Abstract
In Thrombotic thrombocytopenic purpura ADAMTS-13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity is deficient and microangiopathy occurs after a second trigger. In Shiga toxin producing Escherichia coli (STEC-HUS) stimulated release of Weibel-Palade bodies (WBP’s) is presumed to be the first hit in damaging Gb3 positive endothelial cells in the diarrhea phase. The objective role of the release of WBP’s components after admission of the patients is not adequately evaluated. Different mechanisms involved in the pathogenesis are discussed and approaches for treatment proposed. It includes complement system, active coagulation, cytokines/chemokines, cell-free hemoglobin/heme and finally the release of constituents of WPB`s. After admission all evaluated constituents are increased in serum/plasma. Preferred treatment, inhibiting the damaging effect is testing the use of anti-P-selectin antibodies and complete complement inhibition by Pegcetacoplan in order also to prevent long-term sequelae.
Keywords
Weibel-Palade bodies, STEC-HUS, P-selectin, Pegcetacoplan
Introduction
The Weibel-Palade bodies (WPB’s) form a unique secretory carrier for Von Willebrand factor (VWF), and other cargo produced by endothelial cells. The main components, although not complete, are presented in (Table 1). These proteins are ready without de novo synthesis. Related to their presumed role in the clinical phase of hemolytic uremic syndrome secondary to Shiga toxin-producing Escherichia coli (STEC-HUS) selected information is provided. STEC-HUS is characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury. Shiga toxin injures endothelial cells during the first few days of infection.
|
VWF P-selectin CD63 Endothelin Angiopoetin-2 |
Results
Selected information about components of Weibel-Palade bodies
P-selectin is rapidly exposed on the surface of platelets and on endothelial surface after stimulation. Re-expression of functional characterized P-selectin molecules occurs after repeated stimulation by thrombin. It is recycled [1]. P-selectin binds to leucocyte P-selectin glycoprotein ligand (PSGL1) leading to firm adhesion and transmigration of leucocytes through the vessel wall. It also induces expression of tissue factor on monocytes. CD63 is an essential cofactor for P-selectin function.
Endothelin: ET-1, the major endothelial isoform, is secreted on the abluminal surface of endothelial cells. Plasma concentrations of ET-1 do not accurately reflect ET-1 production.
Angiopoetin-2 (Ang-2): Ang-2 results in destabilization of endothelium, rending it responsive to stimulation by inflammatory cytokines such as TNF, interleukin-1, and angiogenic cytokines (VEGF).
VWF: From the large number of WPB’s undergoing exocytosis upon stimulated, released VWF multimers assemble into VWF strings on the apical side of the endothelium. They have increased platelet adhesive capacity compared with the low molecular VWF. Unfolded VWF multimers can be cleaved by ADAMTS-13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). VWF can recruit leucocytes via direct leucocyte binding or by recruiting platelets, which in turn will attract leucocytes.
Weibel-Palade bodies-function in thrombotic thrombocytopenic purpura (TTP) and in diarrhea phase of STEC-HUS
In TTP ADAMTS-13 activity is deficient and systemic aggregation of platelets will occur after a second trigger. In STEC-HUS stimulated release of WPB’s components in the diarrhea phase of the disease can be presumed to be the first hit in the damage of Gb3 positive endothelial cells [2]. The role of the release of WPB’s content in the clinical phase of STEC-HUS needs further evaluation and will be discussed in the next section.
