Abstract
Mood disorders represent a major medical need requiring chronic treatment. About one million people die by suicide worldwide each year, both as a consequence of major depression or not. Multiple deficits, including cell atrophy and loss, were described in the brains of mood disorders affected patients and in experimental animal models. Numerous changes in gene expression and activity were described in limbic and cortical brain regions. Available therapies probably regulate many of these changes. Different signal transduction pathways play a role in the pathogenesis of schizoaffective disorders, namely the cyclic-AMP, phosphoinositides (PI), mitogen-activated protein kinase, and glycogen synthase kinase cascades. Neurobiology studies focused upon abnormalities of signaling mechanisms with special regard to the serotonin system and related PI signaling system. Involvement of PI-specific Phospholipase C (PLC) enzymes was also described. In suicide brains the overall PLC expression was altered due to a complex reorganization of the isoforms, and PLC β1 isoform was suggested to be involved in schizophrenia and bipolar disorder. The knowledge of the complex network of neurobiological molecules and interconnected signal transduction pathways in the brain might help to understand the natural history and the pathogenesis of mood disorders, as well as of the suicidal behavior. Moreover, it might widen the panel of available therapeutic tools, also gaining prognostic suggestions in order to prevent suicide.
Keywords
Phospholipase C, Phosphoinositides, Mood disorders, Schizophrenia, Bipolar disorder, Major depression, Gene expression
The Role of Phosphoinositide-Specific Phospholipases C in Psychiatric Diseases and Suicide
Mood disorders or affective disorders are emotional disturbances manifesting as pervasive feelings of depression, episodes of mania, or both. Mood disorders are categorized into depressive disorders (DDs) and bipolar disorders (BPDs). DDs include major depressive disorder (MDDs), persistent DD, disruptive mood dysregulation disorder, and premenstrual dysphoric disorder [1].
BPDs include bipolar I disorder, bipolar II disorder, cyclothymia, and substance-induced bipolar disorder [1].
DDs are the largely prevalent mood disorders in the general population. The onset of the disease can impair the quality of patient’s life. The National Health Interview Survey in 2019 came to the conclusion that 18.5% of the general population experienced more or less severe symptoms of MDDs [2,3].
MDDs are diagnosed when patients bear a combination of symptoms such as depressed mood, anhedonia, irritability, emptiness, neuro-vegetative symptoms, including abnormal appetite and sleep, reduced energy and interest, and difficulty in concentration [4]. All those features significantly impair patients’ lives. Some features are not specific and can mask the presentation, so that patients are not correctly and immediately referred to psychiatric care [2-4]. BPDs are characterized by biphasic alternating episodes of depression and mania/ hypomania or single episode of mania [5], with estimated prevalence of 2.8%. In the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) BPDs and related disorders were separated from DDs. Despite the availability of psychotropic medications for mood disorders, morbidity and mortality are progressively increasing, especially from suicide [6].
Mood disorders represent a major medical need requiring chronic treatment. Strictly related to mood disorders, one million people die by suicide worldwide each year, both as a consequence of MDDs or not. Taken together, these illnesses produce a disproportionately large economic burden due to expenditures for hospitalization, treatment, rehabilitation, and lost productivity [6]. A great interest was addressed to identify the risk factors and therapeutic strategies in order to reduce that burden.
Evidences supported by a number of studies suggested that psychiatric disorders may be associated with specific neurobiological abnormalities, and changes in gene expression and activity were described in limbic and cortical brain regions. Probably, the therapeutic molecules actually available act by regulating some of these changes. The knowledge of the complex network of molecules and interconnected signal transduction pathways in the central nervous system might help to understand the natural history and the pathogenesis of nervous disorders, with special regard to psychiatric illnesses, as well as provide insights into the suicidal behavior. Moreover, it might widen the panel of available therapeutic tools, and provide prognostic parameters in order to prevent suicide.
Neurobiology studies in post-mortem brains of patients affected with mood disorders demonstrated abnormalities in the serotonergic pathway and related receptor subtypes [7]. Abnormalities in signaling mechanisms were recently analyzed in the post-mortem brains of suicide-dead individuals, exciting great interest into the 5-hydroxytryptamine2A (HT2A) receptor-linked phosphoinositide (PI) signaling system. The involvement of PI-specific Phospholipase C (PLC) enzymes, belonging to the PI system, was suggested in a number of brain disorders, including epilepsy, Huntington's disease and Alzheimer's disease [8-10], schizophrenia [11,12], BPD [13], MDD [14], and suicide [15]. Selected PLC isoforms were suggested to be involved in schizophrenia and BPD [11-13]. In suicide brains the overall PLC expression was altered due to a complex reorganization of the PLC system [15].
PI Signal Transduction Pathway
The signal transduction pathway of PI was ultimately demonstrated to be involved in the pathogenesis of selected psychiatric disorders. The metabolism of PI molecules contributes an important intracellular signaling system involved in a variety of cell functions such as ion-channel activity, membrane trafficking, cytoskeleton regulation, cell growth, cell-cycle control and apoptosis, neurotransmitter signal transduction, and hormone secretion [16]. Evidences also indicated PI involvement in cell and tissue polarity [16,17]. A combination of compartmentalized and temporal changes in the expression of PI-related signaling molecules elicits different cellular responses, including gene expression modulation/regulation, DNA replication, and chromatin degradation. The PI signal transduction pathway is involved in the calcium signaling cascade, also playing a pivotal role during neuronal development, as well as in the maintenance of neural plasticity and in synapse formation.
In the PI pathway cascade, the regulation of phosphatidyl inositol 4,5-bisphosphate (PIP2), a highly versatile signaling molecule [16] acts crucially. PIP2, mainly located in the inner half membrane, is hydrolyzed by the enzymes belonging to the PLC family in response to a wide panel of stimuli, including growth factors (GFs), hormones, and neurotransmitters, that act on specific receptors localized at the plasma membrane [16]. Thus, PLC plays a central role by regulating the spatialtemporal balance of PI metabolism by acting upon PIP2. PLC enzymes are also involved in cell proliferation, differentiation and apoptosis [16-21].
The PI Specific PLC Family of Enzymes
Once activated, PLC cleaves the membrane PIP2 into inositol trisphosphate (IP3) and diacylglycerol (DAG) (Figure 1). IP3, a small hydrophilic molecule, rapidly diffuses to the cytoplasm. IP3 induces calcium release from the endoplasmic reticulum by binding to IP3-gated calcium-release channels. The initial calcium increase induced by IP3 propagates as a wave through the cytoplasm [16-20]. DAG can be further cleaved to release arachidonic acid (AA), which either acts as a messenger or can be used in the synthesis of eicosanoids.
