Commentary
Increasingly prominent roles in interventional oncology are held by various tumor ablation therapies performed by direct applications of local acute trauma-inducing insult to the targeted lesion aiming for its rapid in situ destruction. These therapies include treatments based on various forms of thermal energy delivery (photothermal, cryoablation, microwave ablation, radiofrequency ablation), non-thermal illumination (photodynamic therapy), electric field exposure, or high hydrostatic pressure [1-3]. Common injury inflicted in cells of tumors treated by ablation therapies is the impairment of proteostasis due to accumulation of misfolded/damaged proteins. This is sensed by afflicted cells as a trauma typically associated with thermal or oxidative stress that threatens the integrity and homeostasis at the affected site [4]. These types of stress provoke evolutionary well conserved canonic protection mechanisms based on cellular stress signaling network that serves to re-balance biochemical activities within the cell. They work by conversing the incoming signal (appearance of stressor molecule) towards downstream effector molecules involved in transcriptome reprogramming aimed at activation or inhibition of targeted biochemical tasks [4,5]. The aim of this commentary is to emphasize that the activity of the engaged intracellular signaling pathways determines the fate of involved tumor cells and ultimately the outcome of the applied therapies.
Stress signaling network consist of interlinked intracellular signal transduction programs providing defense mechanisms for mitigating stress challenge expressed as adaptive responses developed to assure restored homeostasis and survival of afflicted cells. This is attained either by improving the resistance to stress or by reducing the stress impact with increased tolerance [6]. The former is attained by enhancing the clearance of induced primary stressor molecules, while the latter is mediated through limiting the stress damage (e.g. by improved protein folding capacity) [7]. While the initial response to stress trauma is engaged to help the cells to defend against the insult and recover from it, in case when the damaging stimulus remains unresolved the cellular signaling cascades are switched towards self-destructive programs leading to cell death or elimination through immune rejection [4,8].
Signal transduction cascades incorporated in the stress signaling network can be grouped in four programs [8]:
i) Rectifying changes in gene and protein activity aimed at suspending global protein synthesis while expanding the expression of protein chaperons, folding enzymes and other stress-relieving proteins. The best known among these cell autonomous pathways are heat shock response (HSR), unfolded protein response (UPR), integrated stress response (ISR), and antioxidant response [9-11];
ii) Repairing damaged cellular membranes in order to maintain cell integrity, primarily through the engagement of sterol regulatory element-binding proteins (SREBs) pathway that mediates the metabolism of cholesterol and fatty acid metabolism control together with the associated caspase-1 activity [12];
iii) Amplifying removal of irreparably damaged proteins and cells with programs including the ER-associated protein degradation response (ERAD), autophagy, as well as apoptosis and other cell death signaling cascades [4,13];
iv) Generation of immunogenic cell death and ensuing immune elimination cascades [8].
In adaptive stress response pathways, key pro-survival roles are held by UPR/ISR sensor kinases PERK and IRE1, transcription factor HSF1 of HSR, and by oxidative proteinfolding controlling enzyme protein disulfide isomerase (PDI) that actively participates in the UPR and antioxidant response [4,8]. Selective specific inhibitors of these signaling elements drastically reduce survival of tumor cells treated by ablation therapies and could be useful clinically to improve the outcome of tumor treatment with these therapies [8].
The adaptive stress responses are additionally fine-tuned by cross-talk with c-Jun amino-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) signaling nodes controlling basic cellular metabolism [6]. The MAPK activity in turn regulates the expression of important master nuclear transcription factors, such as STAT3 and HIF-1, enabling them to play important roles in stress response [14,15]. The activity of HIF-1 markedly influences tumor growth because of the effects on angiogenesis, glucose metabolism, and cell survival [16]. By similarly modifying tumor microenvironment, STAT3 promotes angiogenesis, cell survival and immunosuppression [17]. The effects of specific inhibitors of STAT3 and HIF-1 on tumor cells treated by PDT (as a representative of ablation therapies) were examined in our studies. Tested were benzoic acid derivative NSC 74859 that selectively inhibits activation, dimerization and DNA-binding activity of STAT3, while aryloxyacetylaminobenzoic derivative LW6 was used as it promotes degradation of HIF-1α [18,19]. At the doses showing no detectable toxicity when used alone, both inhibitors significantly reduced the survival of tumor cells treated by PDT [20]. This finding reveals the pro-survival role of STAT3 and HIF- 1 in cells coping with PDT-mediated stress. Their inhibitors are good candidates for use as adjuvants for improving the clinical outcome of PDT and other ablation therapies. Importantly, even stronger reduction of survival was seen with cells treated by PDT and exposed to ceramidase inhibitor LCL521 [20]. This is consistent with the important role of sphingolipid activity in cellular stress signaling [21,22]. Indeed, PDT and other stress-inducing treatments were shown to have a pronounced impact on the sphingolipid profile in treated tumors due to their effects on the genes controlling metabolism of these lipids [23,24].
