Perspective
N4-acetylcytidine (N4A) is an organic compound and a metabolite of transferrable ribonucleic acid. Its molecular formula is C11H15N3O6. Earlier studies suggest that N4A was mainly found on tRNA and 18S rRNA, while recent studies have shown that there is also a large amount of N4A on mRNA, whose abundance is not even lower than the m7G cap modification carried by mRNA. The generation of N4A is catalyzed by N-acetyltransferase 10 (NAT10) or its homologous enzyme. N4A is produced by acetylation in eukaryotic RNA and is the only human enzyme with both acetyltransferase and RNA binding activity [1]. The full transcriptome mapping of N4A show abundant discrete acetylation regions in the coding sequence. The ablation of NAT10 reduced the detection of N4A at mRNA localization sites and was globally correlated with the down-regulation of target mRNA. N4A is widely distributed in the human transcriptome, and most sites occur in the coding sequence. Compared with unmodified cytosine, N4A increases the thermal stability of watsoncracker base pair guanosine, thus affecting the interaction with homologous tRNAs during translation. After the release of N4A from tRNA metabolism, it participates in the body’s immune response [2].
N4A was once considered a biomarker for diabetes and colorectal cancer patients who showed significant signs of oxidative stress. In a variety of oxidative stress processes, transfer RNA (tRNA) may be degraded, and N4A can be released from tRNA metabolism, which normally cannot be reused and further degraded, but excreted in urine [3]. Furman et al. found that the level of endogenous nucleoside metabolite N4A from tRNA degradation was significantly increased in the elderly with high expression level of inflammasome gene. In the process of human aging, the body’s ability to regulate and transfer energy gradually decreases, the body’s metabolism slows down, oxidative stress increases, and metabolites accumulate in plasma. However, the role of these metabolites in the human body is not fully understood [4].
Chronic mild inflammation is related to many aging diseases. Inflammasome can drive chronic inflammation and trigger the maturation of IL-1β under the condition of infectious diseases or cell oxidative stress. N4A can activate the microglia of the central nervous system and induce the activation of NLRC4 inflammasome [5]. After treatment of human monocytes with adenosine, the expression of nlrc4 gene was increased and mature IL-1β was released. Because N4A induces cytokine secretion in the presence of ATP, and N4A increases the expression of NLRC4 gene and protein, it seems that N4A can increase the expression of NLRC4 in monocytes, and adenine can then trigger the activation of nlrc4 inflammatory bodies and the maturation of cytokines [6].
There is a high level of oxidative stress in the central nervous system of patients with neurodegenerative diseases such as Alzheimer’s disease, and N4A may be released from tRNA metabolism in their bodies. In addition, some studies have shown that N-acetyltransferase 10 can prolong the life of aging mice model, while N-acetyltransferase 10 can inhibit the production of N4A [7]. The level of N4A, an endogenous nucleoside metabolite from tRNA degradation, increased significantly in the elderly with high expression activity of inflammatory bodies. N4A induced and activated NLRP3 inflammasome to induce IL-1β [8]. N4A can activate NLRP3 inflammasome by inducing the expression of NLRP3. It can also promote the assembly and subsequent activation of NLRP3 inflammasome by inducing the expression of HMGB1 gene. It is found that N4-acetylcytidine can activate the immune function of microglia and maintain the activation of NLRP3 inflammatory body by inducing the expression of HMGB1 protein [9]; the released HMGB1 can cause N4A to activate NF-κB and induce the expression of NLRP3 gene; the silencing of HMGB1 gene can eliminate the activation of NF-κB gene stimulated by N4A and inhibit the gene table of NLRP3 and HMGB1 There is a high level of HMGB1 in the blood of AD patients, suggesting that HMGB1 may be involved in the process of neuroinflammation caused by the activation of NLRP3 inflammatory bodies (Figure 1).
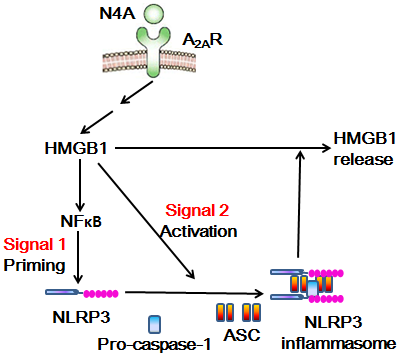
High mobility group protein (HMG) is named for its high migration ability in polyacrylamide gel electrophoresis. HMG can be further divided into three families: HMGA, HMGB and HMGN. HMGB family has HMGB1, HMGB2 and HMGB3 and HMGB1 is the most abundant HMG protein. HMGB1 gene includes 5 exons and 4 introns [11]. Human HMGB1 gene is located on chromosome 13q12. HMGB1 gene has a powerful TATA box promoter and contains a silence element. Under general environmental conditions, the expression of HMGB1 is maintained at the basic level [12]. There is evidence that the proinflammatory effect of HMGB1 is mainly through the late glycation end product receptor (RAGE). NF-κB, mitogen activated protein kinase, protein kinase B, plasminogen activated inhibitor and Cdc42 protein are activated by RAGE [14].