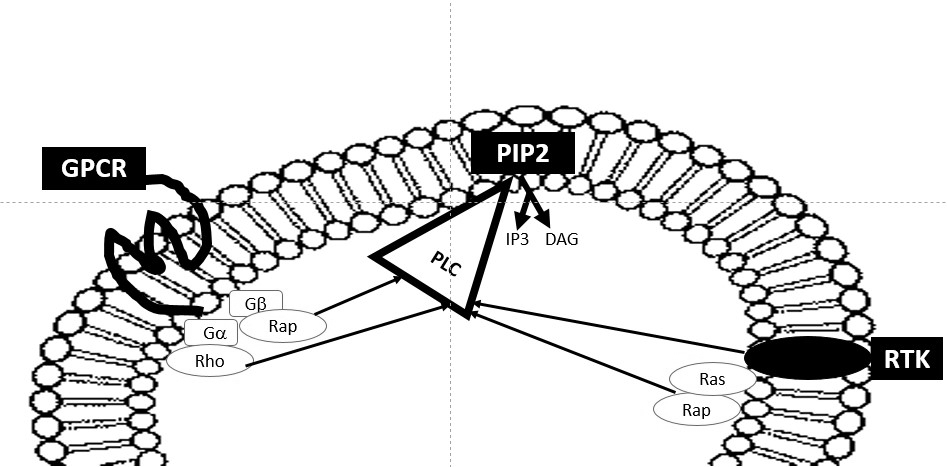
Figure 1: Signaling of the PLC family of enzymes and related pathways. Disruption of PIP2, located within the cell membrane, by PLC in to DAG and IP3. Reciprocal interconnection of PLC signaling, GPCR signaling and RTK signaling.
Thirteen mammalian PLC isoforms were identified, divided into six sub-families: β(1-4), γ(1,2), δ(1,3,4), ε(1), ζ(1), and η(1,2) [16], and classified on the basis of amino acid sequence, domain structure and mechanism of recruitment [16,21,22]. Isoforms within the sub-families share sequence similarity, common domain organization, and regulatory mechanism. PLC isozymes contain catalytic X and Y domains, as well as various regulatory domains, including the protein kinase C conserved region 2 (C2) domain, EF-hand motif, and pleckstrin homology (PH) domain. The PH domain is 110 amino acids long and binds PI. The X and Y domains fold to form the catalytic site. The X domain (65 aminoacids) binds calcium. The Y domain (115 aminoacids) binds primarily the substrate [16].
PLC enzymes are strictly tissue specific and different expression of some isoforms was described in pathological conditions with respect to normal tissues [23-30].
Evidences demonstrated that not all PLC enzymes are contemporarily present within the cell, and that each tissue owns a precise and specific panel of expression. Moreover, the panel of expression of PLC enzymes, analyzed by studying both the mRNA’s transcripts and the corresponding codified protein, varies under different conditions, such as inflammation or tumor enhancement and progression [11-13,29,31-35].
PLC signaling was suggested to be involved in inflammation [35-37]. Moreover, DAG further acts by activating the serine/ threonine calcium dependent protein kinase (PKC) family of enzymes [38]. The increase of calcium induced by IP3 moves PKC to translocate from the cytoplasm to the plasma membrane in order to activate the enzyme. PKC phosphorylates specific serine or threonine residues on a number of target proteins and is actually considered a crucial element in signal transduction by itself. Recently, the mounting evidence that elevated PKC signal transduction pathway may be a pathophysiological feature of BPD and MDD was suggested [39-43].
PLC enzymes act during different events, influencing the activity of several molecules, at several hierarchy of control levels, and act crucially also in the nervous system. PLC enzymes are also involved in the inflammatory activation of the glia [34], as well as in the pathogenesis of rat astrocytoma [44] and in human neuroblastoma [32]. The PLC family of enzymes contributes to the neural development through a complex interaction network in a time-dependent manner. The functional interconnection between the PI signal transduction system and the network of signaling pathways that regulate the neural development recently deserved great attention.
PLC isoforms play specific roles, based on their tissue-specific expression and involvement in diseases affecting the nervous system.
The PLC β sub-family is regulated by the Gq family of GTPbinding proteins [45,46]. With special regard to brain tissue, a modulation of PLC β subfamily enzymes was observed during the cortical development [47].
PLC β1 mediates activity-dependent cortical development and synaptic plasticity [48,49]. PLC β1 is a rate limiting enzyme involved in postnatal cortical development as well as in neuronal plasticity [50,51] and regulates key G proteincoupled signaling pathways in the human cortex [52]. From postnatal stages onwards, PLC β1 is abundant in selected areas of the brain such as cerebral cortex, hippocampus, amygdala, lateral septum and olfactory bulb [19,54]. PLC β1, highly expressed in the cerebral cortex and hippocampus [48,49], is activated by G-protein-coupled receptors that signal through Gq/11. In adult mouse brain, PLC β1 was detected in various grey matter regions, such as layers 2–6 of the cortex, pyramidal and granular layers of the hippocampus, mitral and granule layers of the olfactory bulb, amygdale, caudate, putamen, and lateral septal nucleus [54]. The PLC β1 upregulation during critical periods of development marked by increased cortical plasticity suggests that it might play a role in eliciting structural and functional adaptation [50,51].
Loss of function of PLC β1, following homozygous deletion of the codifying gene [namely PLCB1, OMIM *607120], led to epileptic encephalopathy [55-57]. PLC β2 isoform was suggested to be involved in mental retardation [58,59]. In adult male mice subjected to the forced swim test (FST), an animal model which emulates the behavioral despair paradigm of depression, administration of the PLC inhibitors neomycin and U73122, as well as of the PKC inhibitors calphostin C and chelerytrine, dose-dependently reduced the immobility time in the FST, thus producing an antidepressantlike behavior. Selective knockdown of the PLC β [60] and PKCγ isoforms also induced an antidepressant phenotype. Selective blockade of the PLC β1-PKC γ signaling pathway produced an antidepressant-like phenotype in mice [61-63].
PLC β1 was reported to act in human diseases affecting the nervous system, and it is actually considered the molecular convergence point of several neurotransmitter pathways implicated in schizophrenia [64,65]. The loss-of-function mutation in the PLCB1 gene was associated to homozygous epileptic encephalopathy [55-57].
Plcb1 knockout (KO) mice develop epilepsy, minor hippocampal abnormalities [48], and specific behavioral deficits in location recognition, probably due to the excessive neurogenesis and aberrant migration of adult-born neurons [63]. PLC β1 is also required for activity-dependent regulation of synapse and dendritic spine morphology in the developing barrel cortex [63].
Beside in schizophrenia [11,12,66,67], a role for PLC β1 was also suggested but in BPD [13], and rearrangement of the expression of PLC genes was described in suicide [15]. PLCB1 is constituted from 36 small exons and introns, and was located on the short arm of human chromosome 20 (20p12, nearby markers D20S917 and D20S177) [68,69], a region occasionally rearranged in mental illnesses [70,71]. The activity of pathways involving PLC β1 is modulated by the regulator of G-protein signaling 4 [codified by the RGS4 gene, OMIM *602516], which is considered a candidate gene for schizophrenia [72-78]. RGS4 negatively regulates G protein signaling by acting at Gq11 subunits of the G protein in vitro [74-77]. RGS4, strongly expressed in the brain, directly interacts with PLC β1 [40,76-80].
PLC ε is strictly associated with neuronal lineage differentiation [81]. Its expression persists in terminally differentiated neurons, with no regional specificity. In cultured neural stem cells (NSCs), the expression of PLC ε coincides with the loss of nestin expression, the induction of microtubule-associated protein 2 (MAP2) expression, and the appearance of neuronal morphology. The activity of PLC ε, regulated by association with Ras and Rap, might also play a role in intracellular signaling from receptors for fibroblast growth factor (FGF), and various neurotrophic factors involved in neural development [81,82].