Weibel-Palade bodies- suggested function in the field of other pathogenic factors in the clinical phase of STEC-HUS
Insight into the pathogenesis of STEC-HUS is provided by Tarr et al. [3]. The development of HUS is related to the degree of prothrombotic activation early in infection. Endothelium is a likely target of the absorbed Shiga toxin. A more complete recent view is presented by Michael et al. [4]. Targeting by Stx or Stx associated microvesicles to Gb3-expressing endothelial cells, followed by docking and release of toxin by microvesicles is the key pathogenic mechanism proposed. Other mechanisms of cytotoxicity include increased synthesis of chemokines and adhesion molecules, complement activation, elevated free hemoglobin without referring accurately to the possible role of Weibel-Palade bodies. The different mechanisms will be presented in more detail, as accurate as possible, with a proposal to eliminate or reduce the damaging effect. The damaged effect on glomerular endothelial cells has characteristic pathological features. The endothelial cells are swollen and detached from the basement membrane with debris in the subendothelial space. The different mechanisms presented are the complement system, coagulation, anti-Shiga toxin, cytokines/chemokines, heme followed by the role of Weibel-Palade bodies
Complement system
Systemic complement activation of the alternative pathway of complement is observed in the clinical phase of STEC-HUS. Plasma levels of sC5b-9 and Bb are increased [5,6]. A survey of assumed intracellular events is presented by Zoja et al. [7]. Eculizumab, a humanized monoclonal antibody binds to C5, preventing its cleavage by C5 convertase and the production of both C5a and C5b-90 complex. Inconsistent results in the treatment of STEC-HUS were obtained. In a recent study eculizumab treatment did not appear to be associated with improved renal outcome during the acute phase of the disease but may reduce long-term-kidney sequelae [8]. It is possible that eculizumab may prevent neurological damage [9]. As C3 is cleaved to generate C3b, a small peptide C3a is released. C3a is increased in plasma in the acute phase of the disease. Pegcetacoplan prevents cleavage of C3 blocking formation of all downstream metabolites, an almost complete complement inhibition [10]. C3a is a small anaphylatoxin mediating pro-inflammatory effects by binding to the G protein-coupled complement receptor C3aR. The complement receptor is widely expressed. C3a is also expressed on endothelial and epithelial cells [11]. Stimulation of C3a on endothelial cells induces a rapid mobilization of WPB’s.
Coagulation
In STEC-HUS at admission active coagulation is present (thrombin-antithrombin III complex, prothrombin fragment 1+2 are increased) [12]. Heparin therapy, however, has no benefit [13,14]. Pivotal studies by Weitz et al. [15,16] demonstrated that clot-bound thrombin is protected from inhibition by heparin. This occurs in STEC-HUS at admission. Fibrinopeptide-A was increased in 13 out of 16 children. Only in 3 out of 9 children fibrinopeptide-A normalized after heparin administration [17]. Bivalirudin has the ability to inhibit clot-bound thrombin [18] with a possible role in treatment of STEC-HUS. Due to their small molecular weight direct, oral anticoagulants can be considered, although more evidence is required [19].
Anti-Shiga toxin
The study about the effect of equine anti-Shiga toxin is accomplished [20]. These results will indicate a possible beneficial effect.
Cytokines/chemokines
A survey of the increased serum cytokines/chemokines levels observed after admission in STEC-HUS is reported by Shimuzu [21]. It concerns mainly tumor necrosis factor, Interleukin-1β, IL-6, IL-8. Chemokines (MCP, IL-8) activate leucocytes and promote extravasation [22]. Cytokines generate endothelial cell adhesion molecule expression, slowing blood flow and increase vascular permeability [23]. Blocking of these chemokines and cytokines by antibodies is possible.
Heme
Intravascular hemolysis due to mechanical damage to red blood cells by microthrombi result in cell-free hemoglobin and heme, overwhelming endogenous scavengers haptoglobin and hemopexin. It induces systemic inflammation, vascular dysfunction, thrombophilia and kidney injury as reviewed by Von Avondt et al. [24] and Pinto et al. [25]. Striking is the exposure of P-selectin on endothelial cells [26,27]. Clinical trials are currently underway for the use of hemopexin and haptoglobin.
Weibel-Palade bodies
A large variety of agonists triggering the exocytosis of Weibel-Palade bodies (WPB’s) are well known such as C3a, C5a, heme [2]. A complete exocytosis of all components of the WPB’s is not always occurring (Figure 1). After stimulation and expression of P-selectin on the cell membrane, functional P-selectin is recycled. Resynthesis is required for other components. Intracellular Ang-2 is detectable 6 hrs after complete release and recovered within 16 hrs.
