Abstract
Background: In the past three decades, there has been significant improvements in the diagnosis and management of acute myeloid leukaemia. Cytogenetic and molecular genetic modalities have now enabled physicians to identify and plan appropriate management protocols for patients according to genetic profile.
Case presentation: Here we present case of a 2-year-old boy who initially presented with features that were clinically and haematologically suggestive of juvenile myelomonocytic leukaemia (JMML), but cytogenetic analysis revealed presence of inv(16)/t(16;16) (p13q22) (CBFB/MYH11), which is the recurrent cytogenetic abnormality associated with AML.
Conclusion: To our knowledge this was the first case of acute myeloid leukaemia with inv(16)/t(16;16) (p13q22) diagnosed in a two year old baby. In our patient, cytogenetic analysis helped with correct diagnosis. Cytogenetic and molecular genetic studies now play an important role in diagnosis, as well as management of clonal disorders, including acute leukaemias.
Keywords
AML, inv(16), 2-Year-Old, Diagnostic, Dilemma
Abbreviations
AML: Acute Myeloid Leukaemia; JMML: Juvenile Myelomonocytic Leukaemia; FAB: French American and British; FMIC: French Medical Institute for Mothers and Children; WHO: World Health Organization; AML: Acute Myeloid Leukaemia
Background
Acute myeloid leukaemia (AML) is mainly the disease of adults with median age at diagnosis of 68 years [1]. The incidence of acute myeloid leukaemia is low amongst children with lowest incidence in children between 10- 14 years old [2]. There are incidence of AML increases progressively with age and reaching the peak of incidence at ages 80 years and above [2].
There has been remarkable evolution in the diagnosis as well as management of AML [3,4]. Early-on, in the French-American and British (FAB) classification, the diagnosis was strictly based on clinical and morphological features [5]. This was a blunt approach towards the management of AML, because cytogenetic and molecular markers of prognostic and therapeutic significance were then unheard of [6].
With progressive research in the cytogenetic as well as molecular genetic studies, the profile of management in AML patients has significantly changed, with more patients now achieving complete remission and prolonged survival [7,8]. It would include not only the identification of prognostic and therapeutic targets but also, in some cases, enabling agility in the diagnosis of AML [8].
Considering the FAB classification, it was compulsory to have a blast count equal to or more than 30 percent before labelling the patient as AML [5]. Since, the 2008’s World Health Organization Classification of tumors of haematopoetic and lymphoid tissues, with introduction of the sub-class of AML with recurrent cytogenetic and molecular abnormalities, it became possible to diagnose AML without the consideration of blast count in peripheral blood or bone marrow [9,10].
Patients are now diagnosed and therefore receive therapy at a much early stage of the disease, thus enabling more patients to achieve the therapeutic targets [11]. The established significance that cytogenetics and molecular genetics have achieved in the field of medical diagnostics, cannot be overlooked [12]. Here we present a case of AML in a 2-Year-Old baby boy, who was initially diagnosed as juvenile myelomonocytic leukaemia (JMML), but cytogenetic analysis helped in accurate diagnosis.
Case Presentation
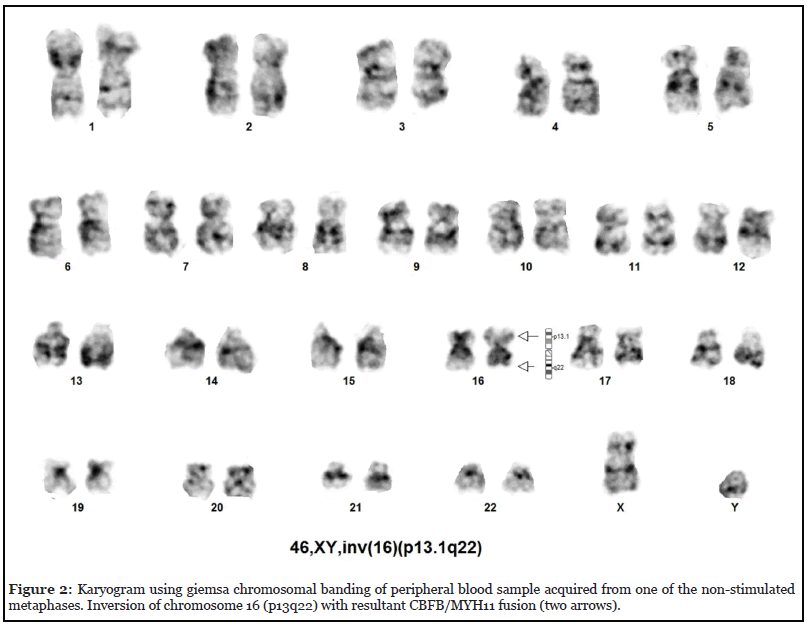
2-Year-Old boy with normal birth and developmental history, presented to pediatrician for rapidly progressive swelling in the neck, abdomen and eye. The patient initially was noted to have lymphadenopathy in the cervical region with monocytosis on blood counts. Accordingly, the patient was prescribed antibiotics and was asked to re-visit after one week. During the visit after a week lymphadenopathy was noted to increase on the second visit, with new lymphadenopathies noted in the axillary region and swelling around the eyes. On general examination the patient was in distress due to pain with bilateral cervical, axillary lymphadenopathy, upper and lower limb ecchymosis and bilateral ocular swelling, more prominent in the right side, with patient unable to close the eyes. Abdominal examination revealed moderate hepatomegaly and huge splenomegaly. The initial CBC revealed anaemia (haemoglobin of 8.6grams/ deciliter) and monocytosis (absolute monocyte count of 18,000/microliter). Peripheral blood film revealed presence of abnormal monocytes that constituted 23% of nucleated cells, having moderate cell sizes, irregular nuclear margins, nuclear membrane clumping, absence of nucleoli and