Although significant progresses have been achieved in studying the PI system and its alterations in the human brain, many issues remain to be addressed with special regard to the relationship with selected diseases. In fact, further studies are required both to refine the analysis of the gene PLCB1, as well as of the PLC β1 protein and to extend the examination to other, more recently discovered molecules involved in the PI signaling.
Although considerable research efforts aimed to delineate the metabolic pathways acting in the nervous system, with special regard to the dysfunctions occurring in mood disorders, further studies are required to fully elucidate the complex interplay among signaling molecules. Besides increasing the knowledge of the events regulating the neural development and the nervous system activities, understanding the role and the timing of action of the signaling pathways acting in the nervous system might allow highlight the pathogenesis and the clinical history of several nervous diseases. That will probably help in formulating diagnoses and prognoses, which are often respectively difficult to characterize and determine, also opening the way to novel molecular therapeutic strategies.
The Lithium Treatment and the PI Pathway
One important clue of the PI involvement in psychiatric disorders is represented by the relationship with lithium metabolism. Lithium salts are actually considered as firstline mood stabilizing agents for their acute antimanic/ antidepressant properties and proven efficacy in the long term relapses prevention. The effects of lithium, more or less successfully used since 1940s in the therapy of schizoaffective disorders, were described along the years, and a number of hypotheses were suggested about its activity. Although the mechanisms whereby lithium exerts its action are not highlighted, evidences support the hypothesis that lithiuminduced enhancement of serotonergic neurotransmission, enhancement or stabilization of cholinergic neurotransmission, and inhibition of the PI pathway might play a relevant role [40].
Previous studies demonstrated that the activity of PLC enzymes was reduced in platelets belonging to euthymic manic-depressive patients on therapeutic lithium doses compared to age/sex matched control group [42]. The activities of prostaglandin E1 (PGE1)-, aluminum/NaF-, and forskolin-stimulated platelet adenylate cyclase activity were also measured in lithium-treated and control subjects [41]. A marked reduction in both post-receptor (aluminum/NaF and forskolin) and receptor-stimulated PGE1 platelet adenylate cyclase activity was observed in the lithium-treated group [42]. Further studies confirmed that the therapeutic efficacy of lithium might follow the inhibitory effect on either PI and/ or cyclic nucleotide metabolism [42]. In PC12 cells lithium, at therapeutic doses, enhanced the PI-mediated FOS (OMIM *164810) expression induced by activating a muscarinic cholinergic pathway. By contrast, it had no effect at tenfold the therapeutic dose on FOS expression induced by receptor or post-receptor activators of cyclic adenosine monophosphate (cAMP) [42], also described in phorbol esters-treated cells, which directly activate PKC. That allowed suggest that lithium interacts with the PI pathway at the post-receptor level [42].
Several predictor features of lithium treatment efficacy were reported by different authors, such as psychopathological, environmental, biological, neurophysiologic and genetic predictors [43]. Elements belonging to the PI pathway were also suggested as possible predictors of lithium efficacy. Interesting findings were described with respect to the gene which encodes for PLC β1, namely PLCG1 (OMIM *172420) [43]. In fact, a higher frequency of PLCG1-5 repeat allele genes was associated to good lithium response [43].
PLC Signaling in Mood Disorders
Mood disorders comprise of a group of diseases characterized in the Diagnostic and Statistical Manual of Mental Disorders classification system featured by disturbance of the mood as the main underlying feature [83]. Mood disorders comprise of both mania/hypomania, depressed mood, including MDD (also called clinical depression, unipolar depression, or major depression), and moods which cycle between mania and depression, BPD (also called manic depression). There are several sub-types of depressive disorders or psychiatric syndromes featuring less severe symptoms such as dysthymic disorder (similar to but milder than MDD) and cyclothymic disorder (similar to but milder than BPD).
Overall mood disorders represent an economic and social cost, as a burden of functional impairment, disability or lost work productivity, and increased use of health services [84]. Different signaling pathways play a role in the pathogenesis of mood disorders, including the cyclic-AMP, PI, MAPK, and glycogen synthase kinase cascades, and changes in selected signaling pathways were described [85]. Therapies actually available seem to reciprocally regulate many of these changes [85].
MDD has a lifetime prevalence of 16.2%, and is one of the most frequent causes of loss of productivity, and suicide than any other affective disorder, significantly contributing to decreased quality and expectancy of life [86,87]. As for other mood disorders, the signal transduction pathway of PIs was also suggested to be involved in the pathophysiology of MDD [88]. Brain regionally selective deficits in G-protein function associated with PI signaling were reported in subjects presenting with MDD [89]. Abnormalities in nerve cell myoinositol levels and/or PI-cycle regulation seem to be involved in the pathogenesis of many psychiatric disorders, also including MDD. Interestingly, the metabolism of myo-inositol is strictly related to the PI-cycle [89]. Some inconclusive reports suggested a role for PLC β1 in mood disorders [11-15].
BPD presents with extreme unprovoked mood changes, shifts in energy and activity levels, and impaired ability to carry out day-to-day tasks. BPD usually presents with severe symptoms which may limit the quality of life, and can result in damaged interpersonal relationships, poor job or school performance, rarely leading to suicidal behavior. BPD affected patients more likely present to clinicians in the depressive phase [90,91]. The lifetime prevalence of BPD spectrum is 4.5% [92,93]. The diagnosis of BPD is often difficult, especially in early presentations, and may require a long term observation of the patient. The clinical presentation of a patient with BPD in the depressive phase may not differ from that of a MDD affected patient, so that, without appropriate screening, BPD patients may be misdiagnosed [94-96]. Therefore, the identification of molecular indicators for BPD diagnosis might be of great interest in order to refine and/or anticipate the diagnosis.
Mood stabilizer molecules used in the treatment of BPD act upon cellular receptors, including G-protein-coupled receptors, glutamate receptors, and tyrosine receptor kinase. The efficacy of mood stabilizers on transcription factors probably allow the regulation of gene expression, strictly related to neuroplasticity and cellular resilience.
Intracellular alterations were described in BPD to second messenger systems, such as cAMP, protein kinase A, PI signal transduction pathways, glycogen synthase kinase-3, protein kinase B, Wnt, and AA [97], which might result in the alteration of different numerous neurotransmitter systems, probably explaining the varied clinical symptoms in BPD.
Previous studies identified deletion of PLCB1 gene in the orbito-frontal cortex biopsy of one patient affected with BPD using molecular cytogenetics analyses [13].
PLC Signaling in Suicide
About one million people die by suicide worldwide each year, making suicide a major public health burden [98]. Evidences supported by a number of studies suggested that suicide may be associated with specific neurobiological abnormalities and scientists focused upon abnormalities of the serotonergic pathway and related receptor subtypes in post-mortem brain of suicide victims [7].
Abnormalities of signaling mechanisms were recently analyzed in the post-mortem brains of suicide dead individuals, and great interest rose around the 5-hydroxytryptamine2A (HT2A) receptor-linked PI signaling system [7]. Recent evidences indicated that PLC enzymes are involved in the complex processes of neurite outgrowth and neurons positioning [8]. Moreover, PLC isoforms were demonstrated to participate in neuron functions mediated by neurotrophins, and to be involved in brain development and synaptic transmission [8].
