Abstract
Liver kinase B1 (LKB1) is a serine/threonine kinase that serves as a central regulator of cellular energy homeostasis, growth control, polarity, and stress adaptation. Acting as a master kinase, LKB1 activates AMP-activated protein kinase (AMPK) and a family of related kinases, thereby restraining mTORC1 signaling and limiting anabolic, energy-consuming processes. Through these functions, LKB1 has classically been regarded as a tumor suppressor. However, accumulating evidence demonstrates that LKB1 signaling is highly context dependent. While LKB1 loss promotes tumor initiation, immune evasion, and metastatic progression in several epithelial cancers, preserved or contextually co-opted LKB1 signaling can paradoxically support metabolic flexibility, stress tolerance, and survival in select malignancies, including hepatocellular carcinoma and hematologic cancers. This review synthesizes current understanding of the upstream regulatory mechanisms and downstream signaling networks governing LKB1 function, emphasizing its dual and lineage-specific roles in cancer biology and highlighting emerging therapeutic vulnerabilities associated with LKB1 pathway dysregulation.
Keywords
LKB1, AMPK, Metabolic checkpoint, mTORC1 signaling, Energy stress, Autophagy, Cancer metabolism, Context-dependent tumor suppression
Abbreviations
LKB1: Liver Kinase B1; mTOR: Mechanistic Target of Rapamycin; AMPK: Adenosine Monophosphate-Activated Protein Kinase; MO25: Mouse Protein-25; STRAD: STE20-Related Adaptor
Introduction
The LKB1 gene encodes a serine/threonine kinase that plays a fundamental role in maintaining cellular homeostasis by integrating signals governing cell growth, energy metabolism, and structural organization [1–3]. Acting as a molecular switch, LKB1 phosphorylates and activates multiple downstream substrates, thereby coordinating cellular responses to metabolic and environmental cues. Thus, serving as a central regulator of cellular integrity and function.
The principal function of LKB1 is the regulation of cellular energy balance. Under conditions of energy stress, LKB1 activates AMP-activated protein kinase (AMPK), a master metabolic sensor that restores energy homeostasis by suppressing anabolic, energy-consuming pathways while promoting catabolic processes that generate ATP [2,4]. Consequently, the LKB1–AMPK axis is widely recognized as a critical safeguard of cellular energy homeostasis and metabolic adaptation.
Beyond metabolism, LKB1 is indispensable for the establishment and maintenance of cell polarity, a process essential for normal tissue architecture and organ function. Loss of cellular polarity disrupts spatial organization and signaling, facilitating abnormal cell behavior and contributing to cancer progression [3,5,6]. Through its coordinated control of metabolism, growth, and polarity, LKB1 functions as a canonical tumor suppressor gene.
Inactivation or mutation of LKB1 leads to dysregulated cell proliferation, metabolic imbalance, and loss of structural order. Such alterations are implicated in a range of malignancies, including lung and cervical cancers, as well as in Peutz–Jeghers syndrome, an inherited disorder characterized by gastrointestinal hamartomatous polyps and a markedly increased lifetime risk of cancer [1,6]. Mechanistically, LKB1 acts as a master kinase, activating a broad family of kinases that includes AMPK and several AMPK-related kinases such as MARKs, NUAKs, SIKs, BRSKs, and SNRK. This activity requires the formation of a heterotrimeric complex with the pseudokinase STRAD and the scaffolding protein MO25, which is essential for LKB1 stability and catalytic function [7–10].
While the AMPK arm of LKB1 signaling has been extensively characterized, many LKB1-dependent kinases remain incompletely understood, underscoring the complexity and breadth of this signaling network [3,7]. This intricate architecture positions LKB1 at the intersection of metabolism, cell structure, and stress responses, making it a focal point of contemporary research in cancer biology and metabolic regulation [3,4].
Importantly, although LKB1 is classically regarded as a tumor suppressor, its biological effects are highly context-dependent. LKB1 activity is finely tuned by upstream regulatory mechanisms that govern its localization, stability, and kinase function. Perturbations at this regulatory level can profoundly reshape downstream signaling outputs and biological consequences. A comprehensive understanding of how upstream signals modulate LKB1 activity is therefore essential to explain its diverse roles in metabolism, cellular growth control, and cancer progression. Thus LKB1–AMPK axis acts as a metabolic checkpoint whose downstream consequences range from growth arrest to stress tolerance in a context-dependent manner.
Upstream Regulation of LKB1
LKB1 activity is not autonomous; it is tightly governed by upstream regulatory mechanisms that control its catalytic activation, subcellular localization, and protein stability. These inputs critically shape its tumor-suppressive capacity and downstream signaling fidelity.
LKB1 is a 433–amino acid kinase comprising an N-terminal regulatory region, a central catalytic domain, and a C-terminal region containing the STRAD/MO25 interaction region and a CAAX prenylation motif (Cys430). Association with the pseudokinase STRAD and scaffolding protein MO25 stabilizes LKB1 in an active conformation and promotes its cytoplasmic localization. The C-terminal CAAX motif mediates membrane association and contributes to polarity regulation. Loss-of-function mutations within the kinase domain impair AMPK activation and downstream mTOR suppression, linking structural perturbations to metabolic dysregulation and tumor progression. The structural domains of LKB1 and their roles in complex formation and activation are illustrated in Figure 1.
