Abstract
Genomic characterization of cancer has transformed understanding of the pathogenesis for all types of cancer in the last two decades, with the study of hematologic malignancies at the forefront of the genomics revolution. With continuous rapid advances, as we envision future possibilities in diagnosis, it is helpful to reflect upon how we reached this point. This review includes a historical perspective, and touches upon significant milestones since the microscope’s invention in 1674 toward the current diagnosis of acute myeloid leukemia (AML), which is an example of an aggressive hematologic malignancy that now requires integration with genomics for diagnosis, prognosis and clinical management. The 2016/2017 World Health Classification (WHO) classification criteria precisely identify AML with recurrent chromosomal abnormalities and the two new, molecularly-defined subtypes, AML with mutated NPM1 (AML-NPM1mut) and biallelic mutated CEBPA (AML-biCEBPAmut). Epidemiologic and hematologic features of the two new molecularly-defined WHO subtypes of AML are highlighted, including for familial AML-NPM1mut and familial AML-biCEBPAmut. Genomic testing, in conjunction with cytomorphologic evaluation, flow cytometric immunophenotypic, and cytogenetic analysis, is now essential for accurate diagnosis of AML. In light of current guidelines for testing, risk stratification, and newer therapies, considerations for integrated genomic testing are discussed toward routine diagnosis by the WHO classification of AML.
Keywords
Diagnostic classification, Acute myeloid leukemia, Familial leukemia, Genomics, Smoking, Epidemiology, Mutations, Cytogenetics, Flow cytometry, World Health Organization
Abbreviations
AML: Acute Myeloid Leukemia; WHO: World Health Organization; AML-NPM1mut: AML with mutated NPM1; AML-biCEBPAmut: AML with biallelic mutated CEBPA; FAB: French-American-British; NCI: National Cancer Institute; WF: Working Formulation; FCI: Flow Cytometric Immunophenotyping; EGIL: European Group for Immunological characterization of Leukemias; FLT3: FMS-like tyrosine kinase 3; ITD: Internal Tandem Duplication; TKD: Tyrosine Kinase Domain; NPM1: Nucleophosmin; CEBPA: CCAAT/Enhancer Binding Protein-alpha; NGS: Next Generation Sequencing; APL: Acute Promyelocytic Leukemia; MPAL: Mixed Phenotype Acute Leukemia; ALAL: Acute Leukemias of Ambiguous Lineage; AML-MRC: AML with Myelodysplasia-related Changes; AML-NOS: AML Not Otherwise Specified; NCCN: National Comprehensive Cancer Network; CAP: College of American Pathologists; ASH: American Society of Hematology
Introduction and Historical Perspective
Significant milestones and seminal discoveries during 1674-1966, by individuals who have made crucial contributions toward progress in the diagnosis of hematologic neoplasms as we understand today are depicted chronologically in Figure 1, with selected references [1-11]. It is notable that the path to progress in the understanding of disease and neoplasms initially took centuries for significant discoveries (17th-18th centuries), and subsequently, many decades (19th-20th centuries) for a breakthrough or a change from the prevailing norm. Further, that progress always required perseverance, dedication, innovation, and collaboration among individuals that were not necessarily recognized by the majority at the time, as exemplified by the respectful collaboration between John Hughes Bennett and Rudolph Virchow, as described by Piller [1].
“Leukemia,” lymphomas, and cancer genetics: first descriptions
As depicted in Figure 1, the first clear description for the thought of “leukemia” as the disease cause appears to have been in 1841 by David Craigie, a physician at Edinburgh Royal Infirmary, who observed a patient with thick blood and an enlarged spleen and who was puzzled by, and questioned, the cause of the clinical features in that patient [1,2]. Three years later, in 1844, Craigie attended to another patient with similar clinical features, and was convinced that the pathological cause was the same in both patients, and not pus and inflammation. John Hughes Bennett, a clinician, and pathologist at Edinburgh Royal Infirmary, who had received prior training with Alfred Donne, a French microscopist, performed an autopsy in Craigie’s second patient and examined the deceased patient’s blood under a microscope [1,2]. Bennett first reported in 1845 that the disease, which would now be recognized as chronic myeloid leukemia, was due to systemic involvement of blood (and not inflammation), along with Craigie’s report of his first patient in the same journal. The second clinicopathologic report of leukemia was by Rudolph Virchow, a demonstrator of anatomy in Berlin, who, at age 24, described the unstained appearances of blood cells in the first report of a patient with chronic lymphatic leukemia. Subsequently, Virchow described the third case, also chronic leukemia with splenic enlargement, and recognized that there were two types of leukemias – splenic and lymphatic. While Bennett preferred the name “leucocythemia,” Virchow coined the name “leukemia” for the disease, importantly, with both in agreement for the microscopic features in blood and the cause of the disease as systemic, instead of inflammation [1,2].

Five decades later, in 1900, the words “myeloblast” and “lymphoblast” were introduced by Naegeli, a Swiss hematologist, for precursor cells in the two types of acute leukemias. That nomenclature followed two crucial milestones: (1) first, in 1868, by Ernst Neumann, a Professor of Pathologic Anatomy at Konigsberg, who had stated that blood originated in the bone marrow, hematopoiesis is a continuous process, and that leukemias originate in the bone marrow, and (2) the seminal work of Paul Ehrlich, who, as a medical student in 1877, had described details of blood cells and developed stains to identify acidophil, basophil, and neutrophil granules in white blood cells [1].
Interestingly, malignant lymph node tumors (lymphomas), which represent the solid counterpart of hematologic lymphoid neoplasms, were first studied by post-mortem examination by Thomas Hodgkin at Guy’s Hospital in London in 1832, a decade before Craigie observed his first patient with chronic leukemia in Edinburgh. Nevertheless, “Hodgkin disease” was first recognized by the medical world in 1865, two decades after Bennett and Virchow described leukemias due to Samuel Wilks, a curator at the same museum where Hodgkin had worked. Three decades later, at the turn of the 19th-20th century, Sternberg and Reed described the characteristic large nucleolated cells by histological examination in Hodgkin disease. Twenty-five years later, during 1925-1927, Brill and Symmers described follicular tumors of germinal centers, and another 30 years later, in 1958, Dennis Burkitt described Burkitt lymphoma, which also arises from the germinal center [3,4].
In parallel with the milestones above in hematology in the second half of the 19th century, the field of the underlying genetics in cancer started with the observation by a German pathologist, Hansemann, who in 1890 first observed mitoses in tissues of malignant tumors. Two decades later, in 1914, Theodor Boveri hypothesized that chromosomal abnormalities caused the transition from benign to malignant. However, it was not until 1955 when Tjio and Levan at the Institute of Genetics in Lund ascertained the correct number of human chromosomes as forty-six [5]. Subsequently, in the late 1950s, the abnormally minute “Philadelphia chromosome” was identified as a causative abnormality in 7 patients with chronic myeloid leukemia in the absence of any observed chromosomal abnormalities in 10 AML patients [2,6].
Significantly, also in the late 1950s and 1960s, although not frequently noted, Baikie in Edinburgh, and Sandberg at Roswell Park Memorial Hospital in Buffalo, had described cytogenetic abnormalities in AML cases and suggested that cytogenetics could classify AML [7-9]. However, cytogenetics was not formally incorporated to classify AML until four decades later, when the 2001 WHO classification of tumors was introduced [12].
