Commentary
Granulocyte-colony stimulating factor (G-CSF) has been in clinical use for over two decades to enhance hematopoiesis and granulopoiesis to increase the numbers of hematopoietic stem/progenitor cells in patients with neutropenia and patients involved in bone marrow transplantation. G-CSF protein or G-CSF gene therapy has also been shown to have both neuroprotective and neurogenesis function and is quite effective in improving neurological functions in Parkinson’s disease [1], stroke [2-4] and Alzheimer’s disease [5]. The neuroprotective function of G-CSF is believed to be mediated through activation of the G-CSF receptor by G-CSF resulting in activation of the STAT3, ERK5/ERK1/2 and PI3K/AKT pathways [2-4,6-8]. The PI3K/AKT pathway plays a major role in the action of G-CSF in neuronal protection since it protects neurons against cellular stresses such as endoplasmic reticular (ER) stress, mitochondrial stress, autophagy stress by reducing the level of stress markers, e.g., CHOP or GRP78 for ER stress, Drp1 for mitochondrial stress, Beclin for autophagy resulting in decreased level of pro-apoptotic markers, such as Bax, Bim and increased level of anti-apoptotic markers such as Bcl2 [2-4,7,8]. It is of interest that the effects of G-CSF in modulating these apoptotic factors were abolished by rapamycin, an inhibitor of mTOR/p70S6K suggesting that the potential mechanisms underlying the neuronal anti-apoptotic effects of G-CSF could be through in part the inhibition of caspase 3 activation and by inhibiting Bax translocation, and by increasing Bcl-2 expression levels partly through the activation of the mTOR/p70S6K signaling pathway.
In addition, the role of G-CSF in regulation of the immune system has received much attention lately. G-CSF exerts a pivotal role in the control of the immune response and acts as an anti-inflammatory cytokine, preventing an overactivation of monocytes and lymphocytes by reducing the release of pro-inflammatory mediators while simultaneously activating the anti-inflammatory defense neutrophils [9]. IL-1ß and TNF-α are early-response inflammatory cytokines that are synthesized and secreted by microglia, astrocytes, and neurons. Increased release of TNF-α can elicit post-ischemic brain damage through its pro-inflammatory effects on endothelium, leading to impaired microcirculatory brain perfusion [10]. Dumbuya et al. [7,8] reported that in the neonatal hypoxia-ischemia (HI) rat model, G-CSF significantly reduced the level of pro-inflammatory cytokines, e.g., TNF-α and IL-1ß and increased the immuno-suppressive cytokines, e.g., IL-10 expression level. G-CSF is associated with increased expression of IL-10 levels thereby reducing TNF-α production by inhibiting TNF-α converting enzyme (TACE) catalytic activity resulting in decreased levels of TNF- α and IL-1ß, and thus attenuating neuroinflammation. Furthermore, the action of G-CSF in immune regulation was found to be regulated via the G-CSF receptor-mediated PI3K/AKT pathway leading to activation of the mTOR/p70S6K signaling pathway. This notion is supported by the observation that the effect of G-CSF on immune modulation is abolished by the mTOR/p70S6K inhibitor, rapamycin [7,8].