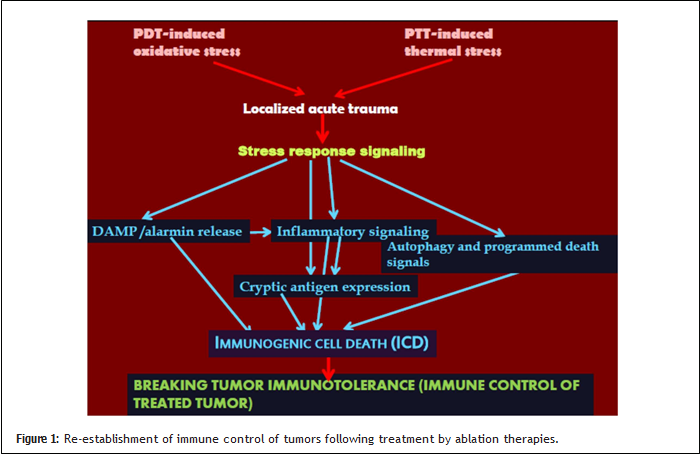
A crucial advantage that can be realized by the use of tumor ablation therapies is the re-establishment of immune control by breaking tumor immunotolerance [2,8]. Stress response signaling triggered by acute trauma in treated tumor site induced by localized thermal, oxidative or other stress caused by ablation therapies includes potent immune signal transduction activities (Figure 1). In addition to the avid non-infectious inflammatory signaling highlighted by the engagement of its key regulators such as nuclear transcription factor NFκB and expression of Toll-like receptors, this is dominated by the danger signaling cascade leading to the release of alarmins and the expression of damage-associated molecular patterns (DAMPs) [4,25]. These molecules from the cells whose appearance on the cell surface or release outside the cell turns them into signals for the mobilization of immune system are critical for the induction of immunogenic cell death [26,27]. The other component important for immune signaling is the abundant availability of tumor antigens derived from programmed cell death pathways with ensuing disposal and processing of dead cell material by phagocytes capable of antigen presentation activity [28,29]. Another decisive element is the stress-induced expression of highly immunogenic cryptic tumor antigens normally hidden within untranslated regions of tumor cell RNA [4,30].
For highly advantageous clinical outcome it is critical that the antitumor immune response orchestrated by tumor ablation therapies remains robust and long-lasting. This can be achieved by optimal choice of adjuvant immune stimulating agents. One such agent is IP-001, a specific variant of the carbohydrate biopolymer N-dihydrogalactochitosan, which is currently evaluated in clinical trials in combination with laserbased thermal ablation of advanced solid tumors [31].
Conflict of Interest
The author declared no conflict of interest.
References
2. Keisari Y. Tumor abolition and antitumor immunostimulation by physic-chemical tumor ablation. Front Biosci Landmark. 2017;22:310-347.
3. Heger U, Hackert T. Can local ablative techniques replace surgery for locally advanced pancreatic cancer? J Gastrointest Oncol. 2021;12(5):2536-2546.
4. Korbelik M. Role of stress signaling networks in cancer cell death and antitumor immune response following proteotoxic injury inflicted by photodynamic therapy. Lasers Surg Med. 2018;50:491-498.
5. Hipp MS, Park S-H, Hartl FU. Proteostasis impairment in protein misfolding and –aggregation diseases. Trends Cell Biol. 2014;24(9):506-514.
