Abstract
The protein tyrosine phosphatase SHP2, encoded by PTPN11, functions as a critical signal transduction regulator and interacts with key signaling molecules in both RAS/ERK and PD-1/PD-L1/ BTLA (B- and T-lymphocyte attenuator) pathways. Targeting SHP2 pharmacologically, therefore, may be a promising therapeutic strategy for many RAS-driven cancers. Multiple small molecule inhibitors of SHP2 (SHP2i) are currently in clinical development, both alone and in combinations. SHP2i combination therapies, including those with inhibitors of EGFR, KRASG12C, BRAFV600E, MEK, ERK, CDK4/6 and PD-1, and other combinations not yet explored, represent targeted strategies with great promise for advanced or refractory RAS-dependent solid tumor malignancies. One recent study demonstrates that combined pharmacologic inhibition of SHP2 and MEK is active in models of NF1-deficient malignant peripheral nerve sheath tumors (MPNST). This article, based on the discovery that a wide range of receptor tyrosine kinases (RTK) are upregulated through the adaptive response to MEKi in this RAS-dysregulated tumor type, adds to the growing body of literature in which SHP2 inhibitors have been combined with other targeted therapies in a range of cancer types. To date, most of these reports have focused on signaling and direct cancer cell related effects of these small molecules. Here, we discuss recent advances in the preclinical and clinical development of SHP2 inhibitors, as well as their potential role in immune signaling modulation as a promising cancer-directed strategy.
SHP2 Structure and Role in Human Diseases
The SHP2 phosphatase consists of one protein tyrosine phosphatase catalytic domain (PTP domain), two tandem Src homology 2 (SH2) domains (N-SH2 and C-SH2), and a C-terminal tail with two tyrosine phosphorylation sites (Tyr542 and Tyr580) [1] (Figure 1A). SHP2 activity is normally auto-inhibited by the binding of the N-SH2 domain with the PTP domain [2]. Upon stimulation of growth factors or cytokines, the N-SH2 domain binds to specific phospho-tyrosine residues and induces a conformational change that leads to exposure of the PTP domain and an increase in the catalytic activity [3] (Figure 1B). Phosphorylated Tyr542 interacts intramolecularly with the N-SH2 domain to relieve steady-state inhibition of the phosphatase, whereas phosphorylated Tyr580 stimulates the phosphatase activity by interaction with the C-SH2 domain [4].
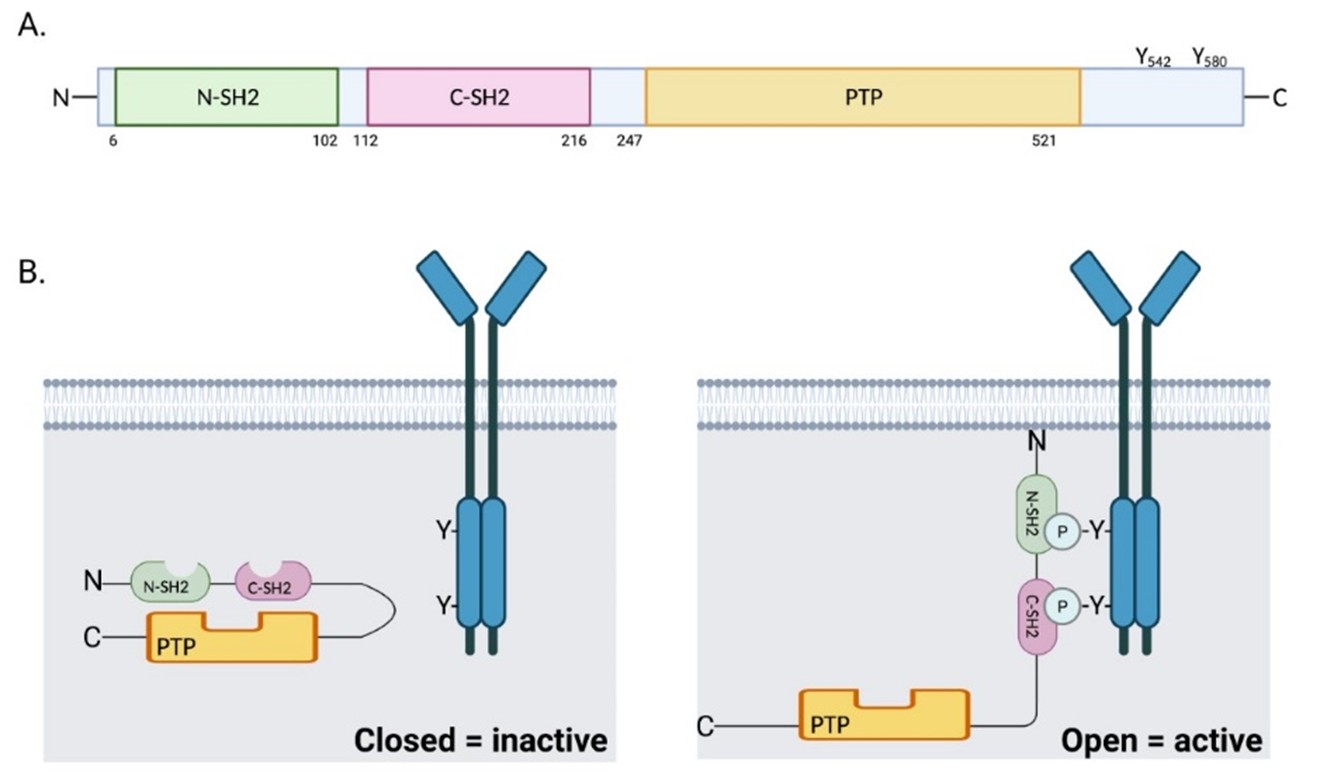
Germline gain of function (GOF) mutations in PTPN11 occur in about 50% of patients with Noonan syndrome [5], which is characterized by abnormal facial features, skeletal malformations, congenital heart disease, short stature and an elevated risk of leukemia and other cancers. In contrast, more than 80% of patients with LEOPARD syndrome (lentigines, EKG abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, and deafness) harbor heterozygous germline inactivating (phosphatase-defective) mutations in PTPN11, despite the overlapping clinical presentations between this syndrome and those with Noonan syndrome [6,7]. Both conditions are associated with an increased risk for malignancies, including leukemia and neuroblastoma [5,8], suggestive of a phosphatase-independent role of SHP2 in cancer pathogenesis. In addition, somatic GOF mutations in PTPN11 are reported in juvenile myelomonocytic leukemia (JMML, 35%), myelodysplastic syndrome (MDS, 10%), sporadic acute myeloid leukemia (AML, 4%) [9] and solid tumors [10], making PTPN11 the first identified proto-oncogene among tyrosine phosphatases [11].