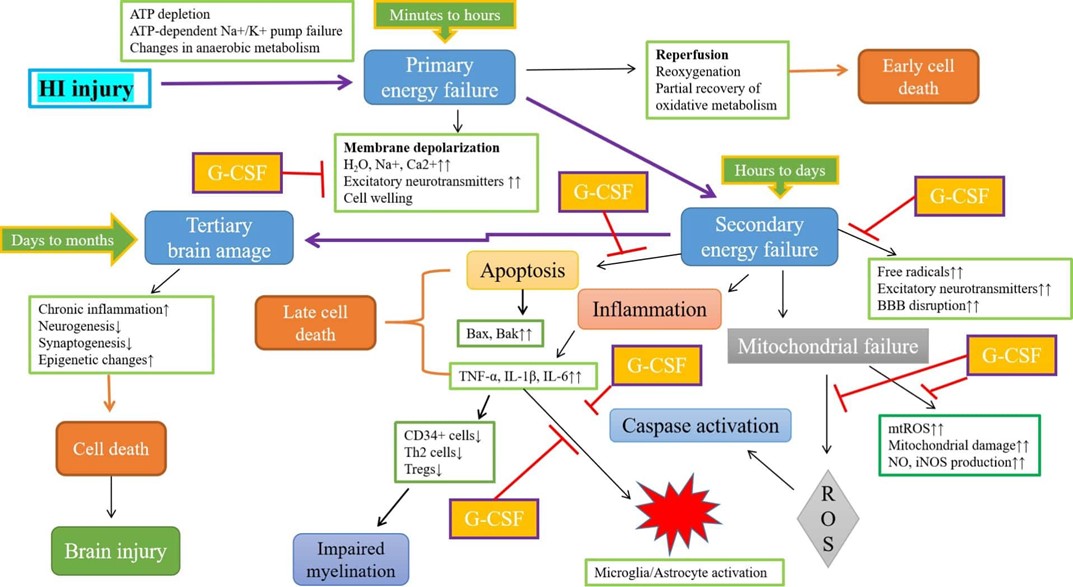
A possible sequence of events from exposure to HI to down regulation of inflammatory response by G-CSF could be postulated as follows: G-CSF could exert it’s action on mitochondria first through its anti-oxidative stress function, followed by anti-apoptotic activity through inhibition of the intrinsic apoptosis pathways such as inhibition of the release of cytochrome C and the related cascade, resulting in increase of pro-survival molecules such as Akt and phosphorylated Akt (p-Akt) and activation of the mTOR/p70SK6 pathway leading to inhibition of microglia activation and reduction of inflammation (Figure 1). Previous studies have demonstrated that decreased activation of S6K, part of mTOR/p70S6K complex, with rapamycin leads to increased astrocyte death during HI, impairment of protein synthesis recovery, imbalance between mitochondrial pro- and anti-apoptotic factors and a rise in ROS levels [11,12]. This concurs with our hypothesis that mTOR inhibition by rapamycin leads to downregulation of S6K rendering it inactive and unphosphorylated, resulting in impaired mRNA translation, protein synthesis and cell cycle arrest [7,8]. Thus, ischemia-induced brain injury downregulates the mTOR/p70S6K pathway, while treatment with G-CSF following ischemic insults potentially reverses this process possibly by counteracting the overexpression of these pro-inflammatory mediators, while activating mTOR/p70S6K leading to phosphorylation of S6K. This implies that, mTOR/p70S6K may be one of the signaling pathways by which G-CSF exerts its neuroprotective effect as well as other important physiological functions including modulation of immune response, gene regulation, protein synthesis and neurogenesis. The role of G-CSF in regulation of the immune system through gene regulation is best illustrated in a recent paper by You et al. [13] using single -cell RNA sequence to decode lymphomyeloid divergence and immune hyporesponsiveness in G-CSF -primed human bone marrow. The authors used single-cell RNA sequencing to compare the transcriptional profile of human hematopoietic cells from two healthy subjects before and after 5-day G-CSF treatment using bioinformatics analysis. The authors found that T and NK cells in G-CSF- primed bone marrow exhibited a higher expression of genes related to immunosuppression such as SOCS1, TSC22D3, ZFP36, and DUSP2 as well as genes involved in Th2 development including IL-4 and IL-3 pathways upon G-CSF stimulation. CD4+ T cells also showed a higher level of expression of signal transducer and activator of transcription 3 (STAT3) target genes in GCSF-primed bone marrow which is required for the Th2 development. STAT3 plays a critical role in B cell differentiation and rheumatoid arthritis since demethylation modification of STAT3 by Jmjd1c restrains B-cell differentiation into plasma cells in rheumatoid arthritis [14] suggesting that G-CSF could also be potentially a valuable therapeutic candidate for autoimmune disorders such as rheumatoid arthritis. You et al [13] further used cell-cell ligand-receptor interaction analysis and found an up-regulation of ligand-receptor pairs such as CTSG:F2R and TGFB1:CXCR4 which can increase the expression of down-stream target genes related to immunosuppression and anti-inflammation, e.g., TSC22D3, AREG and NFKB1A. In addition, the authors also found additional up-regulated ligand-receptor pairs enriched in immunosuppressive pathways such as those associated with reduced NK cell-mediated cytotoxicity and negative regulation of immune effector processes. In summary, You et al. [13] have provided important and insightful information of the mechanism of G-CSF in regulation of immune response using single -cell RNA sequencing technique in G-CSF primed bone marrow. Further studies using a similar approach in microglial cells which are the primary immune regulators in the brain, would be highly desirable since G-CSF has been shown to be a promising and novel therapeutic agent for neurological diseases including Parkinson’s disease [1], stroke [2-4], and Alzheimer’s disease [5].