The morphologic and subsequent immunologic era for hematologic cancers
Ehrlich’s groundbreaking work in 1877 initiated the morphological era for hematology, which has progressed for almost 1½ centuries. Major progress in the diagnosis and classification of leukemias was achieved by careful examination of peripheral blood and bone marrow aspirate smears, including cytochemical-stained smears, by the FAB Co-operative Group. As shown in Table 1, the FAB group classified acute leukemias during 1976-1985, and subsequently, chronic lymphoid and myeloid leukemias [13-16]. Figure 2 shows the AML subtypes by FAB classification, based on the differentiation of the leukemic cells and the extent of myeloid and monocytic maturation, as determined by microscopic evaluation of Wright- Giemsa-stained and cytochemical-stained smears [13,14].

| Lymphomas | Leukemias |
|---|---|
| 1956 Rappaport classification – based on histologic pattern and cytologic features | 1976 FAB Group classification of acute leukemias, updated 1985
|
| 1966 Lukes & Butler – classified Hodgkin disease, modified as the Rye classification | |
| 1974 Lukes & Collins - based on immunological cell-of-origin and cytology of cells (small and large cleaved and non-cleaved) | |
| 1974 Kiel classification, updated 1988 - based on cytologic features to assign grade | |
| 1982 National Cancer Institute Working Formulation for clinical usage | 1989 FAB classification chronic (mature) lymphoid leukemias |
| 1994 Revised European American-Lymphoma (REAL) classification | 1994 FAB classification chronic myeloid leukemias |
| 1995 EGIL Immunological classification of acute leukemias | |
| 2001 World Health Organization (WHO) classification of tumours of the haematopoietic and lymphoid tissues | |
Abbreviations: FAB, French-American-British Co-operative Group; AML, Acute myeloid leukemia; EGIL, European Group for the Immunological characterization of Leukemias
Table 1: Earlier Diagnostic Classifications for Lymphomas and Leukemias [10-23].
The earlier classifications for lymphomas were proposed by multiple groups, as shown in Table 2 [10-23]. Before 1982, in addition to the three non-Hodgkin lymphoma classifications in Table 1, three additional classifications (Dorfman, WHO, and the British National Lymphoma Investigation) were also in use. The NCI-sponsored Working Formulation was developed in 1982 after evaluating those six classifications to classify lymphomas for clinical use, but the WF became most widely used for diagnostic pathology [21]. In 1994, the REAL classification was introduced by a group of expert hematopathologists who described clinicopathologic entities that could be precisely diagnosed, including with immunohistochemistry or flow cytometric immunophenotyping [22]. FCI was used in clinical laboratories at least since the 1980s for lineage determination (myeloid, B- or T-lymphoid) in acute leukemias. Rare bilineal or biphenotypic leukemias were recognized by cytomorphology or FCI, leading to the immunological classification of acute leukemias by the EGIL in 1995 [23].
| Acute myeloid leukemia with recurrent genetic abnormalities |
| Acute myeloid leukemia with balanced translocations/inversions |
| AML with t(8;21)(q22;q22); RUNX1-RUNX1T1 |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 |
| Acute promyelocytic leukemia with PML-RARA |
| AML with t(9;11)(p21.3;q23.3); KMT2A-MLLT3 |
| AML with t(6;9)(p23;q34.1); DEK-NUP214 |
| AML with inv(3)(q21.3q26.2) or t(3;3)(q21;q26.2); GATA2, MECOM |
| AML (megakaryoblastic) with t(1;22)(p13.3;q13.1); RBM15-MKL1 |
| Provisional entity: AML with BCR-ABL1 |
| Acute myeloid leukemia with gene mutations |
| AML with mutated NPM1 |
| AML with biallelic mutation of CEBPA |
| Provisional entity: AML with mutated RUNX1 |
| Acute myeloid leukemia with myelodysplasia-related changes |
| Therapy-related myeloid neoplasms |
| Acute myeloid leukemia, not otherwise specified |
| AML with minimal differentiation |
| AML without maturation |
| AML with maturation |
| Acute myelomonocytic leukemia |
| Acute monoblastic and monocytic leukemia |
| Acute erythroid leukemia |
| Acute megakaryoblastic leukemia |
| Acute basophilic leukemia |
| Acute panmyelosis with myelofibrosis |
| Myeloid sarcoma |
| Myeloid proliferations related to Down syndrome |
| Transient abnormal myelopoiesis associated with Down syndrome |
| Myeloid leukemia associated with Down syndrome |
Abbreviation: AML, acute myeloid leukemia
Table 2:The 2016/2017 World Health Organization classification of acute myeloid leukemia and related precursor neoplasms (Adapted from Arber et al [41] and [42]).
The incorporation of cytogenetics and molecular genetics for diagnosis in acute myeloid leukemia
As noted earlier, as early as the late 1950s, cytogenetics was suggested to classify AML possibly [7-9]. As previously described, the diagnostic paradigm shifted only in 2001 when, based on the principles of the REAL classification, the WHO classification for hematopoietic and lymphoid tumors was introduced, including cytogenetics incorporated for acute leukemia classification [12,24]. That WHO volume represented the 3rd edition, which had included 10 volumes published by the International Agency for Research on Cancer during 2000-2006. The 4th edition (2006-2018) included 12 volumes and 2 revised editions, which had become necessary due to significant genomic advances in tumors of the central nervous system and hematolymphoid tissues. Before 2001, the WHO Classification of Tumors had included the 1st edition (1967-1981) containing a list of accepted terms with short histologic descriptions, and the 2nd edition (1982-2002), which included histologic and immunohistochemical features and one image for each histologic type [25].
Most importantly, the WHO 2001 classification represented a collaboration of pathologists, oncologists, and geneticists worldwide to standardize criteria for the definition and classification of cancer types and standardized nomenclature to ensure progress in clinical cancer care [12].
In the 1990s, large studies had established the role of cytogenetics in establishing prognosis in AML. The Medical Research Council Trial established 3 cytogenetic risk groups: t(8;21), t(15;17), inv(16) as favorable, -5 or -7, del(5q), abn(3q) or complex karyotype as unfavorable, and normal as intermediate risk, with 5-year survival at 65%, 14% and 41%, respectively [26]. In 1999, a landmark study differentiated acute leukemias as myeloid or lymphoid by applying gene expression microarrays [27]. In 2004, Dutch investigators described 16 classes of AML, including chromosomal abnormalities, CEBPA and FLT3 mutations, EVI1 overexpression, and groups with normal cytogenetics in 285 cases, using gene expression profiling, FAB criteria, cytogenetics and clinical outcome [28].
Since normal cytogenetics comprised 45-50% of all AML, molecular genetic abnormalities were investigated for risk stratification in a normal karyotype. That motivation led to studies that established the prognostic significance of abnormalities in the FLT3 and NPM1 genes. The inactive FLT3 receptor is monomeric and is normally present on bone marrow CD34+ stem cells and immature hematopoietic progenitors (myeloid, monocytic, and B-lymphoid) [29]. The FLT3 receptor dimerizes in its active form, with phosphorylation activating downstream pathways for cell proliferation [29]. Mutations in the FLT3 gene, located on chromosome 13q12, are most well-known to occur as internal tandem duplication in the juxtamembrane domain (FLT3ITD) and as point mutations in the tyrosine kinase domain (FLT3TKD), and these mutations lead to constitutive activation of the FLT3 receptor leading to uncontrolled cell proliferation [29]. In 2001, in a study of 854 AML, the presence or absence of FLT3ITD mutations in the intermediate-risk cytogenetics group led to increased or decreased relapsed disease, respectively [30]. In 2002, another study of 224 AML showed the worst survival if FLT3ITD-positive, intermediate if FLT3TKD-positive, and best with absent FLT3 mutations [31]. In 2005, Falini et al. identified insertion mutations in NPM1 that led to abnormal cytoplasmic localization of the nucleolar protein in AML [32]. Subsequently, the prognostic role of FLT3 and NPM1 mutations in AML was ascertained, with the favorable effect of NPM1 mutations present only if FLT3ITD mutations were absent [33].