The Role of SHP2 in Dysregulated RTK/ RAS/ERK Signaling in Cancer
SHP2 has been implicated as a major contributor to regulation of RTK/RAS/ERK signaling [12,13], but how exactly the SHP2 phosphatase promotes activation of RAS/ ERK signaling has been controversial. Functional studies demonstrate that SHP2 enhances RAS activation through activated protein tyrosine kinases or cytokine receptors [14]. SHP2 is required for sustained RAS/ERK signaling activation via multiple phosphatase-dependent and -independent mechanisms. First, SHP2 reverses negative regulators of RAS activation, including dephosphorylation and inactivation of Sprouty, as well as inhibition of RAS-GAP recruitment, both negative regulators of RAS activation [14,15]. Second, dephosphorylation of RAS at tyrosine 32, as a direct target of SHP2, increases RASRAF interaction and enhances downstream signaling [16]. Third, SHP2 dephosphorylates Src-regulatory proteins and leads to Src activation, which in turn promotes RAS/ ERK signaling [17]. More recent evidence implicates the role of SHP2 as a scaffolding adaptor providing the major GRB2 binding site (phosphorylated Tyr542), which forms a functional signaling complex containing SHP2/GRB2/SOS/GAB1, to promote RAS activation by its guanine exchange factor (GEF) [17-19]. However, it has been posited that the association of SHP2 with GRB2 is not sufficient for full ERK activation, and that phosphorylated Tyr580 is also required for sustained ERK signaling in response to some growth factors [20].
Considering the emerging role of SHP2 in cancer, discovery of small molecule inhibitors targeting SHP2 has recently gained significant attention. To our excitement, the first SHP2 allosteric inhibitor SHP099 was developed in 2015 [21], enabling further mechanistic studies on SHP2 function in cancer. Compared with the low selectivity/offtarget effect of catalytic/active-site inhibitors targeting SHP2 among other PTP [22], the SHP2 allosteric inhibitor SHP099 potently and selectively inhibits SHP2 activity through stabilization of wild-type SHP2 in the autoinhibited/ closed conformation [23]. The earliest reports identifying novel small molecule inhibitors of SHP2 (SHP099) suggested that these compounds would be most effective in cancer cells driven by a variety of aberrantly regulated RTK (including ERBB2, FGFR2, EGFR and ALK, among others) [13]. Other studies, however, have demonstrated only modest activity of SHP2i as a single agent [24], suggesting that as a class, SHP2i may realize their full potential as cancer therapeutics when given in combination.
SHP2 inhibition offers a promising therapeutic strategy as a means to prevent RTK-driven adaptive and acquired resistance to targeted therapy. SHP2 inhibition enhances the efficacy of tyrosine kinase inhibitors (TKI), such as ALK/EGFR/FGFR inhibitors, in drug-resistant NSCLC and metastatic breast cancer, in which distinct RTK activation mediates adaptive and acquired resistance [25-28]. In addition, SHP2 inhibition (or depletion) also restores sensitivity to ERK signaling inhibition and has additive/synergistic anti-tumor effects when combined with KRASG12C/RAF/MEK inhibitors in multiple models of cancers driven by hyperactivated RAS, including KRASmutant pancreatic, lung and colorectal cancers, KRASWTamplified gastroesophageal cancer, RASWT triple negative breast cancer (TNBC) and ovarian cancers, NRAS-mutant neuroblastoma, NF1-deficient MPNST, and BRAF-mutant colon and thyroid cancers, among others, through blocking signal transduction from most RTK that are reactivated through loss of negative feedback following ERK pathway inhibition [19,24,26,29-38].
The Role of SHP2 in Modulating Immune Signaling Pathways
Included in the diverse roles of SHP2 is that of modulating immune signaling responses through interactions with PD-1/PD-L1 receptor/ligand signaling. SHP2 is considered a central molecule downstream of inhibitory immune receptors. Inhibitory receptors are expressed by immune cells and regulate their function in diverse contexts. Upon binding of PD-L1, PD-1 becomes phosphorylated at its immunoreceptor tyrosine-based inhibitory motif (ITIM) and immune receptor tyrosine-based switch motif (ITSM), and then ITSM binds C-SH2, recruiting SHP2 to PD-1; while ITIM binds N-SH2, displacing it from the catalytic pocket and activating SHP2 [39]. Extensive investigation has been carried out to identify the mechanistic contribution of PD-1/PD-L1/SHP2 to T-cell inactivation. One recent study demonstrated that the costimulatory receptor CD28 is preferentially dephosphorylated and inactivated by SHP2 in the PD-1/PD-L1/SHP2 microcluster, thereby leading to T-cell inactivation mediated by PD-1 [40] and promoting immune escape by cancer cells (Figure 2). However, the role of SHP2 in PD-1 signaling was challenged by another study, which found that SHP2 is dispensable for establishing T-cell exhaustion as well as for PD-1 signaling in vivo, supported by the evidence that specific SHP2 depletion in CD8+ T lymphocytes only moderately improves their proliferation and results in decreased polyfunctionality and compromised cytotoxicrelated cytokine production upon chronic infection. Mice with SHP2-deficient T-cells showed no significant improvement in controlling immunogenic tumors and demonstrated responses to α-PD-1 treatment similar to controls, suggesting the existence of redundant or alternative mechanisms complementing SHP2/PD-1 signaling [41]. Furthermore, another report provides evidence supporting the existence of compensatory mechanisms by which 1) PD-1 selectively recruits SHP2; and 2) BTLA preferentially recruits SHP1, and both suppress T cell signaling. Intriguingly, PD-1 and BTLA potently inhibit T-cell proliferation and cytokine production in SHP1/2 double-deficient primary T-cells, suggesting that PD-1 and BTLA suppress T-cell signaling only partially through SHP1/2 [42], and SHP2 depletion or inhibition limits the durability of PD-1 signaling. Further studies are needed to investigate the bypass mechanisms and design combinatorial strategies to target T-cell exhaustion. SHP2 has also been implicated in the response to IL-2 and IL- 15 [43-48]. IL-2 is essential for regulatory, effector CD4+, and effector CD8+ T-cells. IL-15 is critical to the survival of memory CD8+ T-cells and for development, survival, and activation of NK cells, two cytotoxic immune cell subsets which are central to immunity against intracellular pathogens and cancers.