References
2. Menzie-Suderam J, Gharibani P, Modi J, Ma Z, Tao R, Prentice H, et al. Granulocyte-colony stimulating factor protects against endoplasmic reticulum stress in an experimental model of stroke. Brain Research. 2018;1682:1-13.
3. Menzie-Suderam J, Modi J, Xu, H, Bent A, Trujillo P, Medley K, et al. Granulocyte-colony Stimulating Factor Gene Therapy as a Novel Therapeutics for Stroke in a Mouse Model. J Biomed Sci. 2020;27:99.
4. Modi J, Menzie-Suderam J, Trujillo P, Medley K, Marshall M, Tao R, et al. Mode of Action of Granulocyte-colony Stimulating Factor (G-CSF) as a Novel Therapy for Stroke in a Mouse Model. J Biomed Sci. 2020;27:19-37.
5. Bhandari S, Sartipi P, Sontag H, Prentice H, Wu J-Y. Granulocyte colony stimulating factor as a potential therapy for Alzheimer’s disease. Abstract, S-14071, Society for Neuroscience, 2022.
6. Wu J-Y, Modi J, Menzie J, Chou HY, Tao R, Morrell A, et al. Granulocyte Colony Stimulating Factor (GCSF) Gene Therapy in Stroke and Alzheimer’s Disease Model. Journal of Neurology & Experimental Neuroscience. 2018;4(S1):S17.
7. Dumbuya JS, Chen L, Shu SY, Ma L, Luo W, Li F, Wu JY, et al. G-CSF attenuates neuroinflammation and neuronal apoptosis via the mTOR/p70SK6 signaling pathway in neonatal Hypoxia-Ischemia rat model. Brain Res. 2020 Jul 15;1739:146817.
8. Dumbuya JS, Chen L, Wu JY, Wang B. The role of G-CSF neuroprotective effects in neonatal hypoxic-ischemic encephalopathy (HIE): current status. J Neuroinflammation. 2021 Feb 21;18(1):55.
9. Boneberg EM, Hartung T. Molecular aspects of anti-inflammatory action of G-CSF. Inflamm Res. 2002;51(3):119-28.
10. Schmitz T, Chew L. Cytokines and Myelination in the Central Nervous System. The Scientific World Journal. 2008;8:1119-1147.
11. Chi OZ, Barsoum S, Vega-Cotto NM, Jacinto E, Liu X, Mellender SJ, et al. Effects of rapamycin on cerebral oxygen supply and consumption during reperfusion after cerebral ischemia. Neuroscience. 2016;316:321-327.
12. Pastor MD, García-Yébenes I, Fradejas N, Pérez-Ortiz JM, Mora-Lee S, Tranque P, et al. mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J Biol Chem. 2009 Aug 14;284(33):22067-22078.
13. You G, Zhang M, Bian Z, Guo H, Xu Z, Ni Y, et al. Decoding lymphomyeloid divergence and immune hyporesponsiveness in G-CSF-primed human bone marrow by single- cell RNA-seq. Cell Discovery. 2022;8:59-73.
14. Yin Y, Yang X, Wu S, Ding X, Zhu H, Long X, et al. Jmjd1c demethylates STST3 to restrain plasma cell differentiation and rheumatoid arthritis. Nat Immnunol. 2022;23(9):1342-1354.