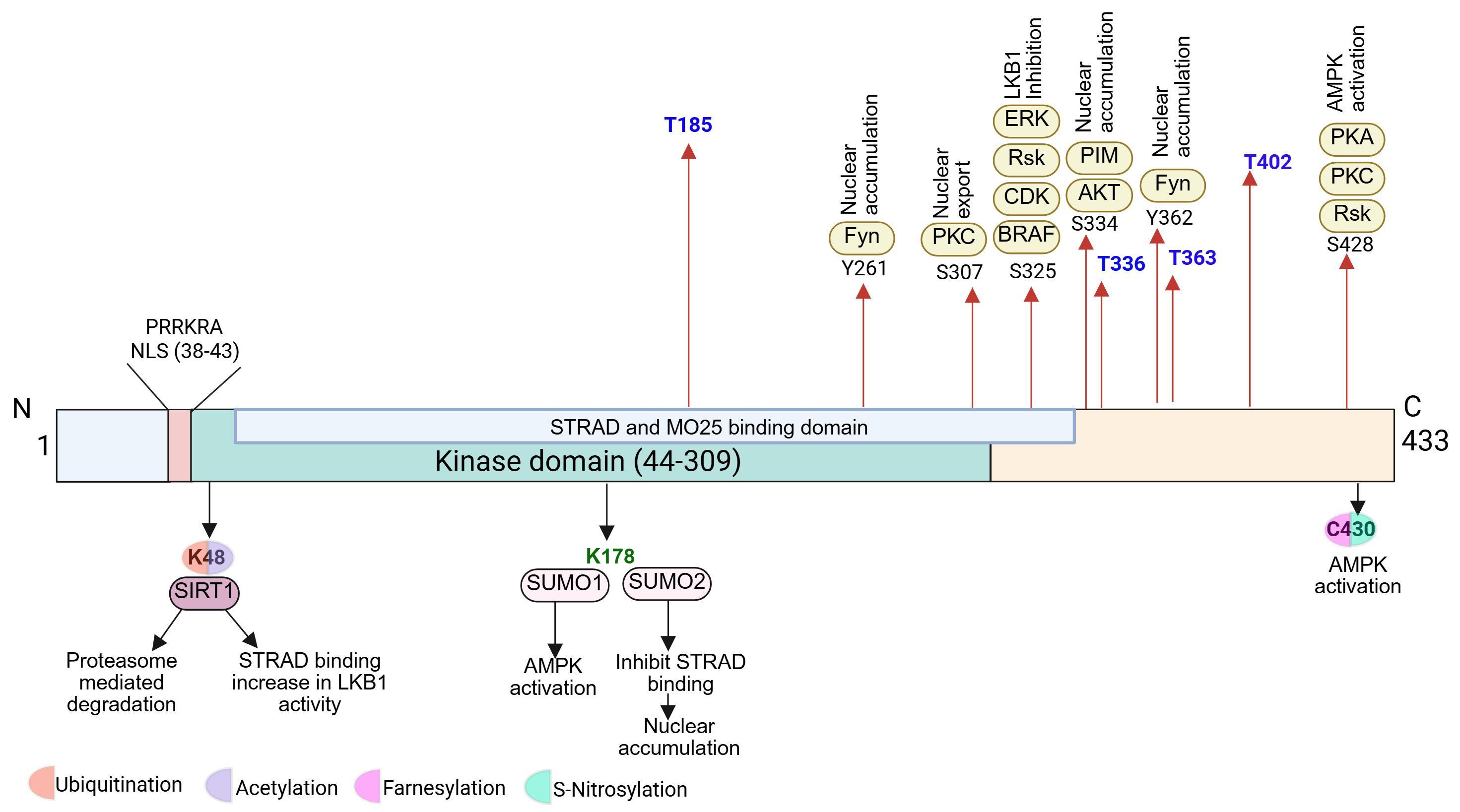
Figure 1. Domains and post-translational modifications sites of LKB1. Schematic representation of the domain organization of LKB1 showing the N-terminal nuclear localization signal (NLS; residues 38–43), the kinase domain (residues 44–309) that mediates binding of STRAD and MO25, and the C-terminal regulatory region. Phosphorylation sites are shown on the upper side. The blue sites represent autophosphorylation. Other sites regulated by kinases including ERK, RSK, CDK, AKT, PKC, PKA, and Fyn regulate LKB1 localization and activity. Other post-translational modifications represented on downside are acetylation, SUMOylation, farnesylation, and ubiquitination.
A fundamental mechanism of LKB1 activation is the formation of the LKB1–STRAD–MO25 complex. In isolation, LKB1 exhibits minimal kinase activity. Full activation requires association with the STRAD and the scaffolding protein MO25. Binding of STRAD induces a conformational rearrangement in LKB1 that exposes its catalytic cleft, while subsequent recruitment of MO25 stabilizes this active configuration. Assembly of this heterotrimeric complex markedly enhances LKB1 kinase activity and facilitates its translocation from the nucleus to the cytoplasm, where many of its downstream substrates, including AMPK, are localized [8–10].
Beyond complex formation, post-translational modifications (PTMs) constitute a major layer of upstream regulation. LKB1 undergoes multiple PTMs, including phosphorylation, acetylation, SUMOylation, and ubiquitination, each of which modulates its function in distinct contexts. Among these, phosphorylation has been most extensively characterized. LKB1 contains several regulatory phosphorylation sites, including Ser431 (human; Ser428 in alternate numbering), Ser325, Thr336, and Thr366, which are targeted by diverse kinases. Notably, phosphorylation of Ser431 by protein kinase A (PKA), protein kinase C (PKC), or p90RSK promotes cytoplasmic localization of LKB1 and has been linked to its roles in cell polarity and energy sensing. Additional regulatory modifications, including acetylation, farnesylation at the C-terminal CAAX motif, and site-specific ubiquitination, further influence LKB1 localization, stability, and complex formation [11]. These major PTMs of LKB1 and their upstream regulatory enzymes are summarized in Figure 1.
Protein stability represents another critical axis of upstream control. LKB1 is predominantly regulated through non-degradative K63-linked ubiquitination, rather than the canonical K48-linked ubiquitination that targets proteins for proteasomal degradation. The E3 ubiquitin ligase Skp2 mediates K63-linked ubiquitination of LKB1, enhancing its stability and reinforcing its interaction with STRAD and MO25. This modification sustains LKB1 signaling output and promotes functional activation rather than protein turnover [12].