In 2001, Pabst et al. described mutations in AML in the CEBPA gene, which encodes for the transcription factor CEBPA, crucial for granulocytic differentiation [34].
Interestingly, germline CEBPA mutations underlying familial AML were identified in 2004 [35], several years after anticipation was observed in one pedigree with the same type of familial AML [36].
After the Human Genome Project was completed in 2003, the role of mutations in AML pathogenesis was intensely investigated, as reviewed earlier [37]. The Cancer Genome Atlas study of 200 de novo AML revealed gene mutations with intricate co-operation patterns and mutual exclusion between and within eight categories of biologically functional genes [38]. Mutations in NPM1 co-occurred with mutations in FLT3 or DNMT3A [38], similar to prior genomic profiling [39], while gene fusions were mutually exclusive of mutations in NPM1, RUNX1, TP53 and CEBPA [38]. Importantly, NPM1 mutations were not detected in age-related clonal hematopoiesis, even with deep sequencing [37]. Subsequently, two types of short-read sequencers enabled analysis of the cancer genome by massively parallel sequencing, termed NGS, in clinical laboratories.
The World Health Organization classification of AML WHO2001-WHO2016/2017
The evolution of the diagnostic criteria for AML was previously described, with a schematic presented in Figure 3 [12-14,24,40-42].

Integrated Genomics for Acute Myeloid Leukemia Diagnosis and Classification
Most significantly, in 2020, we understand AML to be an aggressive clonal hematologic malignancy characterized by marked heterogeneity in genetic and clinical features, with this “single” disease comprised of numerous subtypes, with different prognosis and treatment options. Precise classification of AML, necessary for risk stratification and therapy, is best achieved by the WHO2016/2017 classification [41,42], which requires not only microscopic skills, but also availability and collaboration for multiple modalities of tests, including clinical history, laboratory hematology, FCI, cytogenetics, and molecular genetics.