moderate amount of cytoplasm, as shown in Figure 1a. There was presence of 4% blast cells, having moderate cell sizes, high nucleo-cytoplasmic ratio, open nuclear chromatin and variable number of inconspicuous cytoplasms, as shown in Figure 1b. The patient did not consent for bone marrow examination and therefore considering the age and blood analysis, was managed as juvenile myelomonocytic leukaemia (JMML). After counselling the parents and getting informed consent, cytogenetic analysis was performed on peripheral blood where the karyogram of all 20 metaphases revealed 46, XY with inversion of chromosome 16 (p13.1q22), consistent with acute myeloid leukaemia with inv (16), as shown in Figure 2. Accordingly, the patient was referred to paediatric oncologist for further management.

Discussion
The understanding of AML has shown significant evolution over the past many decades due to the rapid progression in diagnostic and therapeutic modalities. FAB classification being the first to define and characterize different types of haematological malignancies, required presence of equal to or more than 30% blast cells in either of peripheral blood or bone marrow before defining the disease as AML [5]. Initially, it was the morphological and cytochemical staining features that would identify different sub classes of acute myeloid leukaemia.
In early 2000s, the advent of flowcytometric ananlysis, lead to evaluation of minimal residual disease post chemotherapy, that in turn lead to identification of patients requiring special intervention and therefore improved patient outcome [13]. This still was not sufficient and other means of prognostic value were required. With identification and characterization of new cytogenetic and molecular genetic abnormalities associated with AML, new avenues were opened for research and development, that resulted in identification of disease entities that would show novel natural history [14,8]. This led to the identification of disease entities that would show good response to therapeutic regimens and ones that would require special intervention to allow for better patient outcome [15].
Therefore, in 2008, World Health Organization (WHO) Classification of tumors of haematopoetic and lymphoid tissues introduced the sub-class of AML with recurrent cytogenetic and molecular abnormalities [10,9]. Amongst the entities included in the sub-class, were AML with t(8;21) (q22;q22) RUNX1/RUNX1T1, AML inv(16)/t(16;16) (p13q22) (CBFB/MYH11) and AML t(15;17) (q24;q21) PML/RARA [9,10]. It was concluded that harboring the above-mentioned entities would identify the disease to be AML regardless of blast percentage [14]. The reason behind this decision was two folds; first being the fact that patients harboring the mentioned cytogenetic abnormalities would indefinitely end up with AML and secondly, institution of therapy to such patients resulted with better and earlier response [16,14].
In the past decade, with discovery of major molecular markers and progress in the field of molecular genetics, including next generation sequencing and discovery of new molecular therapeutic targets, most of patients who would be deemed incurable in the past, can achieve complete remission with remarkably good long-term survival [17]. Not only this but even healthy individuals with risk of development of acute leukaemia can be identified well early in time to enable proper and timely intervention [18].
Unfortunately, in underdeveloped countries, including Afghanistan, the profile of management for all malignant disorders, including AML, is in a very bad situation due to lack of trained human resource, financial constraints and established diagnostic and interventional institutions [19]. In Afghanistan, diagnosis of AML is solely based on morphological features with lack of even basic cytochemical staining techniques in the whole country. In most instances the diagnostic impression is laid by medical lab technologists. Cytogenetic and molecular genetic evaluation is absolutely lacking in most of the countries recognized hospitals and healthcare facilities. In our patient the initial impression laid down by the paediatric team was juvenile myelomonocytic leukaemia considering the persistence of monocytosis in the peripheral blood and presence of very occasional blast cells. It was the karyotypic evaluation that led us towards the correct diagnosis of AML and thus timely intervention.
In Afghanistan, there is a serious deficiency of diagnostic modalities and trained medical personnel. For better management of patients with malignant disorders, including acute leukaemias development of modern resources including molecular-cytogenetics is indispensable.