Activation of Gq-coupled 5-HT2A receptors results in the PLC catalyzed hydrolysis of membrane PIP2, generating IP3 and DAG. Therefore, abnormalities of 5-HT2A receptors have the potential to alter a diverse set of cellular processes acting via the PI system [99,100]. The most important gene in this aspect is the brain-derived neurotrophic factor (BPDNF), which has been studied in suicide, as well as in schizophrenia. Interestingly, BPDNF and its receptor TrkB induce glutamate release through activation of a pathway involving PLC isoforms in developing cultured cortical neurons [101].
Previous data suggested the involvement of PLC enzymes in suicide, with special regard to the PLC β1 enzyme [102,103]. Recently, the rearrangement of the PLC enzymes was described in post-mortem brains of individuals died by suicide [15]. In the normal control brains casuistry, the panel of expression of the PLC genes, comprising six isoforms (PLCB1, PLCB3, PLCB4, PLCG1, PLCD3 and PLCH1) was different from the panel of expression in the brains of suicide victims. PLCB1 was not expressed in 36%, suicide brains, PLCG1 in 21%, PLCB3 and PLCB4 in less than 50% suicide brains, PLCD3 in 69% and PLCH1 in 79%. By contrast, two isoforms belonging to the PLCD family, unexpressed in normal controls brains, resulted expressed in suicide brains, namely PLCD1 (11%) and PLCD4 (36%). [15]. The authors suggested a relationship between rearrangements and the outcome of patients. Surprisingly, the brain samples lacking PLCB1 expression belonged to the youngest individuals in the analyzed casuistry. Moreover, the youngest patients also lacked the expression of high number of further isoforms. The detection of PLCD1 and PLCD4 in 9 out 19 individuals not expressing PLCD3 might be read as the attempt to discharge the PLC δ3 function. That might indicate that the cooperation and organization of the isoforms belonging to the PLCD family is complex, as suggested by previous literature reports [23].
PLC Signaling in Schizophrenia
Schizophrenia (OMIM #181500) is a long-term mental psychiatric disorder causing a range of different psychological symptoms which affect cognitive functions such as attention, motivation, execution, and emotion. The heterogeneous clinical expression reflects different etiological factors, such as altered expression of one or more genes, complications at birth, biochemical alterations including neurotransmitters imbalance, environmental factors, and immunological abnormalities [104-106]. A large number of studies demonstrated that schizophrenia heritability is 0.70–0.85 and that risk in siblings of affected individuals is 10-fold increased [107]. Evidences suggest that schizophrenia arises from a genetic predisposition affecting neurodevelopmental processes, combined with exposure to environmental risk factors [108]. This is not surprising, as the activity of the cerebral cortex, functionally altered in subjects affected with schizophrenia, requires a strictly regulated genetic program for appropriate development [104-109]. The identification of candidate genes might open the way to improve the diagnosis, to evaluate and, in perspective, to limit the impact of environmental factors. It also might help to provide prognostic tools allowing to identify patients with different clinical outcome, and to arrange personalized therapy approaches.
The genetic predisposition of schizophrenia was supported by a number of notable evidences [105]. An approximately 50% concordance rate in monozygotic twins and 1% prevalence in the general population was reported for schizophrenia [105]. Many susceptibility loci for schizophrenia were identified. Significance levels for linkage to schizophrenia were obtained for chromosome regions 6p24-p22 [110], 1q21-q22 [79], 13q32-q34 [111], 10p14 [112], 10q25.3-q26.3 [113], 8p22-p21 [111,114], 6q21-q25 [115,116], 22q11-q12 [117,118], and 5q21-q33 [119,120]. A further susceptibility locus was identified on the short arm of chromosome 20 (20p) [121]. In particular, a smaller region was identified spanning 20p13-p12.2 [70,71]. PLCB1 is constituted from 36 small exons and introns, and is located on the short arm of human chromosome 20 (20p12, nearby markers D20S917 and D20S177) [69].
The complexity of the mechanisms underlying schizophrenia is witnessed by the involvement of a multitude of molecular pathways, including dopaminergic [122,123], serotoninergic [123,124], muscarinic [125-128], and glutamatergic signaling [129-131]. The phosphoinositide (PI) signaling is thought to represent the point of convergence among the above pathways [132,133]. Previous studies performed to assess the activity of the PI signal transduction system revealed that it is selectively impaired in specific brain regions of subjects with neurological and psychiatric disorders [134]. Further studies suggested that subtle alterations of the activity of the PI signal transduction system influence cognition, mood, and behavior associated with mental disorders, including schizophrenia [135]. Evidences indicated the presence of mono-allelic deletion of the PLCB1 gene in a low percentage of post-mortem brains of schizophrenia affected patients by molecular cytogenetic analyses [11,12]. Independently, another study had demonstrated the reduction of PLCB1 transcript in the post-mortem brains of schizophrenic patients by molecular biology analyses. A possible genetic association of PLCB1 and schizophrenia was reported in linkage studies [121], and abnormal expression patterns in the brains of schizophrenic patients were reported [75,136].
The evidences of the involvement of PLC β1 in schizophrenia might be related to its activity. PLC β1 transduces intracellular signals from specific muscarinic, glutamate and serotonin receptors, all implicated in the pathogenesis of schizophrenia, therefore representing a point of convergence of their activities [21,136].
Corroborating observations were obtained studying experimental animal models. Plc β1 knockout (KO) mice showed abnormal cortical development, synaptogenesis, and dendritic spine maturation [74-76]. In adulthood, PLC β1 KO mice exhibit behavioral abnormalities, such as impaired prepulse inhibition of acoustic startle and hyperactivity in response to a novel environment, phenotypes currently considered representative of schizophrenia features [77,78].
As already cited, the activity of pathways involving PLC β1 is modulated by the regulator of G protein signaling 4 (RGS4). The gene which codifies for RGS4 (RGS4, OMIM *602516) is actually considered a candidate gene for schizophrenia [39,48,79,80,104]. RGS4 negatively regulates the G protein signaling by acting at Gq11 alpha subunits of the G protein in vitro [137-139].
The outcome of disrupted PLC β1 signaling was studied in PLC β1 KO mice [75] which have abnormal cortical maturation including aberrant barrel formation in the somatosensory cortex3, abnormal synapse formation and dendritic spine dysmorphism [76]. Spatial memory deficits [140,141], and behavioral abnormalities were also described [72], as well as deficit in working memory, altered fear conditioning, and motor/sensorimotor gating deficiencies, which all are considered features of relevance to schizophrenia [77]. Furthermore, during development, a specific expression pattern for RGS4 was demonstrated, the results of which dramatically altered in the PLC β1 KO mice [72,140,141].
In normal human brain controls, the authors did not identify PLCB1 deletions or subsequent mRNA transcript reduction. The deletion of PLCB1, as well as the subsequent transcription decrease was identified in a limited number of schizophrenia affected patients, suggesting that the deletion of PLCB1 might be specifically associated to schizophrenia.
Further studies are required to verify whether PLCB1 deletions are causally involved in the etiology and/or pathogenesis of schizophrenia. Although significant progresses were achieved in studying the PI system and its alterations in human brain, many issues remain to be addressed with special regard to its relationship with schizophrenia. In particular, further studies are required both to refine the analysis of PLCB1 gene, the role of the PLC β1 enzyme and to extend the examination to other molecules involved in PI signaling.