Molecular chaperones further contribute to LKB1 regulation by ensuring proper protein folding and stability. The HSP90–CDC37 chaperone complex plays a pivotal role in stabilizing newly synthesized LKB1 and protecting it from misfolding and degradation. Disruption of HSP90 or CDC37 function results in reduced LKB1 stability and diminished kinase activity, underscoring the importance of chaperone-mediated quality control in maintaining LKB1 signaling competence [13].
In addition to these activating mechanisms, LKB1 is subject to negative upstream regulation. Certain proteins suppress LKB1 function by altering its localization or interfering with complex assembly. For instance, the Src family kinase Fyn phosphorylates LKB1 in a manner that promotes its nuclear retention, thereby limiting cytoplasmic activation of AMPK. Similarly, 14-3-3 proteins bind phosphorylated LKB1 and restrict its intracellular mobility, weakening its association with STRAD and MO25 and attenuating downstream signaling [6,14].
Collectively, these upstream regulatory layers integrate structural, biochemical, and spatial cues to fine-tune LKB1 activity. Disruption of any component within this regulatory framework can profoundly reshape LKB1 signaling outputs, providing a mechanistic basis for its context-dependent roles in metabolism, polarity, and cancer progression.
Downstream Regulation of LKB1
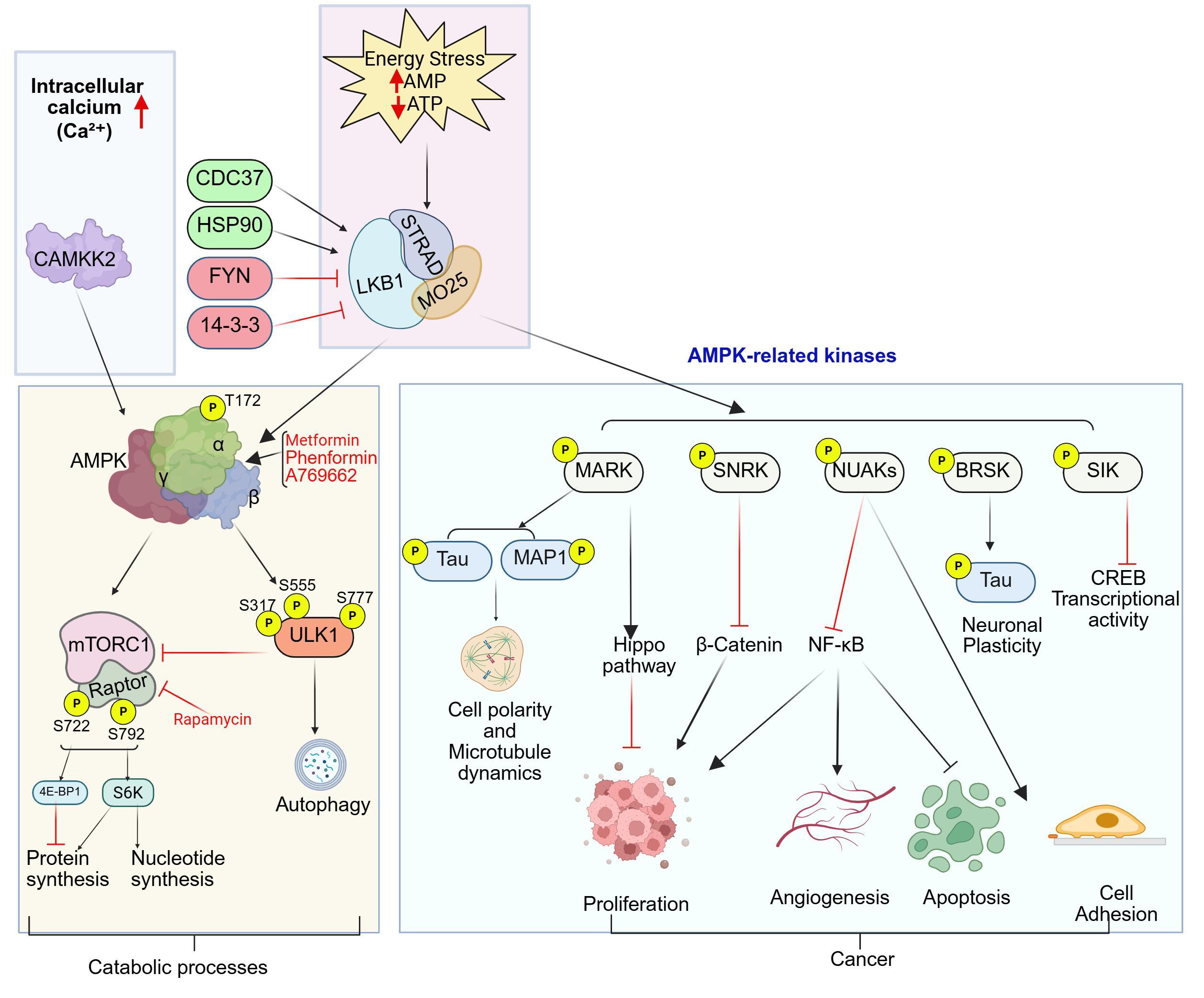
A principal downstream effector of LKB1 is AMP-activated protein kinase (AMPK), a central cellular energy sensor. LKB1 activates AMPK through direct phosphorylation at the conserved Thr172 residue within the AMPK activation loop. Once activated, AMPK functions as a metabolic checkpoint, responding to cellular energy stress characterized by reduced ATP and elevated AMP or ADP levels. Under these conditions, AMPK suppresses anabolic, energy-consuming processes while promoting catabolic pathways that restore energy balance. Specifically, AMPK inhibits lipid and protein synthesis and enhances glucose uptake, glycolysis, fatty-acid oxidation, autophagy, and mitochondrial biogenesis, thereby coordinating a rapid adaptive response to metabolic stress [2,4].