Table 2 shows the WHO2016/2017 AML classification, including seven subtypes based on chromosomal translocations/inversion, AML-NPM1mut, and AMLbiCEBPAmut based on gene mutations, and two provisional subtypes [41,42].
As reported in 2019, careful application of the WHO diagnostic criteria leads to the precise classification of the genetically-defined AML subtypes [24]. Here, features previously reported in text/tables [24] are highlighted in the following Figures: Figure 4A-4C, Study design, methods, and excluded cases; Figure 4D, Distribution of all cases by WHO2008and WHO2016/2017; Figure 4E, WHO2008- classified cases reclassified by WHO2016/2017; Figure 5A, Median age, numbers of males and females in all AML categories. Additionally, the median age for females with AML-NPM1mut was lower (49 versus 62 years) than in males with AML-NPM1mut [24]; Figure 5B, Hematologic findings in all categories; Figure 5C, Cytogenetics findings, and survival outcome in all categories; Figure 6, Smoking status in all molecularly-confirmed AML-NPM1mut; all three non-smokers with familial AML-NPM1mut had a history of female relatives with leukemia [24]; also, one young AMLNOS patient with features suggestive of AML-NPM1mut and with history of familial leukemia, was a non-smoker [24]; Figure 7, Hematologic features and normal cytogenetics in two AML-biCEBPAmut patients, including a 74-yearold with FAB-M2 subtype with Auer rods and familial leukemia consistent with AML-biCEBPAmut [24,35,43,44].
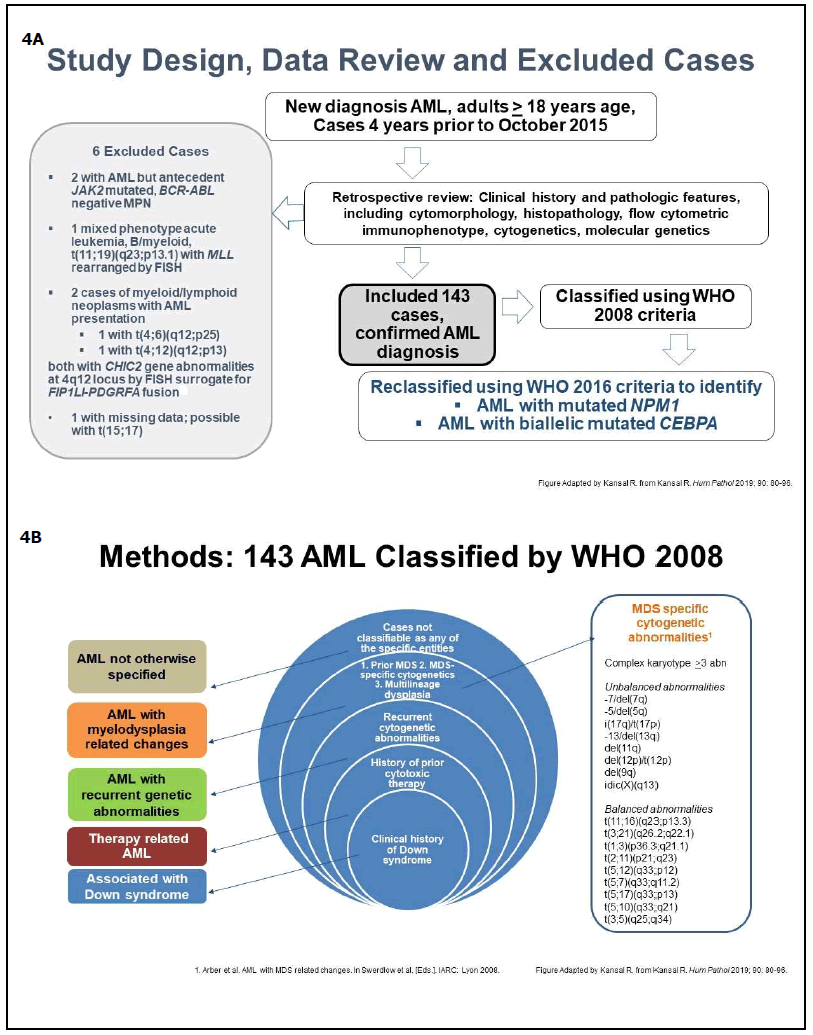
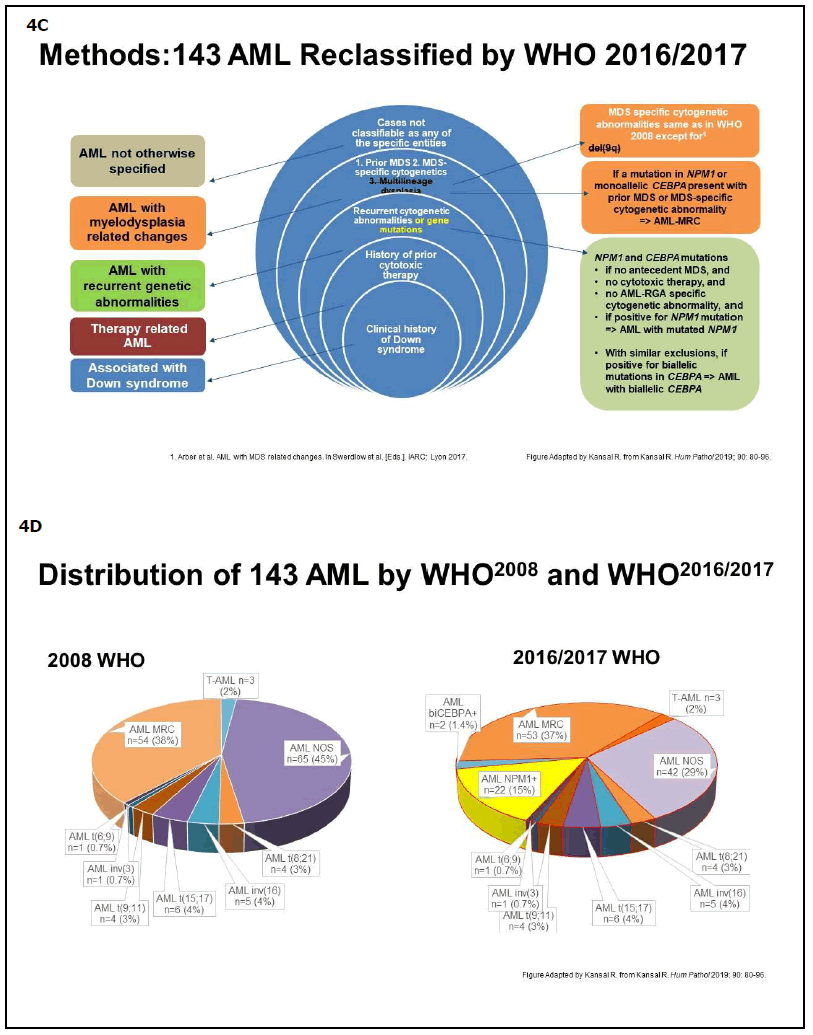
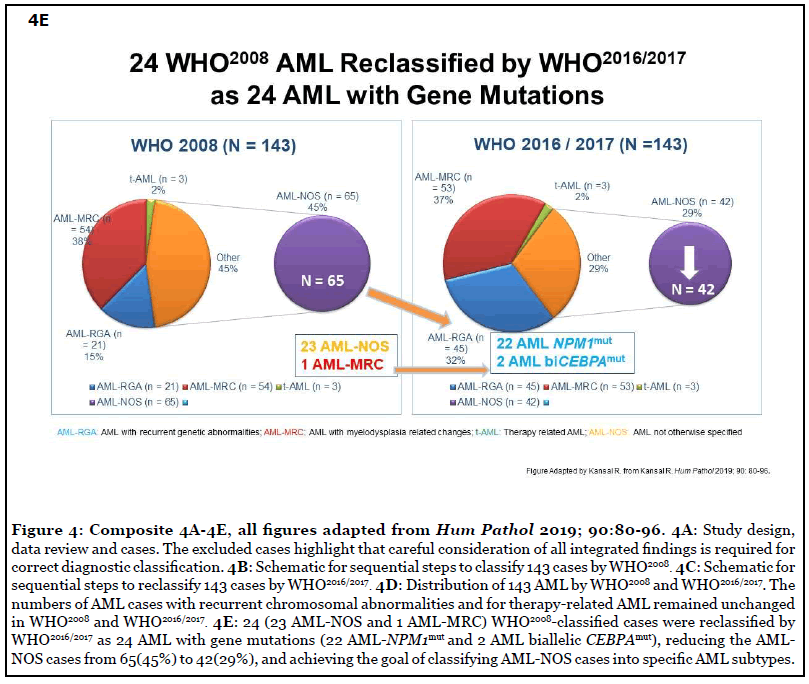
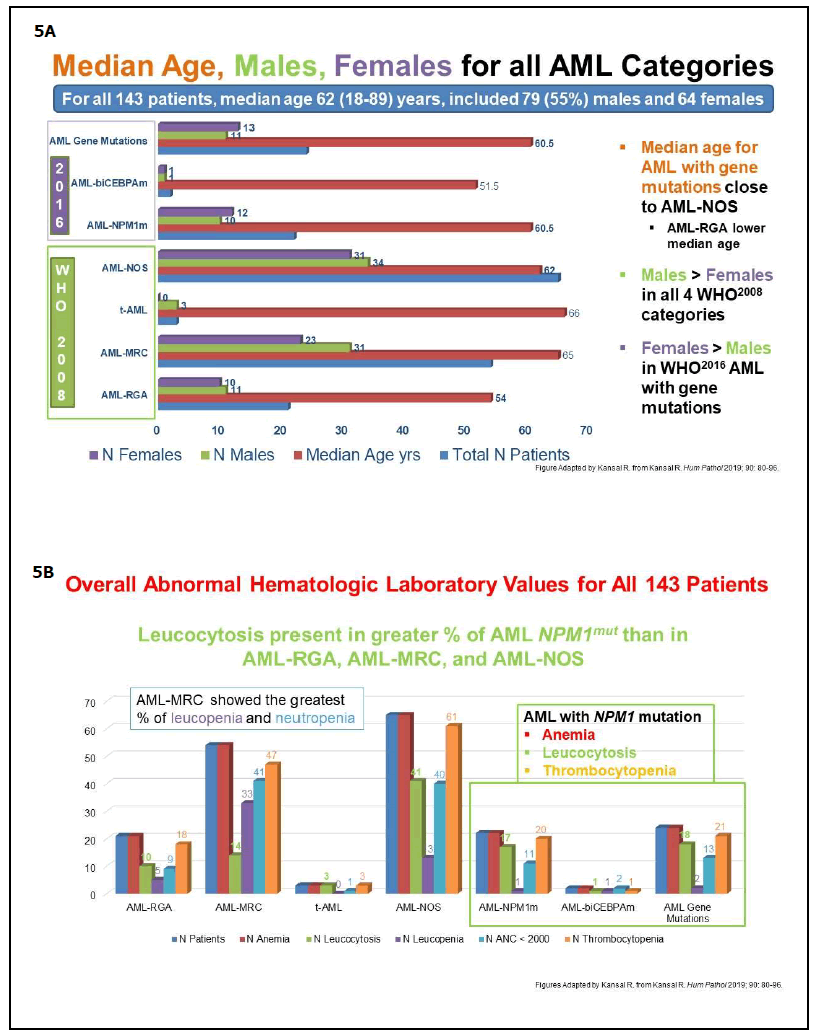
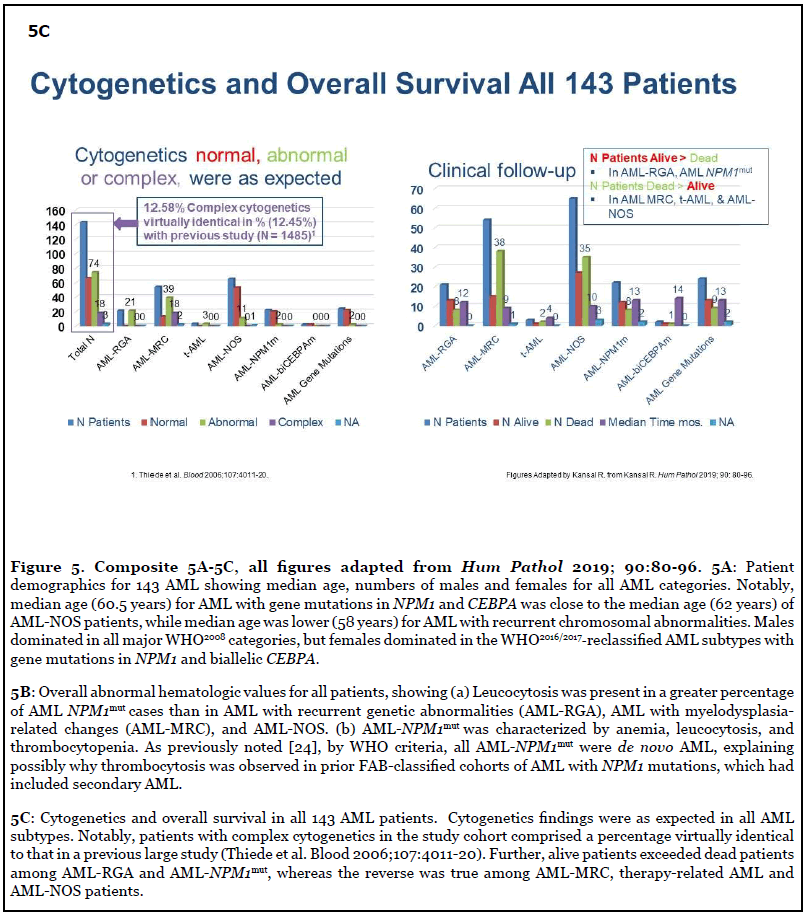
Table 3 is adapted from a previous review to show demographics of prior reported familial AML-CEBPAmut patients (20 males, 19 females, ages 1.75-62 years) from 14 families; 83% (15/18) patients showed normal cytogenetics [44]. Table 4 highlights that in the cohort by Green et al., the percentage of AML-biCEBPAmut patients was highest in the youngest (15-29 years) age group [45].
