) |
1 |
0.43 |
30 |
0.03 |
|
235 |
100 |
86057 |
100 |
|
|
This table was generated using data extracted from the governmental website clinicaltrials.gov and includes all active and/or recruiting clinical trials in Phases 1-4, globally. |
||||
While most participants are enrolled in drug-based clinical trials, a sizeable minority (~20%) are undergoing behavioral training, using biological interventions, or various devices or diets. This diversity of approaches offers some hope and promise to those suffering from AD-related symptoms globally.
Alzheimer’s Disease: Causal Factors
As post-mortem brain tissue analyses reveal, the presence of accumulated plaques is not sufficient to predict LOAD. Conversely, while neural tissue patterning of tau spreading and aggregation strongly correlates with AD neurodegeneration, tau aggregation may manifest at a late and irreversible stage in the disease process.
A consistent presentation among patients diagnosed with LOAD is the rate of disease progression and age-at-onset of disease. That is, a pattern of neurodegeneration emerges in the population after a (somewhat) predictable period of environmental exposure (i.e., living to beyond the age of 70 years). Within a shared environment, some people never develop AD and die beyond the age of 90 from other causes; others carry one or two copies of APOE4 and die of LOAD, while others are heterozygous for alleles with highly penetrant, disease-associated mutations and develop EOAD. Assuming there are no overt yet-unknown environmental causes of AD, the in-between cases – as per the described extremes – must depend on genome sequence identity coupled to gene expression: a variable resistance to the persistent environmental onslaught that is life.
So which events might signify impending neurodegeneration? One’s attention might focus on two potential causes of AD, which are not mutually exclusive: (i) Specific molecular cell surface contacts of Aβ plaques (rather than mere existence of plaques); (ii) Specific molecular interactions of toxic Aβ42 oligomers in equilibrium with Aβ plaques. The interactions in (i) and (ii) include pathologic stabilisation of integral membrane proteins and dysregulated activation of various intracellular signalling cascades. Consequent effects include excitotoxicity, and dysregulated proteostasis and post-translational modifications, which lead to death of cells containing hyperphosphorylated and aggregated tau protein.
References
2. Lyketsos CG, Carrillo MC, Ryan JM, Khachaturian AS, Trzepacz P, Amatniek J, et al. Neuropsychiatric symptoms in Alzheimer's disease. Alzheimer's & Dementia. 2011 Sep 1;7(5):532-9.
3. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Molecular Neurodegeneration. 2019 Dec;14(1):32.
4. Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nature reviews Molecular Cell Biology. 2007 Feb;8(2):101-12.
5. Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harbor Perspectives in Medicine. 2012 Jul 1;2(7):a006338.
6. Jacobs HI, Becker JA, Kwong K, Engels-Domínguez N, Prokopiou PC, Papp KV, et al. In vivo and neuropathology data support locus coeruleus integrity as indicator of Alzheimer’s disease pathology and cognitive decline. Science Translational Medicine. 2021 Sep 22;13(612):eabj2511.
7. Ji X, Wang H, Zhu M, He Y, Zhang H, Chen X, et al. Brainstem atrophy in the early stage of Alzheimer's disease: a voxel-based morphometry study. Brain Imaging and Behavior. 2021 Feb;15(1):49-59.
8. Ferreira-Vieira TH, M Guimaraes I, R Silva F, M Ribeiro F. Alzheimer's disease: targeting the cholinergic system. Current Neuropharmacology. 2016 Jan 1;14(1):101-15.
9. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathologica. 2006 Oct;112(4):389-404.
10. Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Archives of General Psychiatry. 2006 Feb 1;63(2):168-74.
11. Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nature Genetics. 2022 Apr;54(4):412-36.
12. Cuyvers E, Sleegers K. Genetic variations underlying Alzheimer's disease: evidence from genome-wide association studies and beyond. The Lancet Neurology. 2016 Jul 1;15(8):857-68.
13. Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Molecular Psychiatry. 2011 Sep;16(9):903-7.
14. Mol MO, van der Lee SJ, Hulsman M, Pijnenburg YA, Scheltens P, Seelaar H, et al. Mapping the genetic landscape of early-onset Alzheimer's disease in a cohort of 36 families. Alzheimer's Research & Therapy. 2022 Dec;14(1):77.
15. Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nature Genetics. 2021 Sep;53(9):1276-82.
16. Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Research. 1991 Feb 8;541(1):163-6.
17. Kriebs A. APOE4 dysregulates cholesterol homeostasis in human microglia and astrocytes. Nature Aging. 2022 Jul;2(7):565-6.
18. Huang YW, Zhou B, Nabet AM, Wernig M, Südhof TC. Differential signaling mediated by ApoE2, ApoE3, and ApoE4 in human neurons parallels Alzheimer's disease risk. Journal of Neuroscience. 2019 Sep 11;39(37):7408-27.
19. Zhou B, Lu JG, Siddu A, Wernig M, Südhof TC. Synaptogenic effect of APP-Swedish mutation in familial Alzheimer’s disease. Science Translational Medicine. 2022 Oct 19;14(667):eabn9380.
20. Kang SS, Ahn EH, Liu X, Bryson M, Miller GW, Weinshenker D, et al. ApoE4 inhibition of VMAT2 in the locus coeruleus exacerbates Tau pathology in Alzheimer's disease. Acta Neuropathologica. 2021 Jul;142(1):139-58.
21. Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017 Sep;549(7673):523-7.
22. Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021 May 19;109(10):1657-74.
23. Saroja SR, Gorbachev K, TCW J, Goate AM, Pereira AC. Astrocyte-secreted glypican-4 drives APOE4-dependent tau hyperphosphorylation. Proceedings of the National Academy of Sciences. 2022 Aug 23;119(34):e2108870119.
24. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Molecular Medicine. 2016 Jun;8(6):595-608.
25. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992 Apr 10;256(5054):184-5.
26. Ittner LM, Götz J. Amyloid-β and tau—a toxic pas de deux in Alzheimer’s disease. Nature Reviews Neuroscience. 2011 Feb;12(2):67-72.
27. Scheltens P, Blennow K, Breteler MM, De Strooper B, Frisoni GB, Salloway S, et al. Alzheimer's disease. Lancet (London, England). 2016 Feb 24;388(10043):505-17.
28. Therriault J, Pascoal TA, Lussier FZ, Tissot C, Chamoun M, Bezgin G, et al. Biomarker modeling of Alzheimer's disease using PET-based Braak staging. Nature Aging. 2022 Apr 25;2:526-535.