A critical consequence of LKB1–AMPK signaling is the inhibition of the mammalian/mechanistic target of rapamycin complex 1 (mTORC1), a master regulator of cell growth and biosynthesis. AMPK suppresses mTORC1 through multiple mechanisms, including activation of the tumor-suppressor complex TSC1/TSC2, which negatively regulates mTORC1, as well as direct phosphorylation of the mTORC1 component Raptor, leading to reduced complex activity. Inhibition of mTORC1 results in decreased protein translation, ribosome biogenesis, lipid synthesis, and biomass accumulation. Because mTORC1 drives cell growth and proliferation, LKB1-mediated restraint of this pathway constitutes a key mechanism underlying its tumor-suppressive function [15–17].
In addition to mTORC1 regulation, LKB1 promotes cellular survival during metabolic stress by inducing autophagy. This occurs primarily through AMPK-dependent activation of ULK1, initiating autophagosome formation and enabling the recycling of damaged organelles and macromolecules under conditions of nutrient deprivation [18,19]. Beyond AMPK, LKB1 also signals through multiple AMPK-related kinases that regulate cell polarity, cytoskeletal organization, cell adhesion, and tissue architecture, thereby contributing to maintenance of epithelial integrity [7].
Notably, LKB1 signaling exhibits context-dependent and paradoxical effects on cancer. While LKB1-AMPK signaling suppresses anabolic growth pathways and limits proliferation, it simultaneously enhances metabolic flexibility, mitochondrial function, antioxidant defenses, and autophagy. These adaptive responses can allow cancer cells to survive hostile tumor microenvironments characterized by nutrient deprivation, hypoxia, and oxidative stress. In such settings, intact LKB1 signaling may be contextually co-opted to support tumor cell survival and persistence through adaptive stress responses, rather than functioning as a classical growth suppressor [20]. Thus, LKB1 downstream signaling can function either as a growth-restrictive barrier or, under specific metabolic pressures, as a survival-promoting program that facilitates tumor progression.
Post-translational modification further modulates the downstream consequences of LKB1 signaling. SUMOylation of LKB1 reduces its ability to interact with STRAD, resulting in increased nuclear retention and diminished cytoplasmic localization [18,21]. Nuclear-sequestered LKB1 is unable to efficiently activate AMPK and may instead engage alternative nuclear functions. In certain cancer contexts, this shift toward nuclear LKB1 activity has been associated with enhanced cellular stress tolerance rather than growth suppression [18,20–22]. Consequently, LKB1 cannot be viewed as a uniformly acting tumor suppressor. Depending on its post-translational modifications, subcellular localization, and metabolic context, LKB1 may inhibit tumor growth, exert neutral effects, or, in select environments, actively support tumor cell survival.
In addition to AMPK, LKB1 activates a family of AMPK-related kinases, including MARK, NUAK, BRSK, SIK, and SNRK, which extend LKB1 signaling into diverse cellular processes relevant to cancer biology. These kinases regulate cytoskeletal organization through substrates such as Tau and MAP1, modulate transcriptional programs via CREB, and intersect with major oncogenic and inflammatory pathways including Wnt/β-catenin and NF-κB signaling. Although LKB1 represents the principal upstream kinase responsible for AMPK activation, AMPK can also be activated independently through Ca²+-dependent signaling mediated by CAMKK2, while its activity is counterbalanced by phosphatases such as PP2A and P2C that dephosphorylate the activating Thr172 residue. Through these integrated regulatory inputs, LKB1 coordinates cellular energy sensing with translational control, autophagy induction, transcriptional reprogramming, and cytoskeletal remodeling via both AMPK-dependent and AMPK-related kinase pathways. The coordinated interactions among LKB1, AMPK, mTORC1, ULK1, TSC2, Raptor, and associated signaling nodes are summarized in Figure 2.
Methodological Advances Informing LKB1 Signaling in Cancer
Recent genome-wide CRISPR–Cas9 screens have been pivotal in defining context-specific dependencies created by LKB1 loss, uncovering compensatory kinase and stress-response pathways, particularly selective reliance on AMPK-related kinases such as SIK family members [23]. Complementary phosphoproteomic and post-translational regulatory analyses have revealed dynamic rewiring of LKB1–AMPK–mTOR signaling and cytoskeletal pathways under metabolic stress, highlighting how LKB1 output is modulated beyond genetic alteration alone [17,19]. Emerging single-cell and spatial profiling approaches are further resolving lineage- and state-specific consequences of LKB1 dysregulation, including intratumoral heterogeneity in metabolic adaptation, immune exclusion, and therapeutic resistance [24]. Together, these methodological advances have reframed LKB1 from a linear tumor-suppressor pathway to a contextually modulated signaling network with direct translational relevance.
LKB1 in Cancer
Beyond its canonical role as a tumor suppressor, LKB1 pathway dysregulation has emerged as a source of actionable therapeutic vulnerabilities. Recently CRISPR-based functional genomic screens have demonstrated that loss of LKB1 signaling reprograms cellular stress responses and creates selective dependencies on specific downstream kinases and adaptive pathways [23]. Rather than conferring uniform resistance, LKB1 deficiency can render cancer cells uniquely susceptible to targeted perturbations.
In uveal melanoma, combined loss of LKB1 and the AMPK-related kinase SIK2 has been shown to sensitize tumor cells to calcium dysregulation and oxidative stress, unveiling metabolic and redox liabilities that can be therapeutically exploited [25]. Similarly, in acute myeloid leukemia, leukemic cells exhibit a distinct dependency on SIK3-mediated transcriptional programs, where pharmacological inhibition induces selective leukemic cell death while sparing normal hematopoietic progenitors [26]. These observations highlight that downstream LKB1 signaling nodes can become critical survival determinants in LKB1-deficient contexts.
Collectively, these findings challenge the traditional view of LKB1 loss as solely a marker of tumor aggressiveness. Instead, they support a more nuanced model in which LKB1 inactivation rewires cellular dependencies, exposing context-specific vulnerabilities that may be leveraged for precision oncology. Therapeutic strategies targeting these compensatory pathways hold promise for exploiting the metabolic and stress-response fragilities of LKB1-deficient tumors [23].