As HMGB1 has the above structure and function, we hypothesized that N4A can maintain the activation of NLRP3 inflammatory body by inducing the expression and release of HMGB1. The following questions need to be answered: Can N4A induce the sustained expression and release of HMGB1 by activating the immune function of microglia? Does N4A mediated NF-κB signaling and NLRP3 expression require HMGB1 protein expression? Does N4A mediated HMGB1 expression require activation of NLRP3 inflammatory bodies? And so on. The results showed that N4A promoted the production and activation of NLRP3 inflammasome through HMGB1 in microglia, and silencing the expression of NLRP3 eliminated the up regulation of N4A mediated expression and release of HMGB1, indicating that the release of HMGB1 mediated by NLRP3 inflammatory corpuscles is necessary for the sustained expression of HMGB1 in N4A [9]. Reducing the activation of microglia induced by β-amyloid protein (Aβ) is considered to be an effective treatment for AD. Falcao et al. [15] used mouse microglial cell line and solution containing Aβ aggregate mixture to study whether dipeptide vinyl sulfone (DVS) can attenuate Aβ mediated inflammatory response. The results showed that DVS could inhibit the expression of HMGB1, NLRP3 and IL-1 β induced by Aβ.
HMGB1 may play an important role in the pathogenesis of various neurodegenerative diseases, especially AD. Therefore, it is very important to identify the inducer of HMGB1 expression. N4A and its similar chemicals may need to be the focus of monitoring in the future [16]. Through the study of HMGB1 signal transduction and endogenous nucleotide metabolites activating inflammatory bodies in microglia, some new biological basis for the prevention and treatment of AD and other diseases in the future will be provided.
Acknowledgments
I would like to thank professor QF Zhang (Guizhou Medical University in China) for providing the honest help with the schematic diagram.
References
2. Ito S, Horikawa S, Suzuki T, Kawauchi H, Tanaka Y, Suzuki T, et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). Journal of Biological Chemistry. 2014 Dec 26;289(52):35724-30.
3. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’agostino D, Planavsky N, Lupfer C, Kanneganti TD, Kang S. The ketone metabolite ß-hydroxybutyrate blocks NLRP3 inflammasome– mediated inflammatory disease. Nature Medicine. 2015 Mar;21(3):263.
4. Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nature Medicine. 2017 Feb;23(2):174.
5. Bai H, Yang B, Yu W, Xiao Y, Yu D, Zhang Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Experimental Cell Research. 2018 Jan 1;362(1):180-7.
6. Zhang Y, Chen Y, Zhang Y, Li PL, Li X. Contribution of cathepsin B-dependent Nlrp3 inflammasome activation to nicotine-induced endothelial barrier dysfunction. European Journal of Pharmacology. 2019 Dec 15;865:172795.
7. Cekic C, Linden J. Purinergic regulation of the immune system. Nature Reviews Immunology. 2016 Mar;16(3):177.
8. Balmus G, Larrieu D, Barros AC, Collins C, Abrudan M, Demir M, et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nature Communications. 2018 Apr 27;9(1):1700.
9. Duan J, Zhang Q, Hu X, Lu D, Yu W, Bai H. N4-acetylcytidine is required for sustained NLRP3 inflammasome activation via HMGB1 pathway in microglia. Cellular Signalling. 2019 Jun 1;58:44-52.
10. Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014 May 2;344(6183):527- 32.
11. Fonken LK, Frank MG, Kitt MM, D’Angelo HM, Norden DM, Weber MD, et al. The alarmin HMGB1 mediates age-induced neuroinflammatory priming. Journal of Neuroscience. 2016 Jul 27;36(30):7946-56.
12. Jiang S, Zhang Y, Zheng JH, Li X, Yao YL, Wu YL, et al. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacological Research. 2017 Mar 1;117:82-93.
13. An H, Cho MH, Kim DH, Chung S, Yoon SY. Orexin impairs the phagocytosis and degradation of amyloid-ß fibrils by microglial cells. Journal of Alzheimer’s Disease. 2017 Jan 1;58(1):253-61.
14. Arosio B, Casati M, Gussago C, Ferri E, Abbate C, Scortichini V, et al. Adenosine type A 2A receptor in peripheral cell from patients with Alzheimer’s disease, vascular dementia, and idiopathic Normal pressure hydrocephalus: a new/old potential target. Journal of Alzheimer’s Disease. 2016 Jan 1;54(2):417-25.
15. Falcão AS, Carvalho LA, Lido´nio G, Vaz AR, Lucas SD, Moreira R, et al. Dipeptidyl vinyl sulfone as a novel chemical tool to inhibit HMGB1/NLRP3-inflammasome and inflamma-miRs in Aß-mediated microglial inflammation. ACS Chemical Neuroscience. 2017 Jan 18;8(1):89-99.
16. Bhatta A, Atianand M, Jiang Z, Crabtree J, Blin J, Fitzgerald KA. A Mitochondrial Micropeptide Is Required for Activation of the Nlrp3 Inflammasome. The Journal of Immunology. 2020 Jan 15;204(2):428-37.