Conclusion
Considerable research efforts have been made to delineate the metabolic pathways acting in the nervous system, with special regard to the morphology changes occurring in the cortex and to the dysfunctions in the wide number of molecules involved in signal transduction activities. However, we are still far to highlight the complex organization and networks that regulate the activity of the nervous system and the dysfunctions occurring in mental affections.
Further studies are required to fully elucidate the complex interplay among different signal transduction pathways or among selected signaling molecules. Besides increasing the knowledge of the events regulating the neural development and the nervous tissue activity, understanding the role and the timing of action of the signaling pathways acting in the nervous system will contribute to highlight the etiology and the pathogenesis and the clinical history of several nervous diseases, with special regard to mood and schizoaffective disorders.
The promising results of recent reports indicating that the PI system is involved in a number of psychiatric diseases open the way to novel perspectives. That will probably be helpful in diagnosis and prognosis, which often are difficult to characterize, opening the way to novel molecular therapeutic strategies.
References
2. Villarroel MA, Terlizzi EP. Symptoms of Depression Among Adults: United States, 2019. Atlanta: Centers for Disease Control and Prevention, 2020.
3. Datta S, Suryadevara U, Cheong J. Mood Disorders. Continuum (Minneap Minn). 2021 Dec 1;27(6):1712-1737.
4. Suppes T, Ostacher M. Mixed features in major depressive disorder: diagnoses and treatments. CNS Spectr. 2017;22(2):155-160.
5. Angst J, Sellaro R. Historical perspectives and natural history of bipolar disorder. Biol Psychiatry 2000. 48(6):445-457.
6. Whiteford HA, Degenhardt L, Rehm J, et al Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 2013;382(9904):1575-1586.
7. Ressler KJ, Nemeroff CB..Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000;12(Suppl 1):2–19
8. Jang HJ, Yang YR, Kim JK, Choi JH, Seo YK, Lee YH, et al. Phospholipase C-γ1 involved in brain disorders. Adv Biol Regul. 2013;53(1):51-62.
9. Lo Vasco VR. The Phosphoinositide Signal Transduction Pathway in the Pathogenesis of Alzheimer's Disease. Curr Alzheimer Res. 2018;15(4):355-362.
10. Lo Vasco VR. Insights into Brain Signal Transduction can Provide Potential Molecular Targets to Approach and Manage Alzheimer’s Disease. OBM Neurobiology 2020;4(2):22.
11. Lo Vasco VR, Cardinale G, Polonia P. Deletion of PLCB1 gene in schizophrenia-affected patients. J Cell Mol Med. 2012;16(4):844- 51.
12. Udawela M, Scarr E, Hannan AJ, Thomas EA, Dean B. Phospholipase C beta 1 expression in the dorsolateral prefrontal cortex from patients with schizophrenia at different stages of illness. Aust N Z J Psychiatry. 2011;45(2):140-7.
13. Lo Vasco VR, Longo L, Polonia P. Phosphoinositide-specific Phospholipase C ß1 gene deletion in bipolar disorder affected patient. J Cell Commun Signal. 2013;7(1):25-9.
14. Lo Vasco VR, Polonia P. Molecular cytogenetic interphase analysis of Phosphoinositide-specific Phospholipase C β1 gene in paraffin-embedded brain samples of major depression patients. J Affect Disord. 2012;136(1-2), 177-180.
15. Lo Vasco VR, Leopizzi M, Della Rocca C, Fais P, Montisci M, et al. Impairment and reorganization of the phosphoinositide-specific phospholipase C enzymes in suicide brains. J Affect Disord. 2015;15:174:324-8.
16. Suh PG, Park J, Manzoli L, Cocco L, Peak JC, Katan M, et al. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Reports. 2008;41:415-434.
17. Comer FI, Parent CA. Phosphoinositides specify polarity during epithelial organ development. Cell. 2007;128(2):239-240.
18. Berridge MJ. Phosphatidylinositol hydrolysis: a multifunctional transducing mechanism. Mol Cell Endocrinol. 1981;24(2):115-140.
19. Berridge MJ, Irvine RF. Inositol triphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315- 321.
20. Berridge, MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta. 2009;1793(6):933-940.
21. Bunney TD, Katan M. PLC regulation: emerging pictures for molecular mechanisms. Trends Biochem Sci. 2011;36(2):88-96.
22. Nakamura Y, Fukami K. Roles of phospholipase C isozymes in organogenesis and embryonic development. Physiology. 2009;24:332-341.
23. Nakamura Y, Kanemaru K, Fukami K. Physiological functions of phospholipase Cd1 and phospholipase Cd3. Adv Biol Regul. 2013;53:356-362.
24. Lo Vasco VR, Leopizzi M, Puggioni C, Della Rocca C. Ezrin silencing remodulates the expression of Phosphoinositidespecific Phospholipase C enzymes in human osteosarcoma cell lines. J Cell Commun Signal. 2014;8(3):219-29.
25. Lo Vasco VR, Leopizzi M, Puggioni C, Della Rocca C, Businaro R. Neuropeptide Y reduces the expression of PLCB2, PLCD1 and selected PLC genes in cultured human endothelial cells. Mol Cell Biochem. 2014;394(1-2):43-52.
26. Lo Vasco VR, Leopizzi M, Puggioni C, Della Rocca C, Businaro R. Fibroblast growth factor acts upon the transcription of phospholipase C genes in human umbilical vein endothelial cells. Mol Cell Biochem. 2014;388(1-2):51-9.
27. Lo Vasco VR, Leopizzi M, Stoppoloni D, Della Rocca C. Silencing of phosphoinositide-specific phospholipase C ε remodulates the expression of the phosphoinositide signal transduction pathway in human osteosarcoma cell lines. Anticancer Res. 2014;34(8):4069- 75.
28. Lo Vasco VR, Leopizzi M, Chiappetta C, Puggioni C, Di Cristofano C, Della Rocca C. Expression of phosphoinositidespecific phospholipase C enzymes in human skin fibroblasts. Connect Tissue Res. 2013;54(1):1-4.
29. Lo Vasco VR. Phosphoinositide pathway and the signal transduction network in neural development. Neurosci Bull. 2012;28(6):789-800.
30. Yaghobian D, Don AS, Yaghobian S, Chen X, Pollock CA, Saad S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clinical and Experimental Pharmacology and Physiology. 2016 Jan;43(1):56-66.
31. Lo Vasco VR. Signalling in the genomic era. Journal of cell communication and signalling. 2010;4(3):115-117.
32. Lo Vasco VR. 1p36.32 rearrangements and the role of PI-PLC η2 in nervous tumours. J Neurooncol. 2011;103(3):409-16.
33. Lo Vasco VR, Leopizzi M, Chiappetta C, Puggioni C, Di Cristofano C, Della Rocca C. Expression of Phosphoinositidespecific phospholipase C enzymes in human osteosarcoma cell lines. J Cell Commun Signal. 2013;7(2):141-50.
34. Lo Vasco VR, Fabrizi C, Fumagalli L, Cocco L. Expression of phosphoinositide-specific phospholipase C isoenzymes in cultured astrocytes activated after stimulation with lipopolysaccharide. J Cell Biochem. 2010;109(5):1006-12.