Therapeutic Implications of LKB1 Dysregulation
Dysregulation of LKB1 (STK11) signaling reshapes tumor cell dependencies, particularly in LKB1-deficient cancers. Because direct pharmacologic restoration of LKB1 is not currently feasible, therapeutic strategies focus on targeting downstream signaling consequences and compensatory stress-response programs that arise following LKB1 loss [2].
One key consequence of LKB1 deficiency is increased reliance on alternative kinase networks, especially salt-inducible kinases (SIKs). Functional genomic studies demonstrate that LKB1 loss can generate selective dependency on SIK family members (SIK1/2/3), which sustain transcriptional programs required for tumor survival in NSCLC, uveal melanoma, and acute myeloid leukemia [23]. In parallel, impaired LKB1 signaling may increase susceptibility to redox imbalance and calcium stress, revealing metabolic stress sensitivities that can be therapeutically exploited [25]. In NSCLC, LKB1 loss is additionally associated with immune exclusion and primary resistance to immune checkpoint inhibitors (ICI), supporting combination strategies that integrate checkpoint blockade with metabolic or stress-modulating approaches rather than ICI monotherapy [24]. The AMPK–mTOR axis represents another relevant node: despite disruption of canonical AMPK activation, mTORC1 hyperactivation frequently persists, providing a rationale for pathway inhibition in selected LKB1-altered tumors [2,17].
Pharmacologic approaches under investigation include mTOR inhibitors (everolimus, temsirolimus, rapamycin), metabolic modulators such as biguanides (metformin, phenformin), glutaminase inhibitors (CB-839), glycolysis inhibitors (2-deoxyglucose), MEK inhibitors targeting KRAS-driven signaling (trametinib, selumetinib), and immune checkpoint inhibitors. These strategies are summarized in Figure 2.
Collectively, LKB1 alteration serves as an indicator of downstream pathway dependency that may guide rational combination therapy, rather than as a direct drug target.
LKB1 Loss as a Driver of Immune Evasion and Therapy Resistance in NSCLC
Compelling clinical and translational evidence in non-small cell lung cancer (NSCLC) indicates that LKB1 loss is a major determinant of immune evasion and resistance to immune checkpoint blockade. Seminal work by Skoulidis et al. demonstrated that KRAS–LKB1 co-mutant tumors are characterized by an immunologically “cold” tumor microenvironment, marked by profound immune exclusion and defective interferon signaling, which together underpin primary resistance to PD-1/PD-L1–directed therapies [24].
Mechanistically, Hollstein et al. revealed that deletion of LKB1 disrupts AMPK signaling and downstream SIK1/SIK3 kinase activity, leading to transcriptional reprogramming that promotes tumor growth, metastatic potential, and immune suppression within the tumor microenvironment [23]. These molecular alterations impair antitumor immune surveillance and reinforce resistance to immunotherapy.
Clinically, loss or reduced expression of LKB1, as assessed by immunohistochemistry, correlates with inferior survival outcomes in patients with NSCLC, further validating its role as a prognostic and predictive biomarker [27]. Collectively, these findings establish LKB1 deficiency as a central node linking metabolic dysregulation, immune escape, and therapeutic resistance, and highlight the urgent need for alternative or combinatorial treatment strategies in this molecular subset of NSCLC.
Germline and Early Tumor Suppressor Role of LKB1 in Gastrointestinal and Pancreatic Cancers
The tumor-suppressive role of LKB1 was first established through the identification of germline mutations in Peutz–Jeghers syndrome, an inherited disorder characterized by mucocutaneous pigmentation, hamartomatous gastrointestinal polyps, and a markedly elevated lifetime risk of gastrointestinal malignancies [1]. These observations provided the earliest clinical evidence that LKB1 functions as a gatekeeper of epithelial homeostasis in the gastrointestinal tract.
Subsequent mechanistic insights from genetically engineered murine models demonstrated that pancreas-specific deletion of LKB1 cooperates synergistically with oncogenic KRAS to drive pancreatic tumorigenesis. Loss of LKB1 markedly accelerates the progression of pancreatic intraepithelial neoplasia (PanIN) through dysregulated mTORC1 activation, thereby facilitating early neoplastic transformation and disease progression [28]. These findings position LKB1 loss as a critical early permissive event in pancreatic carcinogenesis, lowering the threshold for KRAS-driven malignant progression.
Collectively, germline and tissue-specific genetic studies underscore LKB1 as a foundational tumor suppressor in gastrointestinal and pancreatic cancers, with its loss predisposing epithelial cells to oncogenic signaling, metabolic dysregulation, and accelerated neoplastic evolution.
Context-Dependent Role of LKB1 in Hepatocellular Carcinoma and Cholangiocarcinoma
In contrast to its canonical tumor-suppressive function, LKB1 exhibits marked context-dependent behavior in hepatobiliary malignancies. In hepatocellular carcinoma (HCC), paradoxically elevated LKB1 expression has been associated with adverse clinical outcomes, suggesting AMPK-independent adaptive stress–response functions that may be co-opted by tumor cells, rather than true oncogenic activity [22]. These observations underscore that LKB1 remains a tumor suppressor at the genetic level, but its signaling outputs in HCC reflect contextual co-option of stress-adaptive pathways, rather than oncogenic conversion.
Conversely, cholangiocarcinoma is frequently characterized by epigenetic silencing of LKB1, resulting in disruption of the AMPK–mTOR signaling axis and consequent enhancement of anabolic growth programs, invasive behavior, and aggressive tumor phenotypes [29]. Loss of LKB1 in this context aligns with its traditional tumor-suppressor role, reinforcing uncontrolled cellular proliferation and metabolic reprogramming.
Together, these contrasting patterns highlight the dual and tissue-specific functions of LKB1 in hepatobiliary cancers. They emphasize that the biological and clinical consequences of LKB1 signaling are dictated by cellular lineage, epigenetic landscape, and metabolic context, with important implications for biomarker interpretation and therapeutic targeting.