| Reports for Families with Acute Myeloid Leukemia with mutated CEBPA | Patient Gender, Age in years at onset of Acute Myeloid Leukemia | Cytogenetics |
|---|---|---|
| Smith et al, 2004 | Male, 10 | Not done |
| Male, 30; female, 18 | Both Normal | |
| Male, 2 | ||
| Sellick et al, 2005, De Lord et al, 1997 | Male, 34 | NA |
| Male, 25 | del(6)(q21) | |
| 2 Males, 24 and 4 | Normal | |
| Pabst et al, 2008, pedigree A | Female, 46 | Monosomy 7 |
| Female, 40 | Normal | |
| Pabst et al, 2008, pedigree B | Male, 42 | NA |
| Female, 27 | Normal | |
| Renneville et al, 2009 | Female, 23; male, 5 | Both Normal |
| Nanri et al, 2010 | 2 Males, 39, 26 | NA |
| Taskesen et al, 2011 | Female, 25 | NA |
| Female & male, NA | NA | |
| Taskesen et al, 2011, Stelljes et al, 2011 | 2 Females, 28, 2 | Both Normal |
| Xiao et al, 2011 | Male, 36 | del(9)(q11q34) |
| Debeljak et al, 2013 | 2 Females, 1.75 (21 months), 15 | Both Normal |
| Tawana et al, 2015 | 2 Females, 32 and 3 | NA |
| Female, 18 | NA; failed | |
| Yan et al, 2016 | Male, 33 | del(9)(q13q22) |
| Pathak et al, 2016 | 4 Males, 36, 41, 58, 62; 1 female, 53 | NA for all 5 |
| 3 Females, 11, 20, 22 y | Normal for all | |
| 2 Males, 2.8 (34 months), 6 y | NA for both | |
| Ram et al, 2017 | Female, 36 | Normal |
| Kansal, 2019 | Male, 74 | Normal |
| Abbreviation: NA, Not available | ||
Table 3: Characteristics of previous familial acute myeloid leukemia with mutated CEBPA patients compared with that of study case, adapted from publication [44].
| Characteristics | Biallelic (double) CEBPAmut | |
|---|---|---|
| No. | % | |
| No. of patients | 59 | 4% |
| Age in Years | ||
| 15-29 | 21 | 36 |
| 30-39 | 12 | 20 |
| 40-49 | 15 | 25 |
| 50-59 | 9 | 15 |
| >60 | 2 | 3 |
| Median Age | 35 | |
| Age Range | 16-67 | |
| Sex | ||
| Female | 30 | 51 |
| Male | 29 | 49 |
| Type of AML | ||
| De novo | 58 | 98 |
| Secondary | 1 | 2 |
| Abbreviation: AML, Acute myeloid leukemia | ||
Table 4:Characteristics of AML patients with biallelic mutated CEBPA from the Green et al publication [45].
Epidemiologic studies are required to determine if there is a real increase in AML with gene mutations in NPM1 and CEBPA in young patients, especially women and if yes, why? Why are women more affected by AML with genetic mutations in NPM1 and CEBPA? Further, studies are necessary for familial AML, including AML-NPM1mut, which comprises the most frequent genetic AML subtype to date in European cohorts [24,46], including for molecular epidemiology, which could potentially identify AML subtypes (familial or sporadic) that may be preventable in the future by lifestyle changes [47], including smoking, which increases the risk of AML [48], or by continued advances in precision gene editing [49].
For routine integrated genomics diagnosis, the WHO classification is meant to be applied worldwide, including in countries with limited resources or advanced molecular genetic techniques. Even in the developed world, laboratory hematologic tests with microscopic evaluation of peripheral blood and bone marrow smears are crucial for a prompt diagnosis of AML. Indeed, a specific diagnosis of APL may be preliminarily rendered with blood smear examination even before a bone marrow biopsy and rapid testing for PML-RARA are performed, particularly if combined with rapid myeloperoxidase cytochemical staining that typically shows intense diffuse positivity in APL even in the hypogranular variant, and if present with disseminated intravascular coagulation. The distinction of an APL from a non-APL--AML is the most urgent priority of the pathologist in communication with the clinician, for the correct life-saving therapy. Morphologic smear review is also invaluable to diagnose a possible AML with monocytic or myelomonocytic differentiation, particularly when a neoplasm might not be clinically suspected. Even with minimal monocytosis, a careful smear examination must be performed for abnormal, possibly neoplastic monocytic cells, as illustrated in the WHO books [12,40,42], with bone marrow biopsy performed if needed to rule-out or rule-in an AML, and as described for one AML- NPM1mut patient [24]. The identification of even rare but unequivocal Auer rods by morphologic review, although that may require time, confirms the diagnosis of a myeloid neoplasm.
FCI using a panel of markers that are required to assign lineage in acute leukemias correctly, is crucial to diagnose AML, including to definitively exclude MPALs, which were first classified as ALALs by WHO2008, as depicted in Figure 3. ALALs also include acute undifferentiated leukemia, which is extremely rare and must only be considered after all other leukemia subtypes (including blastic plasmacytoid dendritic cell neoplasm and leukemias of other unusual lineages) and non-hematopoietic neoplasms are definitively excluded [42]. Immunohistochemical positivity of the leukemic blasts for myeloperoxidase, lysozyme, or CD117 may be useful to identify myeloid lineage. The reader is referred to a recent expert review by Porwit and Bene for recommendations (and pitfalls) for lineage determination in acute leukemias and diagnosis of MPALs by the EGIL and WHO2001-WHO2016/2017 classifications [50]. The myeloid antigens, CD13, CD33, and CD117, included by the EGIL and WHO2001 but not by WHO2008, were myeloid lineage-specific for MPALs by WHO2016/2017. Also, while the EGIL classification included CD15 and not CD11c, the WHO2001-WHO2016/2017 included CD11c and not CD15 as myeloid lineage-specific antigens [12,40,42,50]. Importantly, FCI also provides the patientspecific leukemia-associated immunophenotype that serves as a signature for subsequent disease detection, including the expression of CD33 on leukemic cells for anti-CD33 therapy.