35. Lo Vasco VR, Leopizzi M, Di Maio V, Di Raimo T, Cesa S, Masci A, et al. LPS, Oleuropein and Blueberry extracts affect the survival, morphology and Phosphoinositide signalling in stimulated human endothelial cells. J Cell Commun Signal. 2017;11(4):317-327.
36. Di Raimo T, Leopizzi M, Mangino G, Rocca CD, Businaro R, Longo L, et al. Different expression and subcellular localization of Phosphoinositide-specific Phospholipase C enzymes in differently polarized macrophages. J Cell Commun Signal. 2016. 10(4):283- 293.
37. Casoni SD, Romanelli A, Checchi M, Truocchio S, Ferretti M, Palumbo C, et al Expression and Localization of Phosphoinositide- Specific Phospholipases C in Cultured, Differentiating and Stimulated Human Osteoblasts. J Cell Signal. 2022;3(1):44-61.
38. Katan M. New insights into the families of PLC enzymes: looking back and going forward. Biochem J. 2005. 391(Pt 3):e7–e9.
39. Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, BK T, Ferrell RE, Middleton FA, Devlin B. Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet. 2002 Jun 1;11(12):1373-80.
40. Katona CL. Lithium augmentation in refractory depression. Psychiatr Dev. 1988 Summer;6(2):153-71.
41. Ebstein RP, Lerer B, Bennett ER, Shapira B, Kindler S, Shemesh Z, et al. Lithium modulation of second messenger signal amplification in man: inhibition of phosphatidylinositol-specific phospholipase C and adenylate cyclase activity. Psychiatry Res. 1988;24(1):45-52.
42. Divish MM, Sheftel G, Boyle A, Kalasapudi VD, Papolos DF, Lachman HM. Differential effect of lithium on fos protooncogene expression mediated by receptor and postreceptor activators of protein kinase C and cyclic adenosine monophosphate: model for its antimanic action. J Neurosci Res. 1991;28(1):40-8.
43. Rohayem J, Baylé JF, Richa S. Predictors of prophylactic response to lithium. Encephale. 2008;34(4):394-9.
44. Lo Vasco VR, Fabrizi C, Artico M, Cocco L, Billi AM, Fumagalli L, et al. Expression of phosphoinositide-specific phospholipase C isoenzymes in cultured astrocytes. J Cell Biochem. 2007;100(4):952- 959.
45. Frebel K, Wiese S. Signalling molecules essential for neuronal survival and differentiation. Biochem Soc Trans. 2006;34(Pt 6):1287-90.
46. Fukami K, Inanobe S, Kanemaru K, Nakamura Y. Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog Lipid Res. 2010;49(4):429-37.
47. Farias RN, Fiore AM, Pedersen JZ, IncerpiS. Nongenomic Actions of Thyroid Hormones: Focus on Membrane Transport Systems Immun., Endoc Metab Agents in Med Chem. 2006;6:241- 254
48. O’Donovan MC, Williams NM, Owen MJ. Recent advances in the genetics of schizophrenia. Hum Mol Genet. 2003;2:R125-33.
49. Traynor JR, Neubig RR. Regulators of G protein signalling & drugs of abuse. Mol Interv. 2005;5:30-41.
50. Gratacap MP, Payrastre B, Viala C, Mauco G, Plantavid M, Chap H. Phosphatidylinositol 3,4,5-trisphosphate-dependent stimulation of phospholipase C-gamma2 is an early key event in FcgammaRIIA-mediated activation of human platelets. J Biol Chem. 1998;273(38):24314-21.
51. Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M et al PLC-beta1, activated via mGluRs, mediates activitydependent differentiation in cerebral cortex. Nat Neurosci. 2001;4:282-288.
52. Chuang SC, Bianchi R, Wong RKS. Group I mGluR activation turns on a voltage-dependent inward current in hippocampal pyramidal cells. J Neurophysiol. 2000. 83:2844-2853.
53. Fukaya M, Uchigashima M, Nomura S, Hasegawa Y, Kibuchi H, Watanabe M. Predominant expression of phospholipase Cβ1 in telencephalic principal neurons and cerebellar interneurons, and its close association with related signalling molecules in somatodendritic neuronal elements. Eur J Neurosci. 2008;28:1744-1759.
54. Gil OD, Zanazzi G, Struyk AF, Salzer JL. Neurotrimin mediates bifunctional effects on neurite outgrowth via homophilic and heterophilic interactions. J Neurosci. 1998;18(22):9312-25.
55. Kurian MA, Meyer E, Vassallo G, Morgan NV, Prakash N, Pasha S, et al. Phospholipase C beta 1 deficiency is ass Sociated with early-onset epileptic encephalopathy. Brain 2010;133:2964-2970.
56. Poduri A, Chopra SS, Neilan EG, Elhosary PC, Kurian MA, Meyer E, et al. Homozygous PLCB1 deletion associated with malignant migrating partial seizures in infancy Epilepsia. 2012;53(8):e146-50.
57. Ngoh A, McTague A, Wentzensen IM, Meyer E, Applegate C, Kossoff EH, et al. Severe infantile epileptic encephalopathy due to mutations in PLCB1:expansion of the genotypic and phenotypic disease spectrum. Dev Med Child Neurol. 2014;56(11):1124-8.
58. Lo Vasco VR. Role of phosphoinositide-specific phospholipase C η2 in isolated and syndromic mental retardation. Eur Neurol. 2011;65(5):264-269.
59. Fais P, Leopizzi M, Di Maio V, Longo L, Della Rocca C, Tagliaro F, Bortolotti F, Lo Vasco VR. Phosphoinositide-specific phospholipase C in normal human liver and in alcohol abuse. J Cell Biochem. 2019;120(5):7907-17.
60. Ananthanarayanan B, Das S, Rhee SG, Murray D, Cho W. Membrane targeting of C2 domains of phospholipase C-delta isoforms. J Biol Chem. 2002;277:3568-3575.
61. Galeotti N, Ghelardini C. Antidepressant phenotype by inhibiting the phospholipase Cβ(1)--protein kinase Cγ pathway in the forced swim test. Neuropharmacology. 2011;60(6):937-43.
62. Gold SJ, Ni YG, Dohlman HG, Nestler EJ. Regulators of Gprotein signalling (RGS) proteins:Region-specific expression of nine subtypes in rat brain. J Neurosci. 1997;17:8024-37.
63. Nomoto S, Adachi K, Yang LX, Hirata Y, Muraguchi S, Kiuchi K. Distribution of RGS4 mRNA in mouse brain shown by in situ hybridization. Biochem Biophys Res Commun. 1997;241:281-7.
64. Druey KM, Sullivan BM, Brown D, Fischer ER, Watson N, Blumer KJ, et al Expression of GTPase-deficient Gialpha2 results in translocation of cytoplasmic RGS4 to the plasma membrane. J Biol Chem. 1998;273:18405-10.
65. Dowal L, Elliott J, Popov S, Wilkie TM, Scarlata S. Determination of the contact energies between a regulator of G protein signaling and G protein subunits and phospholipase C beta 1. Biochemistry. 2001;40(2):414-21.
66. Koh HY. Phospholipase C-β1 and schizophrenia-related behaviors. Adv Biol Regul. 2013;53(3):242-8.
67. Kim SW, Seo M, Kim DS, Kang M, Kim YS, Koh HY, et al. Knockdown of phospholipase C-β1 in the medial prefrontal cortex of male mice impairs working memory among multiple schizophrenia endophenotypes. J Psychiatry Neurosci.2015;40(2):78-88.