LKB1 Loss and Metastatic Reprogramming in Colorectal and Breast Cancers
Loss of LKB1 is increasingly recognized as a driver of metastatic reprogramming across multiple epithelial malignancies. In colorectal cancer, CRISPR-mediated deletion of LKB1 enhances tumor cell motility and invasive potential through activation of TNIK-driven actin cytoskeletal remodeling, linking LKB1 deficiency directly to altered cell migration dynamics and metastatic competence [30].
Similarly, in HER2-positive breast cancer models, LKB1 loss cooperates with ErbB2 signaling to promote metabolic reprogramming and induction of epithelial–mesenchymal transition (EMT). This process is accompanied by disruption of epithelial polarity and loss of cell–cell adhesion, key prerequisites for local invasion and distant metastatic dissemination [31,32]. Through these coordinated effects on cytoskeletal organization, metabolism, and polarity, LKB1 deficiency facilitates the acquisition of metastatic traits across distinct tissue contexts.
Collectively, these findings position LKB1 as a critical suppressor of metastatic progression, whose loss enables epithelial cancers to overcome structural and metabolic barriers to dissemination.
Invasion and Stress Response in Melanoma
In melanoma, loss of LKB1 (STK11) potentiates invasive and migratory behavior in the setting of both oncogenic and environmental stress. Experimental models demonstrate that LKB1 deficiency cooperates with BRAF activation and ultraviolet (UV)–induced stress to drive cytoskeletal reorganization and enhanced cell motility through activation of FAK/SRC signaling pathways [33]. These findings underscore a critical role for LKB1 in constraining stress-induced cytoskeletal dynamics and limiting invasive plasticity in melanoma.
Therapeutic Vulnerabilities in Hematologic and Rare Cancers
Beyond solid tumors, dysregulation of the LKB1 signaling axis generates distinct therapeutic vulnerabilities in hematologic and rare malignancies. Genome-wide CRISPR screening approaches have identified LKB1–SIK2 signaling as a critical regulator of calcium homeostasis and oxidative stress responses in uveal melanoma, revealing exploitable metabolic and redox dependencies in this otherwise therapeutically challenging disease [25].
Similarly, in acute myeloid leukemia (AML), leukemic cells demonstrate a selective dependency on SIK3-driven transcriptional programs. Pharmacologic inhibition of SIK3 induces preferential leukemic cell death while sparing normal hematopoietic progenitors, highlighting a potential therapeutic window and underscoring the translational relevance of targeting LKB1-associated kinase networks in hematologic malignancies [26].
Collectively, these findings extend the relevance of LKB1 pathway dependencies beyond epithelial cancers and support the development of precision strategies targeting context-specific vulnerabilities in rare and hematologic tumors.
Hormonal and Metabolic Regulation in Gynecologic Cancers
In ovarian cancer, dysregulation of the LKB1–AMPK–mTOR axis intersects mechanistically with hormonal signaling to coordinate metabolic and proliferative outputs. Under physiological conditions, LKB1 activates AMPK, which suppresses mTORC1 through phosphorylation of TSC2 and Raptor, thereby restraining anabolic growth. Loss of LKB1 removes this checkpoint, leading to sustained mTORC1 activation that converges with estrogen receptor (ER)– and gonadotropin-driven PI3K/AKT signaling to amplify translational and biosynthetic programs.
Hyperactive mTORC1 enhances S6K- and 4E-BP1–dependent protein synthesis, including cyclin D1 and other hormone-responsive regulators, while reduced AMPK activity relieves inhibition of SREBP-1c, promoting lipogenesis and cholesterol synthesis required for membrane expansion and steroidogenesis. Concurrent stabilization of HIF-1α and altered FOXO3 activity shift transcription toward glycolytic metabolism and stress tolerance. Crosstalk with insulin/IGF-1 signaling further reinforces PI3K/AKT activation, establishing a feed-forward loop in which metabolic rewiring and endocrine signaling synergize to drive tumor progression [34].
LKB1 in Pituitary Tumors: An Illustrative Knowledge Gap in Endocrine Oncology
Unlike many epithelial and hematologic malignancies, human pituitary tumors remain largely unexplored with respect to LKB1 signaling, representing a notable knowledge gap in endocrine oncology. At present, there is no direct evidence implicating LKB1 as a driver, tumor suppressor, or major molecular modulator in pituitary adenomas or carcinomas. Interpretation of LKB1 function in this context is constrained by the extreme rarity of pituitary carcinomas and the limited depth of molecular profiling studies in pituitary adenomas, which comprise the overwhelming majority of pituitary neoplasms.
Several biological features may explain the apparent divergence of pituitary tumors from other LKB1-driven cancers. Pituitary adenomas are typically characterized by a low proliferative index, relative genomic stability, and distinct lineage-specific transcriptional and metabolic wiring, which may reduce selective pressure on LKB1-dependent stress and metabolic checkpoints. Moreover, pituitary cells operate within a unique endocrine milieu, where hormonal regulation and feedback loops may supersede canonical metabolic growth controls governed by the LKB1–AMPK axis.
To date, the prevalence of LKB1 mutations or loss of LKB1 expression in non-metastatic pituitary adenomas remains undefined, and no dedicated mechanistic studies using pituitary-derived cell lines or in vivo models have been reported. Consequently, robust conclusions regarding the role of LKB1 in pituitary tumorigenesis cannot yet be drawn.
Nevertheless, an isolated case report of an ACTH-secreting pituitary carcinoma harboring an LKB1 mutation, with therapeutic response to mTOR pathway inhibition, highlights the potential clinical relevance of LKB1 pathway alterations in selected aggressive pituitary neoplasms [35]. Although preliminary, this observation suggests that LKB1 mutation or expression profiling may warrant consideration in rare, invasive, or treatment-refractory pituitary tumors.
Future progress in this area will require systematic multi-omics approaches, including genomic, epigenomic, and phosphoproteomic profiling, as well as single-cell analyses to resolve lineage- and state-specific signaling dependencies. Given the rarity of malignant pituitary tumors, international rare-tumor consortia and collaborative datasets will be essential to define whether LKB1 signaling contributes meaningfully to pituitary tumor biology or represents a true exception within endocrine malignancies.