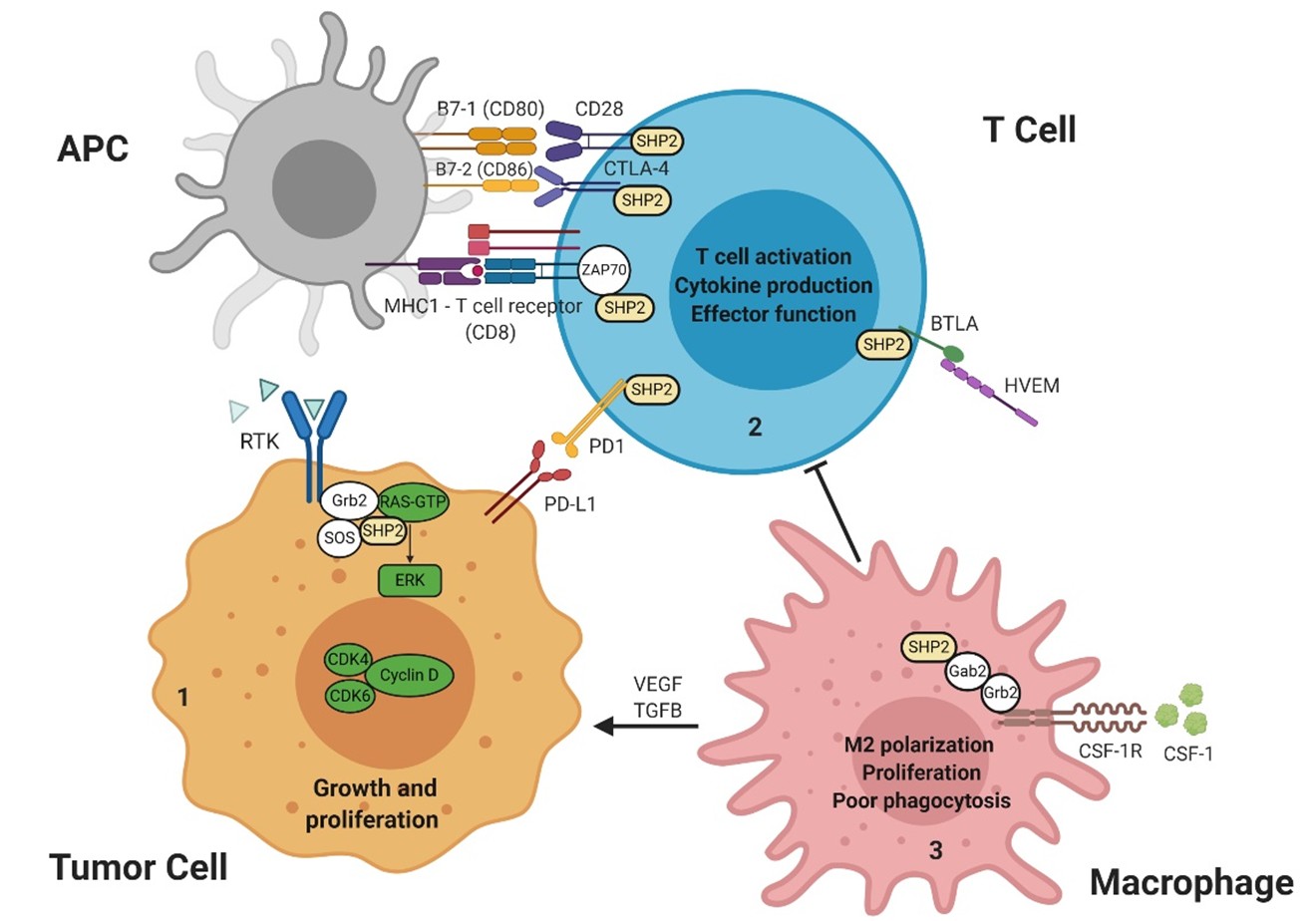
RTK: Receptor Tyrosine Kinase; SHP2: Src Homology region 2 domain-containing Phosphatase-2; Grb2: Growth factor receptor-bound protein 2; Gab2: Grb2 associated binding protein 2; SOS: Son of sevenless, RAS guanine nucleotide exchange factor; GTP: Guanosine-5'-triphosphate; ERK: Extracellular signal–regulated kinase; CDK4/6: Cyclin-dependent kinase 4/6; PD-1: Programmed cell Death-1; PD-L1: Programmed cell Death Ligand 1; APC: Antigen Presenting Cell; MHC1: Major Histocompatibility Complex 1; B7-1/2: Peripheral membrane proteins 1 and 2; CD28: Cluster of Differentiation 28; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; ZAP70: Zeta chain of T cell receptor Associated Protein kinase 70; BTLA: B- and T-lymphocyte-associated protein; HVEM: Herpes Virus Entry Mediator; CSF-1: Colony Stimulating Factor 1; CSF-1R: Colony Stimulating Factor 1 Receptor.
In contrast to studies on T cell-specific SHP2 depletion, however, an abundance of evidence points to additional roles of SHP2 in immunomodulation beyond PD-1 checkpoint signaling in T-lymphocytes. Preclinical studies support the role of SHP2 inhibition in adaptive and innate immunity. SHP2 inhibition provokes increases in intratumoral CD8+ T-lymphocytes and tumor-associated B-lymphocytes, augmenting anti-tumor immunity [38,49]. Recent findings have emerged regarding the role of SHP2 in modulating the myeloid compartment as well [26,38,50]. Myeloid cells are the most abundant white blood cells in the human body and they are present practically in all tissues. They are key regulators of tissue homeostasis and tumor microenvironments [51]. Tumor-associated macrophages (TAM– M2 macrophages) that infiltrate tumor tissues are driven by cancer-derived cytokines to acquire a polarized immunosuppressive phenotype, which in turn de-activates the T cell compartment [52-54]. The salient feature of these cells is their ability to inhibit T cell function and their high density in the tumor microenvironment (TME) is associated with poor prognosis and survival across multiple cancer types [51,55,56]. Moreover, tumor-associated myeloid cell infiltration is associated with clinical resistance to immunotherapy [57]. M2 macrophages exhibit potent T-cell suppressive phenotypes in vitro and in vivo [52,58,59]. SHP2 inhibition in RASdriven cancers results in depletion of immunosuppressive M2 macrophages through attenuation of CSF1R signaling [26,50], which is essential for T-cell suppression by immunosuppressive TAM [60] within the TME. These findings raise the possibility that SHP2 inhibition relieves T cell suppression largely through reduction of M2 macrophages via CSF1R signaling, and partially through PD-1 signaling, where SHP2 appears dispensable due to functionally redundant mechanisms. Interestingly, despite modest effect of PD-1 blockade on M2 TAM, further reduction of M2 macrophages is elicited with combined SHP2i and PD-1 blockade through an undefined mechanism [26,50], which may be the potential basis for additive anti-tumor activity of combined SHP2i and anti-PD-1. Furthermore, combined SHP2i and anti-PD-1 treatment also demonstrates synergistic effects on colon cancer models that are sensitive to checkpoint blockade [49,50]. In models of pancreatic ductal adenocarcinoma (PDAC), a cancer type with limited response to checkpoint blockade, the antitumor immunity and efficacy of SHP2 and KRASG12C inhibition can be enhanced by anti-PD-1 [38,61], implicating a combination benefit of immune checkpoint inhibitors (ICI) in PDAC treatment. A growing understanding of the mechanism of action of SHP2 in these immune regulating cascades is therefore relevant and timely. Overall, the effects of SHP2i on the TME remain to be clarified and could certainly have implications for the development of synergistic combinations in antitumor therapy.