6. Hotamisligil GS, Davis RJ. Cell Signaling and stress responses. Cold Spring Harb Perspect Biol. 2016;8(10):a006072.
7. Goncalves RLS, Hotamisligil GS. TMEM2 modulates ER stress in a non-canonical manner. Cell Metab. 2019;30(6):999-1001.
8. Korbelik M, Hode T, Lam SSK, Chen WR. Novel immune stimulant amplifies direct tumoricidal effect of cancer ablation therapies and their systemic antitumor immune efficacy. Cells. 2021;10:492.
9. Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU. Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol. 2010;2(12):a004390.
10. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374-1395.
11. Kourtis N, Tavernakis N. Cellular stress response pathways and ageing. EMBO J. 20111;30(13):2520-2531.
12. Shimano, H, Sato, R. SREBP-regulated lipid metabolism: Convergent physiology – Divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710-730.
13. Meusser B, Hirsch C, Jarosh E, Sommer T. ERAD: The long road to destruction. Nat Cell Biol. 2005;7(8):766-772.
14. Jain N, Zhang T, Fong SL, Lim CP, Cao X. Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK). Oncogene. 1998;17:3157-3167.
15. Sutton KM, Hayat S, Chau N-M, Cook S, Pouyssegur J, Ahmed A, et al. Selective inhibition of MEK1/2 reveals a differential requirement for ERK1/2 signaling in the regulation of HIF-1 in response to hypoxia and IGF-1. Oncogene. 2007;26:3920-3929.
16. Bardos JI, Ashcroft M. Hypoxia-inducible factor-1 and oncogenic signaling. Bioessays. 2004;26:262-269.
17. Yu H, Lee H, Herrmann A, Beuttner R, Jove R. Revisiting STAT3 signaling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736-746.
18. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391-7396.
19. Lee K, Kang JE, Park S-K, Jin Y, Chung K-S, Kim H-M, et al. LW6, a novel Hif-1 inhibitor, promotes proteosomal degradation of HIF- 1α via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol. 2010;80(7):982-989.
20. Korbelik M, Zhao J, Zeng H, Bielawska A, Szulc ZM. Mechanistic insights into ceramidase inhibitor LCL521-enhanced tumor cell killing by photodynamic and thermal ablation therapies. Photochem Photobiol Sci. 2020;19:1145-1151.
21. Korbelik M. Sphingolipid activity in oxidative stress response and tumor immunity. Austin J Vaccines Immunother. 2016;3(1):1008.
22. Liu F, Li X, Lu C, Bai A, Bielawski J, Marschall B, et al. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induce ER stress to suppress myeloid-derived suppressor cells. Oncotarget. 2016;7:83907-83925.
23. Separovic D, Bielawski J, Pierce JS, Tarca AL, Ogretmen B, Korbelik M. Increased tumor dihydroceramide production after Photofrin-PDT alone and improved tumor response after the combination with the ceramide analogue LCL29. Evidence from mouse squamous cell carcinomas. Br J Cancer. 2009;100:626-632.
24. Yabu T, Shiba H, Shibasaki Y, Nakanishi T, Imamura S, Touhata K, et al. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase by JNK signaling. Cell Death Differ. 2015;22:258-273.
25. Korbelikm M, Zhang W, Merchant S. Involvement of damageassociated molecular patterns in tumor response to photodynamic therapy: surface expression of calreticulin and release of HMGB1. Cancer Immunol Immunother. 2011;69:1431-1437.
26. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018;18(4):e27.
27. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012:12(12):860-875.
28. Korbelik M, Stott B, Sun J. Photodynamic therapy-generated vaccines: relevance of tumour cell death expression. Br J Cancer. 2007:97:1381-1387.
29. Korbelik M. Impact of cell death manipulation on the efficacy of photodynamic therapy-generated cancer vaccines. World J Immunol. 2015;5:95-98.
30. Lu PD, Harding HP, Ron D. Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J Cell Biol. 2004;167(1):27-33.
31. ClinicalTrials.gov. Intratumoral injection of IP-001 following thermal ablation in patients with advanced solid tumors. https://clinicaltrials.gov/ct2/show/NCT03993678 (accessed on 11/24/2021).