A Unifying Model of Context-Dependent LKB1 Signaling in Cancer
Collectively, available evidence supports a context-dependent model of LKB1 signaling in which its biological output is dictated by the integration of subcellular localization, post-translational regulation, lineage specificity, and tumor microenvironmental stress, rather than by linear pathway activity alone [2]. In its canonical state, cytoplasmically localized LKB1 restrains malignant progression by enforcing metabolic checkpoints, suppressing mTORC1-driven anabolic growth, maintaining epithelial polarity, and supporting immune surveillance, thereby functioning as a classical tumor suppressor [6]. Conversely, under conditions of nutrient deprivation, hypoxia, oxidative stress, or therapeutic pressure, intact or rewired LKB1 signaling can promote metabolic flexibility, autophagy, and stress tolerance, enabling tumor cell survival despite growth constraint [20]. The directionality of LKB1 signaling is critically shaped by subcellular localization and post-translational modifications, particularly SUMOylation-driven nuclear retention, which limits AMPK activation and favors alternative stress-adaptive programs [21]. In parallel, tissue lineage and downstream kinase dependency further determine whether LKB1 loss or preservation results in growth suppression, immune evasion, metastatic competence, or therapeutic vulnerability. Thus, LKB1 should not be viewed as a uniformly acting tumor suppressor across all contexts; rather, its downstream signaling outputs are shaped by metabolic and environmental stress, and this signaling plasticity underlies its paradoxical roles across cancer types.
Conclusion
LKB1 is a central regulator of cellular energy homeostasis, growth, and polarity, acting primarily through the AMPK–mTORC1 axis and related kinase networks. Its biological effects are highly context dependent, shaped by post-translational modifications, subcellular localization, and metabolic stress, allowing LKB1 to function either as a tumor suppressor or, under specific conditions, as a facilitator of tumor survival and adaptation. Across cancers, LKB1 dysregulation influences proliferation, immune evasion, metastasis, and therapeutic response, while emerging data reveal exploitable vulnerabilities in LKB1-deficient states. The paucity of evidence in select malignancies, particularly pituitary tumors, highlights important knowledge gaps. A deeper mechanistic understanding of LKB1 signaling will be critical for refining its utility as a biomarker and for developing context-specific therapeutic strategies.
|
Cancer Type |
LKB1 Context |
Common Co-Alterations |
Dominant Signaling Axis |
Biologic/Clinical Consequence |
Therapeutic Relevance |
|
NSCLC |
Loss-of-function |
KRAS, TP53, KEAP1 |
↓AMPK → ↑mTORC1; STING suppression |
Immune-cold phenotype; metastasis; ICI resistance |
Combine immunotherapy with metabolic targeting |
|
PDAC |
Cooperative loss with KRAS |
KRAS |
Enhanced KRAS-driven metabolic rewiring; ↑mTORC1 |
Accelerated tumor growth and progression |
mTOR and metabolic pathway targeting |
|
Cervical cancer |
Viral-mediated degradation |
HPV (E6/E7) |
Loss of AMPK restraint; polarity disruption |
Increased invasion and transformation |
mTOR vulnerability |
|
Melanoma |
Reduced/functionally suppressed |
BRAF, NRAS |
Impaired stress checkpoint; FAK/SRC activation |
Enhanced invasion under stress |
Target stress-response/SRC pathways |
|
Breast cancer (TNBC/HER2+) |
Loss-of-function |
TP53, PIK3CA, ERBB2 |
mTOR activation; polarity loss |
EMT, metastasis, aggressive phenotype |
mTOR/metabolic targeting |
|
Endometrial cancer |
Germline STK11 loss (PJS) |
PTEN |
mTOR hyperactivation |
Early tumorigenesis |
mTOR inhibition in selected cases |
|
Hepatocellular carcinoma |
Dual/context-dependent |
TP53, CTNNB1 |
Early AMPK suppression; late stress adaptation |
Stage-dependent effects (suppressive → adaptive) |
Context-dependent biomarker |
|
Cholangiocarcinoma |
Epigenetic silencing |
Wnt alterations |
Disrupted AMPK–mTOR axis |
Increased proliferation and invasiveness |
mTOR/Wnt targeting |
|
Colorectal cancer |
Loss-of-function |
KRAS, APC |
Loss of energy sensing; cytoskeletal remodeling |
Adenoma progression; metastasis |
mTOR/cytoskeletal targeting |
|
Prostate cancer |
Reduced expression |
PTEN |
PI3K/AKT/mTOR activation |
Enhanced survival signaling |
PI3K/AKT/mTOR inhibitors |
|
AML (hematologic) |
Rare mutation; pathway dependency |
MYC |
SIK kinase dependency |
Metabolic and transcriptional survival |
Emerging SIK inhibitors |
|
Uveal melanoma |
LKB1–SIK2 dependency |
GNAQ/GNA11 |
ROS and Ca²+ stress regulation |
Stress-adaptive survival |
SIK2/redox targeting |
|
This table summarizes how LKB1 functional status, co-occurring genomic alterations, and dominant signaling outputs shape tumor behavior across malignancies. Loss or suppression of LKB1 commonly attenuates AMPK activation and enhances mTORC1 signaling, promoting metabolic reprogramming, immune evasion, and metastatic progression. In select contexts, preserved or rewired LKB1 signaling supports stress adaptation and tumor persistence. In hematologic and rare tumors, LKB1-associated kinase dependencies (e.g., SIKs) represent emerging therapeutic vulnerabilities. |
|||||
Declaration of Conflict of Interest
The author declares no conflict of Interest. All authors have read and approved the final version of the manuscript.
Author Contribution
VK and AS performed the literature survey and contributed in writing the review. MV, AA, SB, SM, contributed to critical analysis and manuscript revision. PD conceptualized the idea, contributed to critical analysis and supervision of the manuscript. AR has conceptualized the idea and prepared the figures along with VK.