The Effects of RAS Pathway Inhibition on Immune Modulation
RAS/ERK signaling activation is associated with significantly reduced levels of tumor-infiltrating lymphocytes (TIL), thereby potentially facilitating immune evasion by the tumor cells [62]. ERK signaling inhibition therefore alleviates a local immunosuppressive phenotype, and promotes TIL homing to the tumor [63]. Distinct effects on immune cell modulation in the TME due to inhibition of different nodes in the RAS/ERK signaling pathway have been observed. Similar to the modulatory effect of SHP2i on TME, KRASG12Ci also improves anti-tumor immunity in KRAS-mutant cancers through an increase in CD8+ T-cells and cytokine production [38,64], and decrease in immunosuppressive CD4+ T-cells and CD11b+ myeloid cells [38]. Likewise, BRAFi induces a favorable TME through multiple mechanisms, enhancing T-cell specific recognition of BRAFV600-mutant melanoma in vitro and in patients, with no obvious effects on lymphocyte function [65-69]. MEKi can also improve tumor immunogenicity via enhancement of MHC-1 expression [70]. However, in contrast to selective mutation-specific inhibitors that exclusively suppress RAS/ERK pathway in tumor cells, the effects of broad ERK signaling inhibition using MEKi on T-cell function have been perplexing and somewhat contradictory, as T-cell immune response is at least partially dependent on RAS/ERK signaling downstream of the TCR. Two early reports revealed that MEKi impairs peripheral blood derived T-cell function through reduction in proliferation and cytokine secretion [65,71]. Nevertheless, this notion was supplemented with followup studies demonstrating that MEKi promotes recruitment of TIL and increases antigen specific CD8+ T cells within the tumor; whereas markedly inhibits naïve CD8+ T-cell priming which rebounds after the early onset of the suppression, in tumor-draining lymph nodes [72,73]. A recent study extended these observations and further explored the mechanistic basis of the immunomodulatory effects of MEKi on TME. MEKi treatment reprograms naïve CD8+ T-cells into stem cell-like memory T-cells with potent antitumor activity, through cell cycle inhibition and metabolic enhancement [74]. Additionally, given the potential inhibitory effect of MEKi on T cell function, an alternative regimen with intermittent, rather than continuous, exposure to MEKi was found to induce T-cell activation and anti-tumor immunity [75]. Despite this apparent paradox, MEKi still further enhances anti- PD-1/PD-L1/CTLA4 immunotherapy through enhanced T-cell activation [62,63,72,75,76]. Based on the effects of SHP2i and the equivocal impact of MEKi on innate and adaptive immunity, consequently, additional studies are needed to further explore the effectiveness and toxicity of combined MEK and SHP2 inhibition, either alone or in combination with immunotherapy. These studies will need to be carefully optimized, using immune-competent syngeneic and/or genetically engineered mouse models, careful study of dose and treatment schedules to achieve optimal efficacy and minimize toxicity, and ultimately select combinations and regimens that hold the most promise for achieving superior anti-tumor responses.
The Combination Partner SHP2 as a Promising Co-Target in MPNST and Other Cancers
NF1 and CDKN2A tumor suppressor losses are genomic hallmarks found in the majority of human MPNST (90%, and 60-80%, respectively) [77]. As loss of NF1 is a major driver of RAS-ERK signaling in many cancers, MEK inhibition seems a logical focus for the design of combination strategies in MPNST and other tumors with loss of NF1. MEKi alone, however, has limited anti-tumor activity, leading to the notion that combinations that target the adaptively upregulated molecules that emerge upon loss of ERK-induced negative feedback should be effective. Our data suggested that a number of tyrosine and serine/threonine kinases become adaptively upregulated in response to MEKi, leading to challenges in the design of MEKi plus TKI combination strategies, as genomic or other predictive biomarkers to identify the adaptively changed RTK are not readily discernible [24,32]. In order to overcome this challenge, we posited that inhibition of the central node of convergence between the RTK and RAS recruitment to the membrane and activation – SHP2 – would serve as a viable strategy. Indeed, in vitro and in vivo analysis of the MEKi/ SHP2i combination demonstrated more profound and durable inhibition of ERK signaling, improved anti-proliferative effects and synergy in vivo [24]. The success of this combination in patients with MPNST remains to be seen.
In addition to CDKN2A deletion, hyperactivation/ and/or upregulation of cyclin dependent kinases (CDK) and D-type cyclins, leading to inactivation of the RB1 tumor suppressor, occurs in the majority of MPNST, and suggests that small-molecule inhibitors of CDK4/6 (CDK4/6i) may be an additional therapeutic strategy [78]. However, CDK4/6i elicits a primarily cytostatic phenotype and has limited efficacy as a single agent, due to early adaptive upregulation of cyclin D1 and subsequent CDK2 hyperactivation, and other bypass mechanisms such as E2F amplification [79-83]. The CDK4/6i ribociclib improves the efficacy of SHP2 inhibition in RTK-driven and a subset of KRAS-mutant NSCLC and colorectal cancer models, and is equally efficacious as the combination of MEKi and SHP2i [26]. Despite similar anti-tumor activity, combined SHP2i and CDK4i may be a better tolerated regimen, with a wider therapeutic index, given the preliminary toxicity data reported for MEKi and SHP2i combinations [84]. According to our understanding, combined SHP2i and CDK4/6i may provide a viable therapeutic approach not yet tested in immunocompetent models of MPNST.
Macrophage infiltrates are abundant in neurofibromas and MPNST, accounting for nearly half of the cells within a tumor [85]. The tumor promoting and immunosuppressive M2 macrophages are dependent on CSF1R signal and CSF1R+ TAM correlate with poor survival in many tumor types, making this receptor an attractive target to decrease these cells [86]. A previous study reported a marked depletion of TAM and a shift from M2 to M1 TAM upon CSF1R inhibition, and demonstrated the combination benefit of co-targeting CSF1R and mTOR using PLX3397 and rapamycin in an MPNST xenograft model [87], which provided a translational basis for the MPNSTspecific prospective phase 2 clinical trial (NCT02584647). Preliminary data from this trial reported objective responses and durable stable disease in MPNST patients treated with PLX3397 and sirolimus [88]. Furthermore, combined SHP2i and CSF1Ri demonstrated additive anti-tumor activity in CT26 colon syngeneic mice, a model known to express high levels of TIL [50]. As such, combined SHP2i and CSF1Ri may hold promise as a potential treatment strategy for MPNST.
Little is known about the potential roles of immune modulating clinical therapeutics in NF1-deficient MPNST.