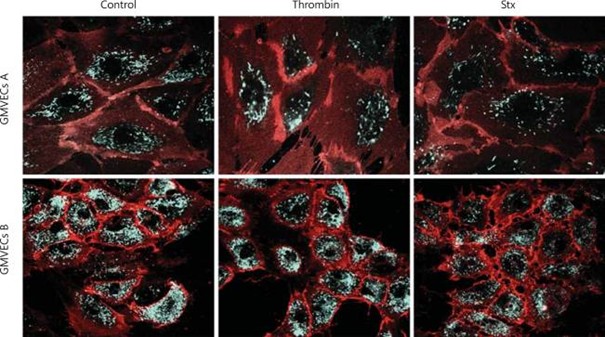
Figure 1. Confocal microscopy of GMVECs incubated with thrombin or Stx1 in flow conditions. GMVEC donors A and B were incubated in flow with thrombin and Stx1 (1 dyne/cm2). The cells were fixed and stained for VWF (green) and CD31 (red). The merge of fluorescent signals is shown. GMVECs B show less effect of thrombin than GMVECs A. A noticeable effect can be detected in both donors after exposure to Stx1. Permission is granted by S. Karger AG, Basel. Nephron Extra 2014.
After admission of STEC-HUS an increase in serum/plasma of all studied components of WPB’s are observed. It concerns Von Willebrand factor (VWF) multimers although with a decreased size [28], P-selectin [29], Ang-2 [30]. Caplacizumab prevents platelet binding of VWF to receptor GPIb-IX-V and the formation of platelet-rich thrombi and is effective in the treatment of TTP. Using new technology ADAMTS-13 activity is only slightly decreased [31]. P-selectin is not only exposed on endothelial cells but also on the surface of activated platelets promoting leucocyte activation. An oral inhibitor famotidine and humanized monoclonal anti-P-selectin antibody (crizanlizumab) are available.
Targeting Ang-2 is an option [32], although it does not directly prove the role of stimulated endothelin secreted on the abluminal surface is expected. ET-1 induces cytoskeletal (actin) remodeling in podocytes and loss of slit-diaphragm proteins such as nephrin. It increases glomerular permeability. Et-1 receptor antagonists inhibit these effects [33].
Our preferred new approaches for treatment are presented in (Table 2).
|
Anti-P-selectin Pegcetacoplan Bivalirudin |
To elucidate the pathogenesis and possible treatment a plethora of models in small animals, imitating STEC-HUS are explored. None, however, reflects STEC-HUS entirely [34]. In addition, the obtained data cannot predict the effect in humans. Gnotobiotic piglets infected orally with enterohemorrhagic E. coli showed thrombotic microangiopathy in the kidneys without, however, hematologic findings [35].
Volume expansion has a mitigating role in STEC-HUS [36]. Other approaches, however, are needed considering the long-term outcome [37]. Fifty-one percent of 281 patients evolved to chronic renal disease (CKD) after 12 years of follow-up and 14% to CKD [2-5].
Conclusion
New approaches for the treatment of STEC-HUS after admission should be evaluated, considering not only the acute mortality but also the long-term prognosis. Targeting WPBs (especially P-selectin) might offer new therapeutic avenue outside the application of pegcetacoplan and bivalirudin.
Conflict of Interest
No conflict of interest.
Funding
No funding for this manuscript.
Acknowledgements
Sebastian Quiroz-Monnens took care of the figure.
References
2. Monnens L. Weibel-Palade bodies: function and role in thrombotic thrombocytopenic purpura and in diarrhea phase of STEC-hemolytic uremic syndrome. Pediatr Nephrol. 2025 Jan;40(1):5-13.
3. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. The Lancet. 2005 Mar 19;365(9464):1073-86.
