Keywords
Bevacizumab, Gemcitabine, Docetaxel, Melphalan, Carboplatin, High-dose chemotherapy, Autologous stem-cell transplantation, Germ-cell tumors
Commentary
Germ-cell tumors (GCT) are among the most curable solid malignancies, with approximately 70% of patients with advanced disease cured by frontline cisplatin-based standard-dose chemotherapy (SDC), with or without surgery [1]. However, outcomes are worse for patients whose tumors relapse or have refractory disease. In this setting, both salvage SDC with regimens such as paclitaxel, ifosfamide, and cisplatin (TIP), and high-dose chemotherapy (HDC) with carboplatin and etoposide (CE) supported by autologous stem-cell transplantation (ASCT), have curative potential [2–5]. Retrospective matched-pair analyses have suggested superior outcomes with HDC compared with SDC in first relapse [6,7], leading to the widespread adoption of multicycle HDC and inclusion in the US and European guidelines [8,9], although patient selection may partly explain these findings. In contrast to the frontline setting, where three randomized trials failed to show benefit of HDC over SDC [10–12], the question of the value of HDC in 1st relapse remains unsettled. The randomized international TIGER trial, comparing SDC with TIP to HDC with CE x 3 in untreated 1st relapse, has completed accrual and should identify the best approach in this scenario [13].
What is clear is that a substantial proportion of relapsed patients derive little benefit from conventional CE-based HDC, either given for 2 cycles (as pioneered at the University of Indiana more than 35 years ago [14]) or 3 cycles (as reported by the group at Memorial Sloan Kettering Cancer Center) [5]. Prognostic models, such as those developed by Beyer [6], and the International Prognostic Factor Study Group (IPFSG), identify distinct risk categories in first relapse with widely divergent relapse-free survival (RFS) rates [7]. For example, the latter model, developed and validated in large patient datasets, discriminates subgroups in first relapse with very low (2-year 75% RFS rate), low (51%), intermediate (40%), high (26%), and very high risk (6%). This highlights the need for more effective strategies for patients with poor prognosis, who can be prospectively identified.
The biological underpinnings of the poor response to CE-based HDC of high-risk groups appear related to dysregulation of DNA damage repair (DDR) mechanisms. While most cases of GCT are characterized by exceptional responsiveness to DNA-damaging agents, particularly cisplatin, through reduced capability of DDR [15,16], the reverse appears also to be true. Resistant GCT cell lines present a functional increase in the repair of DNA double-strand breaks by the homologous recombination repair pathway [17].
Against this background, we studied a novel HDC regimen consisting of gemcitabine, docetaxel, melphalan, and carboplatin (GemDMC), designed to exploit synergistic mechanisms related to DNA damage and inhibition of DDR [18]. Our preclinical work showed synergy between those agents in GCT cell lines with increased DNA damage and apoptosis (Figure 1). Our early clinical experience with this regimen suggested meaningful but anecdotal activity in heavily pretreated, refractory GCT [19,20], which led us to conduct a phase II trial of sequential HDC cycles with GemDMC followed by ICE (ifosfamide/carboplatin/etoposide) in patients with multiply relapsed or poor-risk disease. Given the high expression of vascular endothelial growth factor (VEGF) in GCT, its association with adverse prognosis, and prior reports of synergy between bevacizumab and chemotherapy for other solid tumors [21], we initially combined HDC with bevacizumab in both cycles [22]. Our study’s primary endpoint was two-year RFS, with secondary endpoints including overall response rate (ORR), complete response (CR), and overall survival (OS). We reported long-term outcomes and an off-trial validation in a subsequent cohort [15].