Cytogenetics analysis is essential, with molecular analysis for genetic mutations. The presence of the t(8;21), t(15;17), t(16;16) or inv(16) abnormalities is diagnostic of AML even if blasts are less than 20%, and even if a MPAL is suggested by FCI [12,40-42,50]. MPALs may also show a complex karyotype, which alone does not indicate a diagnosis of AML-MRC [50]. Additionally, MPALs may harbor gene mutations similar to those present in AML [50], further necessitating diagnostic distinction between MPAL and AML. Finally, in communication with the clinician for clinical history and laboratory scientists, the pathologist can best integrate findings from all necessary testing modalities to correctly classify AML, which guides the clinician for risk stratification and therapy.
Figure 8 shows the current NCCN guidelines for risk stratification [51,52]. Table 5 shows the CAP-ASH 2017 guidelines and NCCN guidelines for mutation testing in AML [51-53].

| 2017 CAP-ASH guidelines [53] |
| In pediatric and adult patients with confirmed or suspected AML of any type 1Strong recommendation: FLT3ITD testing 1Strong recommendation: Mutational testing for NPM1, CEBPA, and RUNX1 for AML other than confirmed CBF-AML, APL, or AML with myelodysplasia-related cytogenetic abnormalities 2Recommendation: Mutational testing including, but not limited to, IDH1, IDH2, TET2, WT1, DNMT3A, TP53 |
| 1Strong recommendation: KIT mutation testing in adult patients with confirmed CBF-AML 3Expert consensus opinion: KIT mutation testing in pediatric patients with confirmed CBF-AML |
| 1Strong recommendation: In suspected APL, ensure rapid testing for PML-RARA is performed; ensure appropriate coagulation studies to evaluate for disseminated intravascular coagulation 3Expert consensus opinion: The pathologist may request cytochemical stains to assist in the diagnosis and classification of AML |
| 2Recommendation: For molecular or genetic studies, may use cryopreserved cells or nucleic acid, formalin-fixed, non-decalcified paraffin-embedded (FFPE) tissue, or unstained marrow aspirate or peripheral blood smears obtained and prepared from peripheral blood, bone marrow aspirate or other involved tissues in which the use of such material has been validated. Such specimens must be properly identified and stored under appropriate conditions in a laboratory that is in compliance with regulatory and/or accreditation requirements. |
| 1Strong recommendation: For extramedullary disease without bone marrow or blood involvement (myeloid sarcoma), the pathologist should evaluate and process a tissue biopsy for morphologic, immunophenotypic, cytogenetic, and molecular genetic studies, as recommended for the bone marrow. |
| 1Strong recommendation: The initial pathology report should include laboratory, morphologic, immunophenotypic, and, if performed, cytochemical data, on which the diagnosis is based, along with a list of any pending tests. The pathologist should issue addenda/amended reports when the results of additional tests become available. |
| 1Strong recommendation: Ensure that all tests performed for classification, management, predicting prognosis, and disease monitoring are entered into the patient’s medical records. This information should include the sample source, adequacy, and collection information, as applicable. |
| 1Strong recommendation: Treating physicians and pathologists should use the current WHO classification for diagnosis and classification |
| 1Strong recommendation: To avoid duplicate procedures, associated patient discomfort, and additional costs, if after examination of a peripheral blood smear, it is determined that the patient will require immediate referral to another institution with expertise in the management of acute leukemia for treatment, the initial institution should, whenever possible, defer non-emergent invasive procedures, including bone marrow aspiration and biopsies, to the treatment center. |
| 1Strong recommendation: Provide the treatment center with all laboratory results, pathology slides, flow cytometry data, cytogenetic information, and a list of pending tests, if a patient is referred, at the time of referral, and forward results of pending tests when available. |
| NCCN Guidelines v3.2020 [51,52] |
| Mutational testing for risk stratification: FLT3, NPM1, CEBPA, CKIT, TP53, ASXL1, and RUNX1, with FLT3ITD, allelic ratio (mutant/normal <0.5 low; >0.5 high), as per 2017 European Leukemia Network guidelines [52] |
| Recommend testing for bothFLT3ITD andFLT3ITD |
| Abbreviations:AML, acute myeloid leukemia; CBF-AML, core-binding factor AML (AML with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 or inv(16)(p13.1q22) /t(16;16)(p13.1;q22); CBFB-MYH11); APL, acute promyelocytic leukemia; WHO, World Health Organization 1Supported by convincing (high) or adequate (intermediate) quality of evidence and clear benefit that outweighs any harms [53]. 2Some limitations in quality of evidence (adequate [intermediate] or inadequate [low]), balance of benefits and harms, values, or costs, but panel concluded that there is sufficient evidence and/or benefit to inform a recommendation [53]. 3Serious limitations in quality of evidence (inadequate [low] or insufficient), balance of benefits and harms, values or costs, but panel consensus was that a statement was necessary [53]. Quality of evidence: Convincing, high confidence that available evidence reflects true effect, and further research is very unlikely to change the confidence in the estimate of effect; Adequate, moderate confidence that available evidence reflects true effect, and further research is likely to have an important effect on the confidence in estimate of effect and may change the estimate; Inadequate, Little confidence that available evidence reflects true effect, and further research is very likely to have an important effect on the confidence in the estimate of effect and is likely to change the estimate [53] |
Table 5: The College of American Pathologists-American Society for Hematology (CAP-ASH) guidelines and NCCN guidelines for molecular genetic testing in patients with acute myeloid leukemia, adapted from publications [51-53].
Moreover, AML treatment now includes drugs targeted against FLT3 and IDH mutations, anti-CD33 antibody therapy, and therapies specific for AML-MRC, therapyrelated AML, and AML in the elderly, as shown in Table 6, with several additional drugs, including in combinations, in clinical trials [54,55].
| Drugs targeted to specific genetic mutation | Approved Date | U.S.A. Food and Drug Administration Approved Indication | |
|---|---|---|---|
| Midostaurin | FLT3 ITD, FLT3 TKD | April 2017 | New diagnosis of FLT3-mutated AML in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation |
| Gilteritinib | FLT3 ITD, FLT3 TKD | November 2018 | Relapsed/refractory FLT3-mutated AML |
| Enasidenib | IDH2 | August 2017 | Relapsed/refractory IDH2-mutated AML |
| Ivosidenib | IDH1 | July 2018 | Relapsed/refractory IDH1-mutated AML |
| May 2019 | New diagnosis of IDH1-mutated AML in patients aged > 75 years or with comorbidities precluding induction chemotherapy | ||
| Antibody-drug conjugate targeted against surface antigen | |||
| Gemtuzumab ozogamycin | CD33 | September 2017 | New diagnosis of CD33-positive AML in adults; relapsed/refractory CD33-positive AML in adults and pediatric patients aged > 2 years |
| Drugs approved for specific AML patients | |||
| CPX-351 | Liposomal cytarabine and daunorubicin in a fixed 5:1 molar ratio | August 2017 | New diagnosis of AML with myelodysplasia-related changes or therapy-related AML |
| Venetoclax | BCL2 inhibitor | November 2018 | New diagnosis of AML in patients aged > 75 years or with comorbidities precluding induction chemotherapy |
| Glasdegib | Hedgehog pathway inhibitor | November 2018 | New diagnosis of AML in patients aged > 75 years or with comorbidities precluding induction chemotherapy |
| Abbreviation: AML, Acute myeloid leukemia | |||
Table 6: Drugs approved for acute myeloid leukemia therapy since 2017.