4. Michael M, Bagga A, Sartain SE, Smith RJH. Haemolytic uraemic syndrome. Lancet. 2022 Nov 12;400(10364):1722-40.
5. Ferraris JR, Ferraris V, Acquier AB, Sorroche PB, Saez MS, Ginaca A, et al. Activation of the alternative pathway of complement during the acute phase of typical haemolytic uraemic syndrome. Clin Exp Immunol. 2015 Jul;181(1):118-25.
6. Ahlenstiel-Grunow T, Hachmeister S, Bange FC, Wehling C, Kirschfink M, Bergmann C, Pape L. Systemic complement activation and complement gene analysis in enterohaemorrhagic Escherichia coli-associated paediatric haemolytic uraemic syndrome. Nephrol Dial Transplant. 2016 Jul;31(7):1114-21.
7. Zoja C, Buelli S, Morigi M. Shiga toxin-associated hemolytic uremic syndrome: pathophysiology of endothelial dysfunction. Pediatr Nephrol. 2010 Nov;25(11):2231-40.
8. Garnier A, Brochard K, Kwon T, Sellier-Leclerc AL, Lahoche A, Launay EA, et al. Efficacy and Safety of Eculizumab in Pediatric Patients Affected by Shiga Toxin-Related Hemolytic and Uremic Syndrome: A Randomized, Placebo-Controlled Trial. J Am Soc Nephrol. 2023 Sep 1;34(9):1561-73.
9. Wildes DM, Harvey S, Costigan CS, Sweeney C, Twomey É, Awan A, et al. Eculizumab in STEC-HUS: a paradigm shift in the management of pediatric patients with neurological involvement. Pediatr Nephrol. 2024 Jan;39(1):315-24.
10. Antonucci L, Thurman JM, Vivarelli M. Complement inhibitors in pediatric kidney diseases: new therapeutic opportunities. Pediatr Nephrol. 2024 May;39(5):1387-404.
11. Vandendriessche S, Cambier S, Proost P, Marques PE. Complement Receptors and Their Role in Leukocyte Recruitment and Phagocytosis. Front Cell Dev Biol. 2021 Feb 11;9:624025.
12. Nevard CH, Jurd KM, Lane DA, Philippou H, Haycock GB, Hunt BJ. Activation of coagulation and fibrinolysis in childhood diarrhoea-associated haemolytic uraemic syndrome. Thrombosis and haemostasis. 1997;78(12):1450-5.
13. Vitacco M, Sanchez Avalos J, Gianantonio CA. Heparin therapy in the hemolytic-uremic syndrome. J Pediatr. 1973 Aug;83(2):271-5.
14. Van Damme-Lombaerts R, Proesmans W, Van Damme B, Eeckels R, Binda ki Muaka P, Mercieca V, et al. Heparin plus dipyridamole in childhood hemolytic-uremic syndrome: a prospective, randomized study. J Pediatr. 1988 Nov;113(5):913-8.
15. Weitz JI, Hudoba M, Massel D, Maraganore J, Hirsh J. Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J Clin Invest. 1990 Aug;86(2):385-91.
16. Weitz JI, Leslie B, Hudoba M. Thrombin binds to soluble fibrin degradation products where it is protected from inhibition by heparin-antithrombin but susceptible to inactivation by antithrombin-independent inhibitors. Circulation. 1998 Feb 17;97(6):544-52.
17. Monnens L, van Aken W, de Jong M. "Active" intravascular coagulation in the epidemic form of hemolytic-uremic syndrome. Clin Nephrol. 1982 Jun;17(6):284-7.
18. Capodanno D, De Caterina R. Bivalirudin for acute coronary syndromes: premises, promises and doubts. Thromb Haemost. 2015 Apr;113(4):698-707.
19. Male C, Thom K, O'Brien SH. Direct oral anticoagulants: What will be their role in children? Thromb Res. 2019 Jan;173:178-85.
20. Hiriart Y, Scibona P, Ferraris A, Belloso W, Beruto V, Bournissen F, et al. A phase I study to evaluate the safety, tolerance and pharmakinetics of anti-Shiga toxin hyperimmune equine F[ab`]2 fragments in healthy volunteers. Br J Clin Pharmacol. 2024 Apr;90(4):1142-51.