68. Peruzzi D, Aluigi M, Manzoli L, Billi AM, Di Giorgio FP, Morleo M, et al. Molecular characterization of the human PLC beta1 gene. Biochim Biophys Acta. 2002;1584:46-54.
69. Peruzzi D, Calabrese G, Faenza I, Manzoli L, Matteucci A, Gianfrancesco F, et al. Identification and chromosomal localisation by fluorescence in situ hybridisation of human gene of phosphoinositide-specific phospholipase Cb1. Biochim Biophys Acta. 2000;484, 175-82.
70. Fanous AH, Neale MC, Webb BT, Straub RE, O'Neill FA, Walsh D, et al. Novel linkage to chromosome 20p using latent classes of psychotic illness in 270 Irish high-density families. Biol Psychiatry. 2008;64:121-7.
71. Hovatta I, Lichtermann D, Juvonen H, Suvisaari J, Terwilliger JD, Arajärvi R, et al. Linkage analysis of putative schizophrenia gene candidate regions on chromosomes 3p, 5q, 6p, 8p, 20p and 22q in a population-based sampled Finnish family set. Mol Psychiatry. 1998;3:452-7.
72. McOmish CE, Burrows EL, Howard M, Hannan AJ. PLC-b1 Knockout Mice as a Model of Disrupted Cortical Development and Plasticity:Behavioral Endophenotypes and Dysregulation of RGS4 Gene Expression. Hippocampus. 2008;18:824-34.
73. Kind PC, Kelly GM, Fryer HJ, Blakemore C, Hockfield S. Phospholipase C-beta1 is present in the botrysome, an intermediate compartment-like organelle, and Is regulated by visual experience in cat visual cortex. J Neurosci. 1997;17(4):1471- 80.
74. Hannan AJ, Kind PC, Blakemore C. Phospholipase C-beta1 expression correlates with neuronal differentiation and synaptic plasticity in rat somatosensory cortex. Neuropharmacology. 1998;37:593-605.
75. Lin XH, Kitamura N, Hashimoto T, Shirakawa O, Maeda K. Opposite changes in phosphoinositide-specific phospholipase C immunoreactivity in the left prefrontal and superior temporal cortex of patients with chronic schizophrenia. Biol Psychiatry. 1999;46:1665-71.
76. Spires TL, Molnár Z, Kind PC, Cordery PM, Upton AL, Blakemore C, et al. Activity-dependent regulation of synapse and dendritic spine morphology in developing barrel cortex requires phospholipase C-beta1 signalling. Cereb Cortex. 2005. 15:385-93.
77. McOmish CE, Hannan AJ. Enviromimetics: exploring gene environment interactions to identify therapeutic targets for brain disorders. Expert Opin Ther Targets. 2007;11:899-913.
78. Van Den Buuse M, Garner B, Gogos A, Kusljic S. Importance of animal models in schizophrenia research. Aust N Z J Psychiatry. 2005;39(7):550-7.
79. Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS. Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science. 2000;288(5466):678-82.
80. Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signalling 4 (RGS4)
81. Margolis B, Rhee SG, Felder S, Mervic M, Lyall R, Levitzki A, et al. EGF induces tyrosine phosphorylation of phospholipase C-II:a potential mechanism for EGF receptor signalling. Cell. 1989;57:1101-1107.
82. Schmidt M, Evellin S, Weernink PA, von Dorp F, Rehmann H, Lomasney JW, et al. A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat Cell Biol. 2001;3:1020-1024.
83. Sadock BJ, Sadock VA. Kaplan and Sadock's Synopsis of Psychiatry: Behavioral Sciences/Clinical Psychiatry (9th ed.). Lippincott Williams & Wilkins; 2002
84. Simon GE. Social and economic burden of mood disorders. Biol Psychiatry. 2003;54(3):208-15.
85. Tanis KQ, Duman RS. Intracellular signalling pathways pave roads to recovery for mood disorders. Ann Med. 2007;39(7):531- 44.
86. Collins PY, Patel V, Joestl SS, March D, Insel TR, Daar AS, et al. Grand challenges in global mental health. Nature. 2011;475(7354):27-30.
87. Kupfer DJ, Frank E, Phillips ML. Major depressive disorder:new clinical, neurobiological, and treatment perspectives. Lancet. 2012;379:1045-1055
88. Guo L, Hu S. PI-PLC signal pathway:a possible pathogenesis link post-myocardial infarction to depression. Med Hypotheses. 2009;73(2):156-7.
89. Pacheco MA, Jope RS. Phosphoinositide signalling in human brain. Prog Neurobiol. 1996. 50 (2-3):255-273.
90. Hirschfeld RM, Cass AR, Holt DC, Carlson CA. Screening for bipolar disorder in patients treated for depression in a family medicine clinic. J. Am. Board Fam. Pract. 2005. 18 (4):233-239.
91. Hirschfeld RM. Differential diagnosis of bipolar disorder and major depressive disorder. J Affect Disord. 2014;169 Suppl 1:S12-6.
92. Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder:results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095-3105.
93. Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder:results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095-3105.
94. Hirschfeld RM, Calabrese JR, Weissman MM, Reed M, Davies MA, Frye MA, et al. Screening for bipolar disorder in the community. J Clin Psychiatry. 2003;64(1):53-59.
95. Hirschfeld RM, Lewis L, Vornik LA. Perceptions and impact of bipolar disorder:how far have we really come? Results of the National Depressive and Manic-Depressive Association 2000 survey of individuals with bipolar disorder. J Clin Psychiatry. 2003;64(2):161-174.
96. Hirschfeld RM, Williams JB, Spitzer RL, Calabrese JR, Flynn L, Keck PE Jr, et al. Development and validation of a screening instrument for bipolar spectrum disorder:the Mood Disorder Questionnaire. Am J Psychiatry. 2000;157(11):1873-1875.
97. Gawryluk JW, Young LT. Signal transduction pathways in the pathophysiology of bipolar disorder. Curr Top Behav Neurosci. 2011;5:139-65.
98. Murphy TM, Ryan M, Foster T, Kelly C, McClelland R, O'Grady J, et al. Risk and protective genetic variants in suicidal behaviour:association with SLC1A2, SLC1A3, 5-HTR1B &NTRK2 polymorphisms. Behav Brain Funct. 2011;7,22.
99. Aghajanian GK, Sanders-Bush E. Serotonin In:Davis KL, Charney D, Coyle JT, Nemeroff C (eds). Neuropsychopharmacology - The Fifth Generation of Progress. Lippincott Williams & Wilkins: New York, NY; 2002. pp 15-25.
100. Pandey GN, Dwivedi Y, Pandey SC, Teas SS, Conley RR, Roberts RC. Low phosphoinositide-specific phospholipase C activity and expression of phospholipase C β1 protein in the prefrontal cortex of teenage suicide subjects. Am J Psychiatry. 1999;156:1895-1901.
101. Numakawa T, Matsumoto T, Ooshima Y, Chiba S, Furuta M, Izumi A, et al. Impairments in brain-derived neurotrophic factor-induced glutamate release in cultured cortical neurons derived from rats with intrauterine growth retardation:possible involvement of suppression of TrkB/phospholipase C-γ activation. Neurochem Res. 2014;39(4),785-92.