All of the above advances now increasingly necessitate comprehensive, integrated diagnostics, including genomics for AML diagnosis, prognosis, and therapy. Considerations to implement such testing for AML include (1) first, correct diagnostic classification of AML with combined testing modalities and multidisciplinary teams as described above, with the pathologist best able to integrate all findings, (2) cost of testing, which is likely to be much less than treatment costs [56], and (3) turnaround time, which can take much longer by NGS than single-gene PCR assays, especially for FLT3 and IDH1/IDH2 mutations. Apart from institution-specific assays [57], commercial assays to allow clinical NGS results in possibly two days from sample collection will soon be available for AML [58]. (4) Several commercially available NGS assays include bulk RNA sequencing for detecting gene fusions [58,59]. (5) Clonal hematopoiesis may co-exist with myeloid neoplasms, including AML, with mutant clone detection dependent upon genomic methods, leading to challenges for interpretation of the significance of genetic mutations identified by NGS [37,60-62]. (6) Technically, in addition to sequencing depth and coverage, variability arising from the entire multi-step NGS process, including nucleic acid extraction, target enrichment, and library preparation, bioinformatics analysis, data analysis, and reporting for genomic aberrations must be considered, including for multi-institutional research, given the variability in NGS methods (including manual versus automated) used in different laboratories [63-68]. A recent study showed genomic sequencing results to be institution-specific, with inter-institution heterogeneity in sequencing methods [ 69]. (7) Particularly for the AML classification, a technical challenge pertains to the detection of mutations in CEBPA [70], a GC-rich single-exon gene, and FLT3ITD mutations by short-read sequencers, as per the experience of many laboratories. Since NGS results for both targets are methoddependent, both genes are often preferentially analyzed by separate PCR-based assays. (8) Differences in NGS assays alone, including customized for academic institutions and commercially available (off-the-shelf or customized), add to the variability in sensitivity and specificity that might vary for different gene targets. Necessarily, if mutations in the genes necessary for precise diagnostic classification might be variably detected inter-institutionally, then ultimately, that would affect the collective diagnoses rendered universally, leading to inadequate or inaccurate assessment of the overall “true” AML subtypes. Similar to the standardized nomenclature required by the WHO classification, standardization of genomic testing for clinical diagnosis, albeit challenging, would be a major step forward towards the very challenging task of integrating genomics in AML for routine clinical care worldwide.
In conclusion, due to progressively rapid advances in technology and genomics, the last two decades have shown exponential progress for depth in our understanding of AML pathogenesis. The WHO2016/2017 classification of AML represents a significant milestone for the care of patients with AML and has paved the way for future continued collaboration and innovation toward further progress for patient care.
Conflict of Interest
None.
References
2. Geary CG. The story of chronic myeloid leukaemia: Historical review. British Journal of Haematology. 2000 Jul;110(1):2-11.
3. Rosenberg SA, Diamond HD, Jaslowitz B, Craver LF. Lymphosarcoma: a review of 1269 cases. Medicine. 1961 Feb 1;40(1):31-84.
4. Lennert K. Classification of Non-Hodgkin’s lymphoma. In: Lennert K, Stein H, Mohri N, Kaiserling E, Muller- Hermelink HK (Editors). Malignant lymphomas other than Hodgkin’s disease. Springer-Verlag New York Heidelberg Berlin 1978. p. 83-110.
5. Tjio JH, Levan A. The chromosome number of man. Hereditas. 1956 May;42:1-6.
6. Hungerford DA, Nowell PC. A minute chromosome in human chronic granulocytic leukemia. Science. 1960;142:1497-9.
7. Sandberg AA, Koepf GF, Crosswhite LH, Hauschka TS. The chromosome constitution of human marrow in various developmental and blood disorders. American Journal of Human Genetics. 1960 Sep;12(3):231-249.
8. Sandberg AA, Ishihara T, Crosswhite LH, Hauschka TS. Chromosomal dichotomy in blood and marrow of acute leukemia. Cancer Research. 1962 Jul 1;22(6):748-756.
9. Sandberg AA. The chromosomes and causation of human cancer and leukemia. Cancer Research. 1966 Sep 1;26(9 Part 1):2064-81.
10. Lukes RJ, Butler JJ. The pathology and nomenclature of Hodgkin’s disease. Cancer Research. 1966 Jun 1;26(6 Part 1):1066-81.
11. Lukes RJ, Craver LF, Hall TC, Rappaport H, Ruben P. Report of the nomenclature committee. Cancer Research. 1966 Jun 1;26(6 Part 1):1311.
12. Jaffe ES, Harris NL, Stein H, Vardiman JW, (Eds). IARC Press; Lyon, France: 2001. Pathology and Genetics: Tumours of Haematopoietic and Lymphoid Tissues (World Health Organization Classification of Tumours).
13. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HT, et al. Proposals for the classification of the acute leukemias. Br J Haematol. 1976 Aug;33(4):451-458.
14. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposed revised criteria for the classification of acute myeloid leukemia: a report of the French-American-British Cooperative Group. Annals of Internal Medicine. 1985 Oct 1;103(4):620-625.
15. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French- American-British (FAB) Cooperative Group. Journal of Clinical Pathology. 1989 Jun 1;42(6):567-584.
16. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, et al. The chronic myeloid leukaemias: guidelines for distinguishing chronic granulocytic, atypical chronic myeloid, and chronic myelomonocytic leukaemia: Proposals by the French-American-British Cooperative Leukaemia Group. British Journal of Haematology. 1994 Aug;87(4):746-754.
17. Rappaport H, Winter WJ, Hicks EB. Follicular lymphoma. A re-evaluation of its position in the scheme of malignant lymphoma based on a survey of 253 cases. Cancer. 1956 Jul;9(4):792-821.
18. Lukes RJ, Collins RD. Immunologic characterization of human malignant lymphomas. Cancer. 1974 Oct;34(S8):1488-1503.
19. Bennett M, Farrer-Brown G, Henry K, Jelliffe AM, Gerard-Marchant R, Hamlin I, et al. Classification of non-Hodgkin’s lymphomas. The Lancet. 1974 Aug 17;304(7877):405-408.
20. Stansfield AG, Diebold J, Kapanci Y, Kelenyi G, Lennert K. Updated Kiel classification for lymphomas. Lancet. 1988;1(8580):292-293.
21. Non-Hodgkin’s Lymphoma Pathologic Classification Project. National Cancer Institute sponsored study of classifications of non-Hodgkin’s lymphomas: summary and description of a working formulation for clinical usage. Cancer. 1982;49(10):2112-2135.
22. Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994 Sep 1;84(5):1361-1392.
23. Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A, et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia. 1995 Oct;9(10):1783-1786.
24. Kansal R. Classification of acute myeloid leukemia by the revised fourth edition World Health Organization criteria: a retrospective single-institution study with appraisal of the new entities of acute myeloid leukemia with gene mutations in NPM1 and biallelic CEBPA. Human Pathology. 2019 Aug 1;90:80-96.