Characterization of TME on a series of MPNST revealed overall low PD-L1 expression but significant CD8+ TIL presence [89,90]. Given the genomic heterogeneity of this tumor type, in several cases, PD-L1 copy number gain/ amplification were also reported [91,92]. There have been three single-patient case reports describing anti-tumor response to immune checkpoint inhibition in patients with MPNST: 1) a patient with metastatic NF1-MPNST harboring PD-L1 genomic amplification had a partial response to nivolumab [92]; 2) a patient with metastatic MPNST, with PD-L1 positivity in the tumor, achieved a complete metabolic response to pembrolizumab [93]; and 3) a patient with MPNST with significant PD-L1 expression (tumor proportion score of 90%) had a complete tumor response to pembrolizumab and procarbazine [94], suggestive of a potential clinical benefit to immune checkpoint blockade in a molecularly-defined subset of MPNST. Three phase 1/2 clinical trials are ongoing using immune checkpoint inhibitors (anti-PD-1 and anti-CTLA4) in patients with MPNST, including the use of pembrolizumab (NCT02691026), or the combination of nivolumab and ipilimumab (NCT02834013 and NCT04465643). Given the preclinical additive combination benefits of SHP2i/ KRASG12Ci plus anti-PD-1 in KRAS-mutant PDAC [38] and SHP2i plus anti-PD-1/anti-CTLA-4 in immunocompetent CT26 colon mouse model [50], additional investigation into SHP2i plus anti-PD-1/anti-CTLA-4 in MPNST is also of significant interest and an area of current pre-clinical investigation in MPNST.
SHP2 Inhibitor-based Therapeutics in Clinical Development
At the time of this manuscript preparation, seven type II SHP2 allosteric inhibitors, based on the structure of SHP099, are currently under clinical assessment for adult advanced/metastatic solid tumors, including: TNO155 (NCT03114319, NCT04000529, NCT04330664, NCT04699188, NCT04294160; Novartis) [26,95], RMC- 4630 (NCT03634982, NCT03989115, NCT04185883, NCT04418661; Revolution Medicines/ Sanofi) [18,50,84], JAB-3068 (NCT03565003 and NCT03518554) and JAB 3312 (NCT04121286 and NCT04045496) (Jacobio/ AbbVie), RLY-1971 (NCT04252339; Relay Therapeutics), BBP-398 (NCT04528836; Navire Pharma/ BridgeBio) [28], and ERAS-601 (NCT04670679; Erasca). Multiple clinical trials of SHP2 inhibitors as single agents, and several which combine SHP2i with inhibitors of semiautonomous oncogenic partners, such as EGFR mutants, MEK, CDK4/6, PD-1, and KRASG12C, are ongoing (Table 1). Recently, anti-tumor activity of dual intermittent dosing of RMC-4630 (SHP2i) and cobimetinib (MEKi) in patients with KRAS mutant colorectal cancer has been studied, with acceptable tolerability based on preliminary reports. Among these, preliminary evidence revealed an unconfirmed partial response (30% reduction in tumor burden at end of cycle 2; 25% reduction at end of cycle 4; progressive disease at 6 months) in a patient with KRASG12D colorectal cancer [84]. Future studies of SHP2 combinations may ideally then focus on optimal dosing schedules and potential combinations to achieve the fine balance needed between efficacy and toxicity in patients with RAS-driven cancers.
| SHP2 inhibitor | Combination partner (target) | Phase | Identifier | Condition | Sponsor |
|---|---|---|---|---|---|
| TNO155 | single agent; nazartinib [EGFR] | 1 | NCT03114319 | Advanced EGFR-mutant or KRAS G12-mutant NSCLC, Esophageal SCC, HNSCC, Melanoma | Novartis |
| spartalizumab [PD-1]; ribociclib [CDK4/6] | 1b | NCT04000529 | NSCLC, HNSCC, Esophageal SCC, GIST, CRC |
Novartis | |
| MRTX849 [KRAS G12C] | 1/2 | NCT04330664 | Advanced solid tumors with KRAS G12C mutation | Mirati Therapeutics; Novartis | |
| JDQ443 [KRAS G12C]; spartalizumab [PD-1] plus JDQ443 [KRAS G12C] |
1/2 | NCT04699188 | Advanced solid tumors with KRAS G12C mutation | Novartis | |
| dabrafenib [BRAF V600E] plus LTT462 [ERK1/2] | 1 | NCT04294160 | Advanced or metastatic BRAF V600 CRC | Novartis | |
| RMC-4630 | single agent | 1 | NCT03634982 | Advanced relapsed or refractory solid tumors | Revolution Medicines/Sanofi |
| cobimetinib [MEK]; osimertinib [EGFR] | 1b/2 | NCT03989115 | Relapsed/refractory solid tumors; advanced or metastatic EGFR-mutant NSCLC | Revolution Medicines/Sanofi |
|
| sotorasib [KRAS G12C] | 1 | NCT04185883 | Advanced solid tumors with KRAS G12C mutation | Amgen | |
| pembrolizumab [PD-1] | 1 | NCT04418661 | Advanced or metastatic solid tumors with KRAS mutations and amplifications, BRAF class 3 mutations, or NF1 LOF mutations |
Sanofi/Revolution Medicines |
|
| JAB-3068 | single agent | 1/2a | NCT03565003 | NSCLC, HNSCC, Esophageal SCC, other metastatic solid tumors |
Jacobio/AbbVie |
| single agent | 1 | NCT03518554 | NSCLC, HNSCC, Esophageal SCC, other metastatic solid tumors |
Jacobio/AbbVie | |
| JAB-3312 | single agent | 1 | NCT04121286 | Advanced solid tumors including NSCLC, CRC, PDAC, Esophageal SCC, HNSCC and breast cancer | Jacobio/AbbVie |
| single agent | 1 | NCT04045496 | Advanced solid tumors including NSCLC, CRC, PDAC, Esophageal SCC, HNSCC and breast cancer | Jacobio/AbbVie | |
| RLY-1971 | single agent | 1 | NCT04252339 | Advanced or metastatic solid tumors | Relay Therapeutics |
| BBP-398 | single agent | 1/1b | NCT04528836 | Advanced KRAS G12C or EGFR-mutant NSCLC; solid tumors with other MAPK-pathway alterations. |
Navire Pharma/ BridgeBio |
| ERAS-601 | single agent; a MEK inhibitor (MEK) | 1/1b | NCT04670679 | Advanced or metastatic solid tumors with specific molecular alterations | Erasca |
| NSCLC: Non-small Cell Lung Cancer; SCC: Squamous Cell Cancer; HNSCC: Head and Neck Squamous Cell Cancer; GIST: Gastrointestinal Stromal Tumors; CRC; Colorectal Cancer; PDAC: Pancreatic Ductal Carcinoma; LOF: Loss of Function. | |||||
Closing Remarks and Future Perspective
SHP2 has recently taken a well-deserved center-stage role in cancer therapeutics, owing to its multifaceted roles directed toward growth-promoting signaling pathways within the tumor cells, as well as the surrounding immunosuppressive microenvironment. Further investigation into the various functional roles of SHP2 will be critically instructive in realizing its full potential as an anti-cancer therapy. Future studies of combinations, as well as preclinical and clinical investigation focused on the mechanisms of resistance to allosteric SHP2 inhibitors, will be critically informative as these agents advance in clinical trials. Preclinical studies demonstrate relative insensitivity to SHP2i associated with oncogenic RAS or RAF mutations; and intrinsic or feedback activation of FGFR in response to inhibition of ERK signaling [13,19,96]. FGFR signal may promote the open active conformation of SHP2, leading to resistance to allosteric SHP2 inhibitors [96]. An alternative therapeutic concept is the utilization of RAS-SOS1 interaction inhibitors, such as BI-3406, RMC- 5845 and BAY-293 [97,98] to block adaptive activation of FGFR upon ERK signaling inhibition in some cellular context. Overall, biochemical and genomic exploration of the preclinical and clinical mechanisms that limit the anti-tumor efficacy of SHP2 inhibitors, promises to inform future development of novel combination strategies and new generations of allosteric SHP2 inhibitors.