21. Shimizu M. Pathogenic functions and diagnostic utility of cytokines/chemokines in EHEC-HUS. Pediatr Int. 2020 Mar;62(3):308-15.
22. Luster AD. Chemokines--chemotactic cytokines that mediate inflammation. N Engl J Med. 1998 Feb 12;338(7):436-45.
23. Holdsworth SR, Gan PY. Cytokines: Names and Numbers You Should Care About. Clin J Am Soc Nephrol. 2015 Dec 7;10(12):2243-54.
24. Van Avondt K, Nur E, Zeerleder S. Mechanisms of haemolysis-induced kidney injury. Nat Rev Nephrol. 2019 Nov;15(11):671-92.
25. Pinto VM, Mazzi F, De Franceschi L. Novel therapeutic approaches in thalassemias, sickle cell disease, and other red cell disorders. Blood. 2024 Aug 22;144(8):853-66.
26. Vallelian F, Buehler PW, Schaer DJ. Hemolysis, free hemoglobin toxicity, and scavenger protein therapeutics. Blood. 2022 Oct 27;140(17):1837-44.
27. Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013 Jul 11;122(2):282-92.
28. Tsai HM, Chandler WL, Sarode R, Hoffman R, Jelacic S, Habeeb RL, et al. von Willebrand factor and von Willebrand factor-cleaving metalloprotease activity in Escherichia coli O157:H7-associated hemolytic uremic syndrome. Pediatr Res. 2001 May;49(5):653-9.
29. Katayama M, Handa M, Araki Y, Ambo H, Kawai Y, Watanabe K, et al. Soluble P-selectin is present in normal circulation and its plasma level is elevated in patients with thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome. Br J Haematol. 1993 Aug;84(4):702-10.
30. Page AV, Tarr PI, Watkins SL, Rajwans N, Petruzziello-Pellegrini TN, Marsden PA, et al. Dysregulation of angiopoietin 1 and 2 in Escherichia coli O157: H7 infection and the hemolytic-uremic syndrome. The Journal of Infectious Diseases. 2013 Sep 15;208(6):929-33.
31. Emirova K, Orlova O, Chichuga E, Muzurov A, Avdonin PP, Avdonin PV. A moderate decrease in ADAMTS-13 actvity correlates with the severity of STEC-HUS. Biomolecules. 2023;13:1671-82.
32. Samuel C. Targeting angiopoetin-2 as a novel treatment option for kidney fibrosis. Kidney Int. 2022;102:683-99.
33. Benigni A, Buelli S, Kohan DE. Endothelin-targeted new treatments for proteinuric and inflammatory glomerular diseases: focus on the added value to anti-renin-angiotensin system inhibition. Pediatr Nephrol. 2021 Apr;36(4):763-75.
34. Mayer CL, Leibowitz CS, Kurosawa S, Stearns-Kurosawa DJ. Shiga toxins and the pathophysiology of hemolytic uremic syndrome in humans and animals. Toxins (Basel). 2012 Nov 8;4(11):1261-87.
35. Gunzer F, Hennig-Pauka I, Waldmann KH, Sandhoff R, Gröne HJ, Kreipe HH, et al. Gnotobiotic piglets develop thrombotic microangiopathy after oral infection with enterohemorrhagic Escherichia coli. Am J Clin Pathol. 2002 Sep;118(3):364-75.
36. Böckenhauer J, Schild R, Kemper MJ, Henne T, Stein MV, Oh J, et al. Volume expansion mitigates Shiga toxin-producing E. coli-hemolytic uremic syndrome in children. Pediatr Nephrol. 2024 Jun;39(6):1901-7.
37. Alconcher LF, Lucarelli LI, Bronfen S, Villarreal F. Kidney sequelae in 281 Shiga toxin-producing Escherichia coli-hemolytic uremic syndrome patients after a median follow-up of 12 years. Pediatr Nephrol. 2024 Apr;39(4):1221-8.