25. International Agency for Research on Cancer World Health Organization: WHO Classification of Tumors. http://whobluebooks.iarc.fr/about/history.php. Website last accessed May 29, 2020.
26. Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. Blood, The Journal of the American Society of Hematology. 1998 Oct 1;92(7):2322-2333
27. Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999 Oct 15;286(5439):531-7.
28. Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, van Doorn-Khosrovani SB, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. New England Journal of Medicine. 2004 Apr 15;350(16):1617- 1628.
29. Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncology Reviews. 2012 Mar 5;6(1).
30. Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood, The Journal of the American Society of Hematology. 2001 Sep 15;98(6):1752-1759.
31. Fro¨hling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood, The Journal of the American Society of Hematology. 2002 Dec 15;100(13):4372-4380.
32. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. New England Journal of Medicine. 2005 Jan 20;352(3):254- 266.
33. Do¨hner K, Schlenk RF, Habdank M, Scholl C, Ru¨cker FG, Corbacioglu A, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005 Dec 1;106(12):3740-3746.
34. Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-a (C/EBPa), in acute myeloid leukemia. Nature Genetics. 2001 Mar;27(3):263-70.
35. Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. New England Journal of Medicine. 2004 Dec 2;351(23):2403-7.
36. Lord D. Familial acute myeloid leukaemia: four male members of a single family over three consecutive generations exhibiting anticipation. British Journal of Haematology. 1998 Mar;100(3):557-60.
37. Kansal R. Acute myeloid leukemia in the era of precision medicine: recent advances in diagnostic classification and risk stratification. Cancer Biology & Medicine. 2016 Mar;13(1):41.
38. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine. 2013 May 30;368(22):2059-2074.
39. Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. New England Journal of Medicine. 2012 Mar 22;366(12):1079-89.
40. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., (Eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th edition. Lyon: IARC; 2008.
41. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016 May 19;127(20):2391- 2405.
42. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al., (Eds). WHO classification of tumours of haematopoietic and lymphoid tissues (Revised 4th edition). IARC: Lyon. 2017.
43. Tawana K, Wang J, Renneville A, Bödör C, Hills R, Loveday C, et al. Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood, The Journal of the American Society of Hematology. 2015 Sep 3;126(10):1214-1223.
44. Kansal R. Familial acute myeloid leukemia. In: Liu D (Editor): Handbook of Tumor Syndromes. Taylor & Francis CRC Press; 2020; 545-57.
45. Green CL, Koo KK, Hills RK, Burnett AK, Linch DC, Gale RE. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. Journal of Clinical Oncology. 2010 Jun 1;28(16):2739-47.
46. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. New England Journal of Medicine. 2016 Jun 9;374(23):2209-2221.
47. Song M, Vogelstein B, Giovannucci EL, Willett WC, Tomasetti C. Cancer prevention: molecular and epidemiologic consensus. Science. 2018 Sep 28;361(6409):1317-1318.
48. Fircanis S, Merriam P, Khan N, Castillo JJ. The relation between cigarette smoking and risk of acute myeloid leukemia: An updated meta-analysis of epidemiological studies. American Journal of Hematology. 2014 Aug;89(8):E125-32.
49. Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020 Apr 17;368(6488):290-296.
50. Porwit A, Béné MC. Multiparameter flow cytometry applications in the diagnosis of mixed phenotype acute leukemia. Cytometry Part B: Clinical Cytometry. 2019 May;96(3):183-194.
51. NCCN. Clinical Practice Guidelines in Oncology. Acute Myeloid Leukemia (version 3.2020). https://www. nccn.org/professionals/physician_gls/pdf/aml.pdf.Last accessed May 31, 2020.
52. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017 Jan 26;129(4):424-447.
53. Arber DA, Borowitz MJ, Cessna M, Etzell J, Foucar K, Hasserjian RP, et al. Initial diagnostic workup of acute leukemia: guideline from the College of American Pathologists and the American Society of Hematology. Archives of Pathology & Laboratory Medicine. 2017 Oct;141(10):1342-1393.
54. Burnett A, Stone R. AML: New drugs but new challenges. Clinical Lymphoma Myeloma and Leukemia. 2020 Jun 1;20(6):341-350.
55. Kurtz SE, Eide CA, Kaempf A, Khanna V, Savage SL, Rofelty A, et al. Molecularly targeted drug combinations demonstrate selective effectiveness for myeloid-and lymphoid-derived hematologic malignancies. Proceedings of the National Academy of Sciences. 2017 Sep 5;114(36):E7554-E7563.
56. Cardarelli WJ. The evolution of care for acute myeloid leukemia and the challenges of defining value. Journal of Clinical Pathways. 2018;4(suppl 1):S28-S34.
57. Patel KP, Ruiz-Cordero R, Chen W, Routbort MJ, Floyd K, Rodriguez S, et al. Ultra-rapid reporting of GENomic targets (URGENTseq): Clinical next-generation sequencing results within 48 hours of sample collection. The Journal of Molecular Diagnostics. 2019 Jan 1;21(1):89- 98.
58. Thermofisher Scientific www.thermofisher.com website last accessed May 29, 2020.
59. Wang Y, Mashock M, Tong Z, Mu X, Chen H, Zhou X, et al. Changing technologies of RNA sequencing and their applications in clinical oncology. Frontiers in Oncology. 2020 April 9;10:477.
60. Watson CJ, Papula AL, Poon GY, Wong WH, Young AL, Druley TE, et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science. 2020 Mar 27;367(6485):1449-1454.
61. Curtis C. Quantifying mutations in healthy blood. Science. 2020 Mar 27;367(6485):1426-27.
62. Tomasetti C. Mutated clones are the new normal. Science. 2019 Jun 7;364(6444):938-9.
63. Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nature Reviews Genetics. 2014 Feb;15(2):121-132.
64. Bacher U, Shumilov E, Flach J, Porret N, Joncourt R, Wiedemann G, et al. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer Journal. 2018 Nov 12;8(11):113.
65. Leisch M, Jansko B, Zaborsky N, Greil R, Pleyer L. Next generation sequencing in AML—on the way to becoming a new standard for treatment initiation and/or modulation?. Cancers. 2019 Feb;11(2):E252.
66. Aigrain L, Gu Y, Quail MA. Quantitation of next generation sequencing library preparation protocol efficiencies using droplet digital PCR assays-a systematic comparison of DNA library preparation kits for Illumina sequencing. BMC Genomics. 2016 Dec 1;17(1):458.
67. Chung J, Lee KW, Lee C, Shin SH, Kyung S, Jeon HJ, et al. Performance evaluation of commercial library construction kits for PCR-based targeted sequencing using a unique molecular identifier. BMC Genomics. 2019 Dec 1;20(1):216.
68. Aguilera-Diaz A, Vazquez I, Ariceta B, Mañú A, Blasco- Iturri Z, Palomino-Echeverría S, et al. Assessment of the clinical utility of four NGS panels in myeloid malignancies. Suggestions for NGS panel choice or design. PloS one. 2020 Jan 24;15(1):e0227986.
69. Borges MG, Rocha CS, Carvalho BS, Lopes-Cendes I. Methodological differences can affect sequencing depth with a possible impact on the accuracy of genetic diagnosis. Genetics and Molecular Biology. 2020;43(2);E20190270.
70. Ng CW, Kosmo B, Lee PL, Lee CK, Guo J, Chen Z, et al. CEBPA mutational analysis in acute myeloid leukaemia by a laboratory-developed next-generation sequencing assay. Journal of Clinical Pathology. 2018 Jun 1;71(6):522-531.