Conclusion
To our knowledge this is the first case of AML with inv(16) (p13q22) (CBFB/MYH11) in Afghanistan, which on its own has never been reported worldwide. Our patient’s prompt and correct diagnosis, that could have been delayed, was led by karyotype examination. It is to be noted that the role of cytogenetic and molecular genetic analysis is very significant and when available, then must be instituted.
Acknowledgement
We would like to acknowledge the role played by Agha Khan Development Network (AKDN), not only in establishing the country’s first cytogenetic laboratory but also facilitating and financing cytogenetic testing of patients who cannot afford the expenses.
Authors’ Contributions
HAM and AMH conceived the idea. JAG, AMH, AJR and SRN helped with the final draft of the manuscript. AMH, HAM, RS, AJR, ASZ and SHR diagnosed the case. SS performed cytogenetic studies. AMH and JAG did the literature review. ALK, MH and HAM acquired the microscopic images of the peripheral blood film. AMH, EE, ASI, RS, NL and HAM were the major contributors for critically revising the manuscript for important intellectual content. HAM, JAG and AMH have given expert opinion and final approval of the version to be published. All authors read and approved the final manuscript.
Availability of Data and Materials
All the generated data are included in this article.
Consent for Publication
Written informed consent was obtained from patient’s legal guardian for publication of this case report and all the accompanying figures. A copy of the written consent shall be availed to the Editor-in-Chief of this journal.
Competing Interests
The Authors declare that they have no competing interests.
References
2. Dong Y, Shi O, Zeng Q, Lu X, Wang W, Li Y, et al. Leukemia incidence trends at the global, regional, and national level between 1990 and 2017. Experimental Hematology & Oncology. 2020 Dec;9(1):1-1.
3. Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, et al. Acute myeloid leukaemia. Nature Reviews Disease Primers. 2016 Mar 10;2(1):1-22.
4. Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. The Lancet. 2018 Aug 18;392(10147):593-606.
5. Walter RB, Othus M, Burnett AK, Löwenberg B, Kantarjian HM, Ossenkoppele GJ, et al. Significance of FAB subclassification of “acute myeloid leukemia, NOS” in the 2008 WHO classification: analysis of 5848 newly diagnosed patients. Blood, The Journal of the American Society of Hematology. 2013 Mar 28;121(13):2424-31.
6. Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood, The Journal of the American Society of Hematology. 2012 Oct 18;120(16):3187-205.
7. Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood, The Journal of the American Society of Hematology. 1998 Oct 1;92(7):2322-33.
8. Grimwade D, Mrózek K. Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematology/Oncology Clinics. 2011 Dec 1;25(6):1135-61.
9. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Swerdlow SH, editor. Lyon, France: International Agency for Research on Cancer; 2008 Sep 20.
10. Swerdlow, SH, Campo, E, Harris, NL, et al. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues, revised 4th edition. IARC, Lyon, 2017. 2017. 30 p.
11. Ferrara F, Palmieri S, Leoni F. Clinically useful prognostic factors in acute myeloid leukemia. Critical Reviews in Oncology/Hematology. 2008 Jun 1;66(3):181-93.
12. Braoudaki M, Tzortzatou-Stathopoulou F. Clinical cytogenetics in pediatric acute leukemia: an update. Clinical Lymphoma Myeloma and Leukemia. 2012 Aug 1;12(4):230-7.
13. Reinhardt D, Langebrake C, Creutzig U, Vormoor J, Brune C, Thorwesten M, et al. Minimal residual disease in acute myeloid leukemia in children--standardization and evaluation of immunophenotyping in the AML-BFM-98 study. Klinische Padiatrie. 2002 Jul 1;214(4):179-87.
14. Foucar K, Anastasi J. Acute myeloid leukemia with recurrent cytogenetic abnormalities. American Journal of Clinical Pathology. 2015 Jul 1;144(1):6-18.
15. Mrózek K, Bloomfield CD. Chromosome aberrations, gene mutations and expression changes, and prognosis in adult acute myeloid leukemia. ASH Education Program Book. 2006;2006(1):169-77.
16. Wang HY, Rashidi HH. The new clinicopathologic and molecular findings in myeloid neoplasms with inv (3) (q21q26)/t (3; 3)(q21; q26. 2). Archives of Pathology & Laboratory Medicine. 2016 Dec;140(12):1404-10.
17. Kayser S, Levis MJ. Advances in targeted therapy for acute myeloid leukaemia. British Journal of Haematology. 2018 Feb;180(4):484-500.
18. Abelson S, Collord G, Ng SW, Weissbrod O, Cohen NM, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018 Jul;559(7714):400-4.
19. Yeoh AE, Tan D, Li CK, Hori H, Tse E, Pui CH. Management of adult and paediatric acute lymphoblastic leukaemia in Asia: resource-stratified guidelines from the Asian Oncology Summit 2013. The lancet Oncology. 2013 Nov 1;14(12):e508-23.