Disclosure of Competing Interest
C.A. Pratilas reports personal fees from Genentech and research funding from Kura Oncology outside the submitted work, as well as research grants from Novartis; in addition, C.A. Pratilas has a patent for Grant US- 7812143-B2 issued. No potential conflicts of interest were disclosed by the other authors.
Acknowledgements
The authors acknowledge support from Hyundai Hope on Wheels (to C.A. Pratilas), the Neurofibromatosis Therapeutic Acceleration Program (NTAP; to C.A. Pratilas), the Children’s Cancer Foundation (to C.A. Pratilas and N.J. Llosa).
References
2. Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998 Feb 20;92(4):441-50.
3. Eck MJ, Pluskey S, Trüb T, Harrison SC, Shoelson SE. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nature. 1996 Jan;379(6562):277-80.
4. Lu W, Gong D, Bar-Sagi D, Cole PA. Site-specific incorporation of a phosphotyrosine mimetic reveals a role for tyrosine phosphorylation of SHP-2 in cell signaling. Molecular Cell. 2001 Oct 26;8(4):759-69.
5. Bentires-Alj M, Paez JG, David FS, Keilhack H, Halmos B, Naoki K, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Research. 2004 Dec 15;64(24):8816-20.
6. Keren B, Hadchouel A, Saba S, Sznajer Y, Bonneau D, Leheup B, et al. PTPN11 mutations in patients with LEOPARD syndrome: a French multicentric experience. Journal of Medical Genetics. 2004 Nov 1;41(11):e117-.
7. Tajan M, Batut A, Cadoudal T, Deleruyelle S, Le Gonidec S, Saint Laurent C, et al. LEOPARD syndrome-associated SHP2 mutation confers leanness and protection from dietinduced obesity. Proceedings of the National Academy of Sciences. 2014 Oct 21;111(42):E4494-503.
8. Miura K, Wakayama Y, Tanino M, Orba Y, Sawa H, Hatakeyama M, et al. Involvement of EphA2-mediated tyrosine phosphorylation of Shp2 in Shp2-regulated activation of extracellular signal-regulated kinase. Oncogene. 2013 Nov;32(45):5292-301.
9. Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nature Genetics. 2003 Jun;34(2):148-50.
10. Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer and Metastasis Reviews. 2008 Jun;27(2):179-92.
11. Chan RJ, Feng GS. PTPN11 is the first identified protooncogene that encodes a tyrosine phosphatase. Blood. 2007 Feb 1;109(3):862-7.
12. Grossmann KS, Rosário M, Birchmeier C, Birchmeier W. The tyrosine phosphatase Shp2 in development and cancer. Advances in Cancer Research. 2010 Jan 1;106:53- 89.
13. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia- Fortanet J, Acker MG, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016 Jul;535(7610):148-52.
14. Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domaincontaining tyrosine phosphatases in cell signaling. Trends in Bbiochemical Sciences. 2003 Jun 1;28(6):284-93.
15. Hanafusa H, Torii S, Yasunaga T, Matsumoto K, Nishida E. Shp2, an SH2-containing protein-tyrosine phosphatase, positively regulates receptor tyrosine kinase signaling by dephosphorylating and inactivating the inhibitor Sprouty. Journal of Biological Chemistry. 2004 May 1;279(22):22992-5.
16. Bunda S, Burrell K, Heir P, Zeng L, Alamsahebpour A, Kano Y, et al. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nature Communications. 2015 Nov 30;6(1):1-2.
17. Dance M, Montagner A, Salles JP, Yart A, Raynal P. The molecular functions of Shp2 in the Ras/Mitogenactivated protein kinase (ERK1/2) pathway. Cellular Signalling. 2008 Mar 1;20(3):453-9.
18. Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1-and RAS-driven cancers. Nature Cell Biology. 2018 Sep;20(9):1064-73.
19. Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, et al. SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-dependent tumors. Cell Reports. 2019 Jan 2;26(1):65-78.
20. Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. Journal of Biological Chemistry. 2003 Oct 24;278(43):41677-84.
21. Chen CH, Chen Z, Fortanet JG, Grunenfelder D, Karki R, Kato M, et al. 1-Pyridazin-/triazin-3-yl-piper (-azine)/ idine/pyrolidine Derivatives and Compositions Thereof for Inhibiting the Activity of SHP2. United States Patent US 10,093,646. 2018 Oct 9.
22. Tsutsumi R, Ran H, Neel BG. Off-target inhibition by active site-targeting SHP 2 inhibitors. FEBS Open Bio. 2018 Sep;8(9):1405-11.
23. Garcia Fortanet J, Chen CH, Chen YN, Chen Z, Deng Z, Firestone B, et al. Allosteric inhibition of SHP2: identification of a potent, selective, and orally efficacious phosphatase inhibitor. Journal of Medicinal Chemistry. 2016 Sep 8;59(17):7773-82.
24. Wang J, Pollard K, Allen AN, Tomar T, Pijnenburg D, Yao Z, et al. Combined inhibition of SHP2 and MEK is effective in models of NF1-deficient malignant peripheral nerve sheath tumors. Cancer Research. 2020 Dec 1;80(23):5367-79.
25. Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, et al. SHP2 inhibition restores sensitivity in ALKrearranged non-small-cell lung cancer resistant to ALK inhibitors. Nature Medicine. 2018 Apr;24(4):512.
26. Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clinical Cancer Research. 2021 Jan 1;27(1):342-54.
27. Chen H, Libring S, Ruddraraju KV, Miao J, Solorio L, Zhang ZY, et al. SHP2 is a multifunctional therapeutic target in drug resistant metastatic breast cancer. Oncogene. 2020 Dec;39(49):7166-80.