102. Pandey GN. Biological basis of suicide and suicidal behavior. Bipolar Disord. 2013;15(5):524-41.
103. Pandey GN. Signal transduction abnormalities in suicide:focus on phosphoinositide signalling system. CNS Neurol Disord Drug Targets. 2013;12(7):941-53.
104. Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology:On the matter of their convergence. Mol Psychiatry. 2005;10:40-68.
105. Jarskog LF, Glantz LA, Gilmore JH, Lieberman JA. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:846-58.
106. Pérez-Neri I, Ramírez-Bermúdez J, Montes S, Ríos C. Possible mechanisms of neurodegeneration in schizophrenia. Neurochem Res. 2006;31:1279-94.
107. Levinson DF, Mowry BJ, Escamilla MA, Faraone SV. The Lifetime Dimensions of Psychosis Scale (LDPS): description and interrater reliability. Schizophrenia Bull. 2002 Jan 1;28(4):683-95.
108. Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593-624.
109. Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia:convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372-6.
110. Straub RE, MacLean CJ, O'Neill FA, Burke J, Murphy B, Duke F, et al. A potential vulnerability locus for schizophrenia on chromosome 6p24-22:evidence for genetic heterogeneity. Nat Genet. 1995;11:287-93.
111. Blouin JL, Dombroski BA, Nath SK, Lasseter VK, Wolyniec PS, Nestadt G, et al. Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet. 1998;20:70-3.
112. DeLisi LE, Shaw SH, Crow TJ, Shields G, Smith AB, Larach VW, et al. A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am J Psychiatry. 2002;159:803-12.
113. Williams NM, Rees MI, Holmans P, Norton N, Cardno AG, Jones LA, et al. A two-stage genome scan for schizophrenia susceptibility genes in 196 affected sibling pairs. Hum Mol Genet. 1999;8:1729-39.
114. Kendler KS, MacLean CJ, O'Neill A, Burke J. Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry. 1996;153:1534-40.
115. Cao Q, Martinez M, Zhang J, Sanders AR, Badner JA, Cravchik A, et al Suggestive evidence for a schizophrenia susceptibility locus on chromosome 6q and a confirmation in an independent series of pedigrees. Genomics. 1997;3:1-8.
116. Lindholm E, Ekholm B, Shaw S, Jalonen P, Johansson G, Pettersson U, et al A schizophrenia-susceptibility locus at 6q25, in one of the world's largest reported pedigrees. Am J Hum Genet. 2001;69:96-105.
117. Pulver AE, Karayiorgou M, Wolyniec PS, Lasseter VK, Kasch L, Nestadt G, Antonarakis S, et al. Sequential strategy to identify a susceptibility gene for schizophrenia:report of potential linkage on chromosome 22q12-q13.1:Part 1. Am J Med Genet. 1994;54:36- 43.
118. Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8. Additional support for schizophrenia linkage on chromosomes 6 and 8:a multicenter study. Am J Med Genet. 1996;67:580-94.
119. Bassett AS. Chromosomal aberrations and schizophrenia. Autosomes. Br J Psychiatry. 1992;161:323-34.
120. Paunio T, Ekelund J, Varilo T, Parker A, Hovatta I, Turunen JA, et al. Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet. 2001;10:3037-48.
121. Arinami T, Ohtsuki T, Ishiguro H, Ujike H, Tanaka Y, Morita Y, et al. Japanese Schizophrenia Sib-Pair Linkage Group. Genomewide high-density SNP linkage analysis of 236 Japanese families supports the existence of schizophrenia susceptibility loci on chromosomes 1p, 14q, and 20p. Am J Hum Genet. 2005;77:937-44.
122. Seeman P, Lee T. Antipsychotic drugs:Direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217-9.
123. Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481-3.
124. López-Figueroa AL, Norton CS, López-Figueroa MO, Armellini-Dodel D, Burke S, Akil H, et al. Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biol Psychiatry. 2004;55:225-33.
125. Bennett JP, Enna SJ, Bylund DB, Gillin JC, Wyatt RJ, Snyder SH. Neurotransmitter receptors in frontal cortex of schizophrenics. Arch Gen Psychiatry. 1979;36:927-34.
126. Watanabe S, Nishikawa T, Takashima M, Toru M. Increased muscarinic cholinergic receptors in prefrontal cortices of medicated schizophrenics. Life sci. 1983;33(22):2187-96.
127. Dean B, Crook JM, Opeskin K, Hill C, Keks N, Copolov DL. The density of muscarinic M1 receptors is decreased in the caudateputamen of subjects with schizophrenia. Mol Psychiatry. 1996;1:54-8.
128. Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2006;12:232-46.
129. Noga JT, Hyde TM, Herman MM, Spurney CF, Bigelow LB, Weinberger DR, et al. Glutamate receptors in the post-mortem striatum of schizophrenic, suicide, and control brains. Synapse. 1997;27:168-76.
130. Ohnuma T, Augood SJ, Arai H, McKenna PJ, Emson PC. et al Expression of the human excitatory amino acid transporter 2 and metabotropic glutamate receptors 3 and 5 in the prefrontal cortex from normal individuals and patients with schizophrenia. Brain Res Mol Brain Res. 1998;56:207-17.
131. Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci Lett. 2004;372:173-7.
132. Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315-25.
133. Fisher SK. Homologous and heterologous regulation of receptor-stimulated phosphoinositide hydrolysis. Eur J Pharmacol. 1995;288:231-50.
134. Wallace MA, Claro E. Transmembrane signalling through phospholipase C in human cortical membranes. Neurochem Res. 1993;18:139-45.
135. Jope RS, Song L, Li PP, Young LT, Kish SJ, Pacheco MA, et al. The phosphoinositide signal transduction system is impaired in bipolar affective disorder brain. J Neurochem. 1996;66:2402-9.
136. Shirakawa O, Kitamura N, Lin XH, Hashimoto T, Maeda K. Abnormal neurochemical asymmetry in the temporal lobe of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:867-77.
137. Jope RS, Song L, Powers R. (3H)PtdIns hydrolysis in postmortem human brain membranes is mediated by the G-protein Gq/11 and phospholipase C-b. Biochemistry. 1994;304:655-659.
138. Jose PA, Yu PY, Yamaguchi I, Eisner GM, Mouradian MM, Felder CC, et al. Dopamine D1 receptor regulation of phospholipase C. Hypertens Res. 1995;8(Suppl 1):S39-S42.
139. Jung H, Kim HJ, Lee SK, Kim R, Kopachik W, Han JK, et al. Negative feedback regulation of Wnt signalling by Gbetagammamediated reduction of Dishevelled. Exp Mol Med. 2009;41(10):695- 706.
140. Böhm D, Schwegler H, Kotthaus L, Nayernia K, Rickmann M, Köhler M, et al Disruption of PLC-beta 1-mediated signal transduction in mutant mice causes age-dependent hippocampal mossy fiber sprouting and neurodegeneration. Mol Cell Neurosci. 2002;21:584-601.
141. Shin J, Kim D, Bianchi R, Wong RK, Shin HS. Genetic dissection of theta rhythm heterogeneity in mice. Proc Natl Acad Sci USA. 2005;102:18165-70.