References
2. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009 Aug;9(8):563–75.
3. Sanchez-Cespedes M. The role of LKB1 in lung cancer. Fam Cancer. 2011 Sep;10(3):447–53.
4. Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond). 2008 Sep;32 Suppl 4:S7–12.
5. Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, et al. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003 Jun 16;22(12):3062–72.
6. Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H. LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev. 2009 Jul;89(3):777–98.
7. Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004 Feb 25;23(4):833–43.
8. Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, et al. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003 Oct 1;22(19):5102–14.
9. Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2(4):28.
10. Dorfman J, Macara IG. STRADalpha regulates LKB1 localization by blocking access to importin-alpha, and by association with Crm1 and exportin-7. Mol Biol Cell. 2008 Apr;19(4):1614–26.
11. Collins SP, Reoma JL, Gamm DM, Uhler MD. LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo. Biochem J. 2000 Feb 1;345 Pt 3(Pt 3):673–80.
12. Lee SW, Li CF, Jin G, Cai Z, Han F, Chan CH, et al. Skp2-dependent ubiquitination and activation of LKB1 is essential for cancer cell survival under energy stress. Mol Cell. 2015 Mar 19;57(6):1022–33.
13. Boudeau J, Deak M, Lawlor MA, Morrice NA, Alessi DR. Heat-shock protein 90 and Cdc37 interact with LKB1 and regulate its stability. Biochem J. 2003 Mar 15;370(Pt 3):849–57.
14. Nakano A, Takashima S. LKB1 and AMP-activated protein kinase: regulators of cell polarity. Genes Cells. 2012 Sep;17(9):737–47.
15. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003 Nov 26;115(5):577–90.
16. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008 Apr 25;30(2):214–26.
17. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018 Feb;19(2):121–35.
18. Hu L, Liu M, Tang B, Li Q, Pan BS, Xu C, et al. Posttranslational regulation of liver kinase B1 in human cancer. J Biol Chem. 2023 Apr;299(4):104570.
19. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011 Feb;13(2):132–41.
20. Jeon SM, Hay N. The double-edged sword of AMPK signaling in cancer and its therapeutic implications. Arch Pharm Res. 2015 Mar;38(3):346–57.
21. Zubiete-Franco I, García-Rodríguez JL, Lopitz-Otsoa F, Serrano-Macia M, Simon J, Fernández-Tussy P, et al. SUMOylation regulates LKB1 localization and its oncogenic activity in liver cancer. EBioMedicine. 2019 Feb;40:406–21.
22. Tan X, Liao Z, Liang H, Chen X, Zhang B, Chu L. Upregulation of liver kinase B1 predicts poor prognosis in hepatocellular carcinoma. Int J Oncol. 2018 Nov;53(5):1913–26.
23. Hollstein PE, Eichner LJ, Brun SN, Kamireddy A, Svensson RU, Vera LI, et al. The AMPK-Related Kinases SIK1 and SIK3 Mediate Key Tumor-Suppressive Effects of LKB1 in NSCLC. Cancer Discov. 2019 Nov;9(11):1606–27.
24. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018 Jul;8(7):822–35.
25. Proteau S, Krossa I, Husser C, Guéguinou M, Sella F, Bille K, et al. LKB1-SIK2 loss drives uveal melanoma proliferation and hypersensitivity to SLC8A1 and ROS inhibition. EMBO Mol Med. 2023 Dec 7;15(12):e17719.
26. Tarumoto Y, Lu B, Somerville TDD, Huang YH, Milazzo JP, Wu XS, et al. LKB1, Salt-Inducible Kinases, and MEF2C Are Linked Dependencies in Acute Myeloid Leukemia. Mol Cell. 2018 Mar 15;69(6):1017–27.e6.
27. Sumbly V, Landry I. Unraveling the Role of STK11/LKB1 in Non-small Cell Lung Cancer. Cureus. 2022 Jan 10;14(1):e21078.
28. Morton JP, Jamieson NB, Karim SA, Athineos D, Ridgway RA, Nixon C, et al. LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology. 2010 Aug;139(2):586–97.
29. Wang J, Zhang K, Wang J, Wu X, Liu X, Li B, et al. Underexpression of LKB1 tumor suppressor is associated with enhanced Wnt signaling and malignant characteristics of human intrahepatic cholangiocarcinoma. Oncotarget. 2015 Aug 7;6(22):18905–20.
30. Hu G, Huang N, Zhang J, Zhang D, Wang S, Zhang Y, et al. LKB1 loss promotes colorectal cancer cell metastasis through regulating TNIK expression and actin cytoskeleton remodeling. Mol Carcinog. 2023 Nov;62(11):1659–72.
31. Dupuy F, Griss T, Blagih J, Bridon G, Avizonis D, Ling C, et al. LKB1 is a central regulator of tumor initiation and pro-growth metabolism in ErbB2-mediated breast cancer. Cancer Metab. 2013 Aug 14;1(1):18.
32. Li J, Liu J, Li P, Mao X, Li W, Yang J, et al. Loss of LKB1 disrupts breast epithelial cell polarity and promotes breast cancer metastasis and invasion. J Exp Clin Cancer Res. 2014 Sep 2;33(1):70.
33. Zhang W, Yin L, Song G, Han X, Yin Z, Luo D. LKB1 loss cooperating with BRAF V600E promotes melanoma cell invasion and migration by up-regulation MMP-2 via PI3K/Akt/mTOR pathway. Oncotarget. 2017 Dec 5;8(69):113847–57.
34. Kang J, Gallucci S, Pan J, Oakhill JS, Sanij E. The role of STK11/LKB1 in cancer biology: implications for ovarian tumorigenesis and progression. Front Cell Dev Biol. 2024 Oct 31;12:1449543.
35. Donovan LE, Arnal AV, Wang SH, Odia Y. Widely metastatic atypical pituitary adenoma with mTOR pathway STK11(F298L) mutation treated with everolimus therapy. CNS Oncol. 2016 Oct;5(4):203–9.