28. Sun Y, Meyers BA, Czako B, Leonard P, Mseeh F, Harris AL, et al. Allosteric SHP2 Inhibitor, IACS-13909, Overcomes EGFR-Dependent and EGFR-Independent Resistance Mechanisms toward Osimertinib. Cancer Research. 2020 Nov 1;80(21):4840-53.
29. Prahallad A, Heynen GJ, Germano G, Willems SM, Evers B, Vecchione L, et al. PTPN11 is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Reports. 2015 Sep 29;12(12):1978-85.
30. Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discovery. 2018 Oct 1;8(10):1237-49.
31. Lu H, Liu C, Velazquez R, Wang H, Dunkl LM, Kazic-Legueux M, et al. SHP2 inhibition overcomes RTK-mediated pathway reactivation in KRAS-mutant tumors treated with MEK inhibitors. Molecular Cancer Therapeutics. 2019 Jul 1;18(7):1323-34.
32. Wang J, Pollard K, Calizo A, Pratilas CA. Activation of receptor tyrosine kinases mediates acquired resistance to MEK inhibition in malignant peripheral nerve sheath tumors. Cancer Research. 2021 Feb 1;81(3):747-62.
33. Ryan MB, de la Cruz FF, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clinical Cancer Research. 2020 Apr 1;26(7):1633-43.
34. Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nature Medicine. 2018 Jul;24(7):968-77.
35. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nature Medicine. 2018 Jul;24(7):954-60.
36. Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nature Medicine. 2018 Jul;24(7):961-7.
37. Valencia-Sama I, Ladumor Y, Kee L, Adderley T, Christopher G, Robinson CM, et al. NRAS Status Determines Sensitivity to SHP2 Inhibitor Combination Therapies Targeting the RAS–MAPK Pathway in Neuroblastoma. Cancer Research. 2020 Aug 15;80(16):3413-23.
38. Fedele C, Li S, Teng KW, Foster CJ, Peng D, Ran H, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. Journal of Experimental Medicine. 2021;218(1).
39. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Science Advances. 2020 Jan 1;6(5):eaay4458.
40. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science. 2017 Mar 31;355(6332):1428-33.
41. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Reports. 2018 Apr 3;23(1):39-49.
42. Xu X, Hou B, Fulzele A, Masubuchi T, Zhao Y, Wu Z, et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. Journal of Cell Biology. 2020 Jun 1;219(6).
43. Gesbert F, Guenzi C, Bertoglio J. A new tyrosinephosphorylated 97-kDa adaptor protein mediates interleukin-2-induced association of SHP-2 with p85- phosphatidylinositol 3-kinase in human T lymphocytes. Journal of Biological Chemistry. 1998 Jul 17;273(29):18273- 81.
44. Gadina M, Sudarshan C, O’Shea JJ. IL-2, but not IL-4 and other cytokines, induces phosphorylation of a 98- kDa protein associated with SHP-2, phosphatidylinositol 3′-kinase, and Grb2. The Journal of Immunology. 1999 Feb 15;162(4):2081-6.
45. Gadina M, Sudarshan C, Visconti R, Zhou YJ, Gu H, Neel BG, et al. The docking molecule gab2 is induced by lymphocyte activation and is involved in signaling by interleukin-2 and interleukin-15 but not other common γ chain-using cytokines. Journal of Biological Chemistry. 2000 Sep 1;275(35):26959-66
46. Adachi M, Ishino M, Torigoe T, Minami Y, Matozaki T, Miyazaki T, et al. Interleukin-2 induces tyrosine phosphorylation of SHP-2 through IL-2 receptor β chain. Oncogene. 1997 Apr;14(13):1629-33.
47. Gadina M, Stancato LM, Bacon CM, Larner AC, O’Shea JJ. Cutting edge: involvement of SHP-2 in multiple aspects of IL-2 signaling: evidence for a positive regulatory role. The Journal of Immunology. 1998 May 15;160(10):4657- 61.
48. Nelson BH, McIntosh BC, Rosencrans LL, Greenberg PD. Requirement for an initial signal from the membraneproximal region of the interleukin 2 receptor γc chain for Janus kinase activation leading to T cell proliferation. Proceedings of the National Academy of Sciences. 1997 Mar 4;94(5):1878-83.
49. Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G, et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharmaceutica Sinica B. 2019 Mar 1;9(2):304-15.
50. Quintana E, Schulze CJ, Myers DR, Choy TJ, Mordec K, Wildes D, et al. Allosteric inhibition of SHP2 stimulates antitumor immunity by transforming the immunosuppressive environment. Cancer Research. 2020 Jul 1;80(13):2889-902.
51. Awad RM, De Vlaeminck Y, Maebe J, Goyvaerts C, Breckpot K. Turn back the TIMe: targeting tumor infiltrating myeloid cells to revert cancer progression. Frontiers in Immunology. 2018 Aug 31;9:1977.
52. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nature reviews Clinical Oncology. 2017 Jul;14(7):399.
53. Chen L, Oke T, Siegel N, Cojocaru G, Tam AJ, Blosser RL, et al. The immunosuppressive niche of soft-tissue sarcomas is sustained by tumor-associated macrophages and characterized by intratumoral tertiary lymphoid structures. Clinical Cancer Research. 2020 Aug 1;26(15):4018-30.
54. Koo J, Hayashi M, Verneris MR, Lee-Sherick AB. Targeting Tumor-Associated Macrophages in the Pediatric Sarcoma Tumor Microenvironment. Frontiers in Oncology. 2020;10.
55. Fridman WH, Zitvogel L, Sautès–Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nature reviews Clinical Oncology. 2017 Dec;14(12):717.
56. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nature reviews Drug Discovery. 2018 Dec;17(12):887-904.
57. Pathria P, Louis TL, Varner JA. Targeting tumorassociated macrophages in cancer. Trends in Immunology. 2019 Apr 1;40(4):310-27.
58. Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, et al. Defining GM-CSF–and macrophage-CSF–dependent macrophage responses by in vitro models. The Journal of Immunology. 2012 Jun 1;188(11):5752-65.
59. Liu M, Tong Z, Ding C, Luo F, Wu S, Wu C, et al. Transcription factor c-Maf is a checkpoint that programs macrophages in lung cancer. The Journal of Clinical Investigation. 2020 Mar 16;130(4).
60. Stanley ER, Chitu V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harbor perspectives in biology. 2014 Jun 1;6(6):a021857.
61. Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Current opinion in immunology. 2013 Apr 1;25(2):200-5.
62. Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clinical Cancer Research. 2016 Mar 15;22(6):1499-509.
63. Hu-Lieskovan S, Mok S, Moreno BH, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Science Translational Medicine. 2015 Mar 18;7(279):279ra41-.
64. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019 Nov;575(7781):217- 23.
65. Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Research. 2010 Jul 1;70(13):5213-9.
66. Donia M, Fagone P, Nicoletti F, Andersen RS, Høgdall E, Straten PT, et al. BRAF inhibition improves tumor recognition by the immune system: potential implications for combinatorial therapies against melanoma involving adoptive T-cell transfer. Oncoimmunology. 2012 Dec 1;1(9):1476-83.
67. Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical Cancer Research. 2013 Mar 1;19(5):1225-31.
68. Reddy SM, Reuben A, Wargo JA. Influences of BRAF inhibitors on the immune microenvironment and the rationale for combined molecular and immune targeted therapy. Current Oncology Reports. 2016 Jul 1;18(7):42.
69. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunology Research. 2014 Jan 1;2(1):70-9.
70. Kang SH, Keam B, Ahn YO, Park HR, Kim M, Kim TM, et al. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology. 2019 Jan 2;8(1):e1515057.
71. Vella LJ, Pasam A, Dimopoulos N, Andrews M, Knights A, Puaux AL, et al. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunology Research. 2014 Apr 1;2(4):351-60.
72. Ebert PJ, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016 Mar 15;44(3):609-21.
73. Dushyanthen S, Teo ZL, Caramia F, Savas P, Mintoff CP, Virassamy B, et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nature Communications. 2017 Sep 19;8(1):1-8.
74. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. MEK inhibition reprograms CD8+ T lymphocytes into memory stem cells with potent antitumor effects. Nature Immunology. 2021 Jan;22(1):53-66.
75. Choi H, Deng J, Li S, Silk T, Dong L, Brea EJ, et al. Pulsatile MEK inhibition improves anti-tumor immunity and T cell function in murine Kras mutant lung cancer. Cell Reports. 2019 Apr 16;27(3):806-19.
76. Bommareddy PK, Aspromonte S, Zloza A, Rabkin SD, Kaufman HL. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Science Translational Medicine. 2018 Dec 12;10(471).
77. Lemberg KM, Wang J, Pratilas CA. From Genes to-Omics: The Evolving Molecular Landscape of Malignant Peripheral Nerve Sheath Tumor. Genes. 2020 Jun;11(6):691.
78. Kohlmeyer JL, Kaemmer CA, Pulliam C, Maharjan CK, Samayoa AM, Major HJ, et al. RABL6A is an essential driver of MPNSTs that negatively regulates the RB1 pathway and sensitizes tumor cells to CDK4/6 inhibitors. Clinical Cancer Research. 2020 Jun 15;26(12):2997-3011.
79. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discovery. 2016 Apr 1;6(4):353-67.
80. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacology & Therapeutics. 2017 May 1;173:83-105.
81. O’leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nature reviews Clinical Oncology. 2016 Jul;13(7):417-30.
82. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor–positive breast cancer. Cancer Research. 2016 Apr 15;76(8):2301-13.
83. Vilgelm AE, Saleh N, Shattuck-Brandt R, Riemenschneider K, Slesur L, Chen SC, et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Science Translational Medicine. 2019 Aug 14;11(505).
84. Bendell J, Ulahannan S, Koczywas M, Brahmer J, Capasso A, Eckhardt SG, et al. Intermittent dosing of RMC- 4630, a potent, selective inhibitor of SHP2, combined with the MEK inhibitor cobimetinib, in a phase 1b/2 clinical trial for advanced solid tumors with activating mutations of RAS signaling. European Journal of Cancer. 2020 Oct 1;138:S8-9.
85. Prada CE, Jousma E, Rizvi TA, Wu J, Dunn RS, Mayes DA, et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathologica. 2013 Jan 1;125(1):159- 68.
86. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. Journal for Immunotherapy of Cancer. 2017 Dec;5(1):1-3.
87. Patwardhan PP, Surriga O, Beckman MJ, De Stanchina E, Dematteo RP, Tap WD, et al. Sustained inhibition of receptor tyrosine kinases and macrophage depletion by PLX3397 and rapamycin as a potential new approach for the treatment of MPNSTs. Clinical Cancer Research. 2014 Jun 15;20(12):3146-58.
88. Manji GA, Van Tine BA, Lee SM, Raufi A, Patwardhan P, Blumberg LE, et al. Phase 1 combination therapy with pexidartinib (PEX) and sirolimus (S) to target tumorassociated macrophages in pigmented villonodular synovitis, malignant peripheral nerve sheath tumors, and other soft tissue sarcomas. Journal of Clinical Oncology. 2019; 11055-11055.
89. Shurell E, Singh AS, Crompton JG, Jensen S, Li Y, Dry S, et al. Characterizing the immune microenvironment of malignant peripheral nerve sheath tumor by PD-L1 expression and presence of CD8+ tumor infiltrating lymphocytes. Oncotarget. 2016 Sep 27;7(39):64300.
90. Haworth KB, Arnold MA, Pierson CR, Choi K, Yeager ND, Ratner N, et al. Immune profiling of NF1- associated tumors reveals histologic subtype distinctions and heterogeneity: implications for immunotherapy. Oncotarget. 2017 Oct 10;8(47):82037.
91. Budczies J, Mechtersheimer G, Denkert C, Klauschen F, Mughal SS, Chudasama P, et al. PD-L1 (CD274) copy number gain, expression, and immune cell infiltration as candidate predictors for response to immune checkpoint inhibitors in soft-tissue sarcoma. Oncoimmunology. 2017 Mar 4;6(3):e1279777.
92. Özdemir BC, Bohanes P, Bisig B, Missiaglia E, Tsantoulis P, Coukos G, et al. Deep response to Anti-PD-1 therapy of metastatic neurofibromatosis type 1-associated malignant peripheral nerve sheath tumor with CD274/PDL1 amplification. JCO Precision Oncology. 2019 Apr;3:1-6.
93. Davis LE, Nicholls LA, Babiker HM, Liau J, Mahadevan D. PD-1 inhibition achieves a complete metabolic response in a patient with malignant peripheral nerve sheath tumor. Cancer Immunology Research. 2019 Aug 5.
94. Payandeh M, Sadeghi M, Sadeghi E. Complete response to pembrolizumab in a patient with malignant peripheral nerve sheath tumor: the first case reported. Journal of Applied Pharmaceutical Science. 2017;7(10):182-4.
95. LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry. 2020 Sep 10;63(22):13578-94.
96. Lu H, Liu C, Hung Huynh TB, LaMarche MJ, Mohseni M, Engelman JA, et al. Resistance to allosteric SHP2 inhibition in FGFR-driven cancers through rapid feedback activation of FGFR. Oncotarget. 2020 Jan 21;11(3):265.
97. Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proceedings of the National Academy of Sciences. 2019 Feb 12;116(7):2551-60.
98. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a potent and selective SOS1–KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discovery. 2021 Jan 1;11(1):142-57.