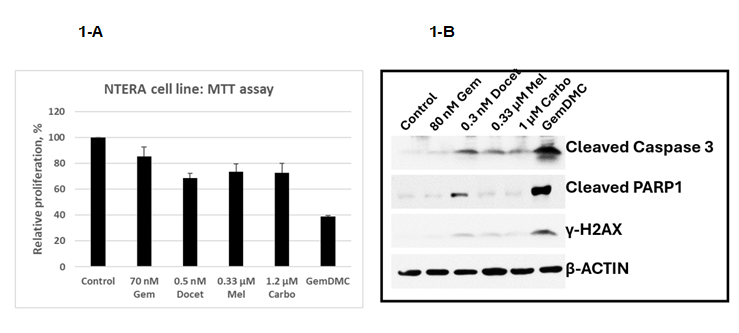
Figure 1. In vitro experiments in an embryonal carcinoma cell line (NTERA-2). 1-A: Cell proliferation (MTT) assay with cells exposed for 48 hours to 10% inhibitory concentrations (IC10) of each drug and the four combined (GemDMC). Error bars denote the standard deviation after 3 experiments. No statistical comparisons were performed. 1-B: Representative Western blot showing increased expression of protein associated with DNA damage (γ-H2AX) and apoptosis (cleaved caspase 3, cleaved PARP1) after exposure to GemDMC as compared to after each individual drug. The β-actin protein served as internal control. The antibodies used for immunoblotting are listed on Supplemental Table 1. Gem: Gemcitabine; Docet: Docetaxel; Mel: Melphalan; Carbo: Carboplatin.
Eligible patients were adolescents or adults with seminomatous or nonseminomatous GCT in second or later relapse, or in first relapse with intermediate- or high-risk features by the Beyer model (the one available at the time we designed our trial), who had adequate end-organ function, and who had previously collected a sufficient number of peripheral blood progenitor cells for two HDC cycles.
Between 2008 and 2017, we enrolled 65 male patients across three protocol-defined cohorts. These patients were heavily pretreated, with a median of three prior lines of therapy (range, 2–8), and the majority had cisplatin-refractory (defined as evidence of tumor progression within four weeks of the last cisplatin-based SDC cycle) or absolutely cisplatin-refractory disease (progressive disease as best response to cisplatin-based SDC). Most had high-risk features according to contemporary prognostic models, including very-high-risk disease by the IPFSG classification. Following completion of the trial, its results were prospectively validated in an additional cohort of 100 consecutive male patients treated off protocol with GemDMC (at reduced doses, as defined during the course of the trial) followed by ICE or carboplatin/etoposide, both without bevacizumab (as was also determined during the study). The validation cohort was broadly comparable to the trial population in baseline characteristics and risk stratification, although it included a higher proportion of patients with absolutely cisplatin-refractory disease and more extensive metastatic burden.
Treatment-related toxicity was substantial but manageable. GemDMC was associated primarily with mucositis, rash, and transient liver enzyme elevations and hyperbilirubinemia without evidence of venoocclusive disease. Treatment-related mortality (TRM) occurred in ten patients overall, predominantly due to infectious complications. The addition of bevacizumab increased toxicity, including longer duration of severe mucositis and higher TRM, without improving efficacy. A total of 123 patients were treated without bevacizumab (23 in study cohort 3 and 100 in validation cohort 4) with a 4% TRM rate. The second high-dose cycle using ifosfamide-containing regimens led to a significant incidence of late chronic kidney disease (CKD), attributed to ifosfamide nephrotoxicity, similar to what was also described by others [23]. The risk of CKD correlated with higher baseline creatinine (P=0.00001) and prior nephrectomy (P<0.001). Elimination of ifosfamide from the 2nd HDC cycle eliminated this complication without compromising antitumor activity. Hematopoietic recovery was prompt in most patients, although bevacizumab modestly delayed platelet engraftment. Late toxicities, besides the aforementioned ifosfamide-induced CKD, included peripheral neuropathy, hypogonadism, and hearing loss. A small number of second primary malignancies were observed during long-term follow-up.
Among patients with evaluable disease at the start of HDC, the ORR exceeded 80%, with the majority achieving a CR or partial response with normalized tumor markers (PRm-). Outcomes did not differ between bevacizumab-treated and non-bevacizumab-treated patients. Many patients underwent surgical resection of residual disease following completion of HDC, most commonly revealing necrosis or teratoma, although viable GCT was found in a subset and was associated with poor subsequent outcomes (Table 1).
|
Site |
N |
Pathological findings |
|
Lung |
38 |
|
|
Retroperitoneal lymph nodes |
32 |
|
|
Mediastinal mass |
18 |
|
|
Liver |
9 |
|
|
Kidney |
4 |
|
|
Abdominopelvic mass |
4 |
|
|
Peripheral lymph nodes |
3 |
|
|
Adrenal gland |
2 |
|
|
Testicle |
7 |
|
At a median follow-up of five years across all treated patients, including those enrolled in the trial and those in the validation cohort, the RFS and OS rates at both two and five years were approximately 57–60% (Figure 2). Trial patients followed for a median of 11 years demonstrated durable outcomes, with five-year RFS and OS exceeding 50%. These results were closely mirrored in the validation cohort (5-year RFS rate of 58%), confirming the reproducibility of the findings in routine clinical practice. Relapse after HDC typically occurred early, within months of treatment, and post-relapse survival was short, underscoring the limited efficacy of further salvage therapy. Even though the validation cohort included all consecutive patients who would have been eligible for the trial, we acknowledge the potential biases inherent to treating patients off protocol. While this might have influenced results, the outcomes of this cohort were very similar to those of the trial patients, validating the study results.
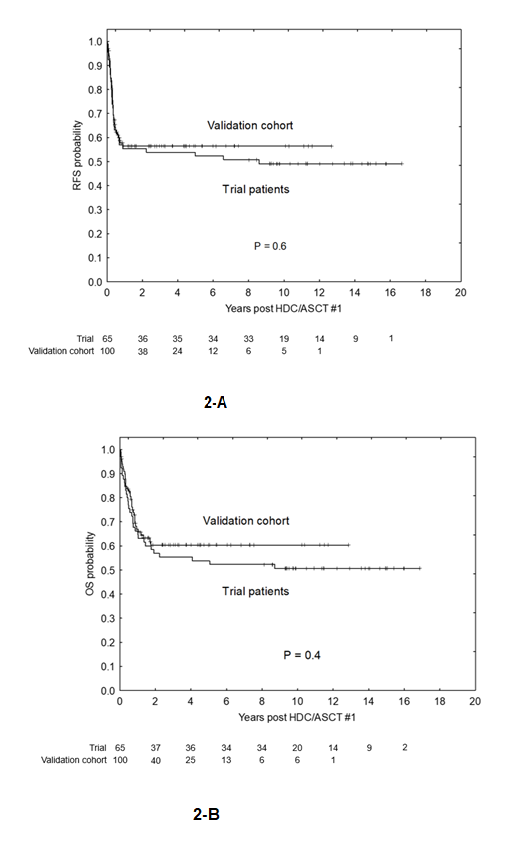
Figure 2. Patient outcomes. 2-A: Relapse-free survival (RFS) of trial patients (N=65) and the validation cohort (N=100). 2-B: Overall survival (OS) of trial patients and the validation cohort. Statistical comparisons used the log-rank test.
Established prognostic models retained their discriminatory power in this advanced population, including patients beyond first relapse. Higher-risk categories by Beyer, IPFSG, and Indiana models were associated with progressively worse RFS, yet outcomes within each risk group compared favorably with historical expectations based on prior HDC or SDC regimens. Particularly notable were the results in patients with extragonadal primaries, including primary mediastinal nonseminomatous germ-cell tumors (PMNSGCT), a group traditionally associated with very poor prognosis (Figure 3A). Approximately one-third of such patients in this study achieved long-term remission despite extensive prior therapy and frequent tumor progression at the time of HDC. Similarly, patients with brain involvement, many of whom had multiple prior relapses in the central nervous system, achieved outcomes comparable to those without brain disease when managed with a multimodal approach incorporating local control with stereotactic radiosurgery (SRS) (Figure 3B).
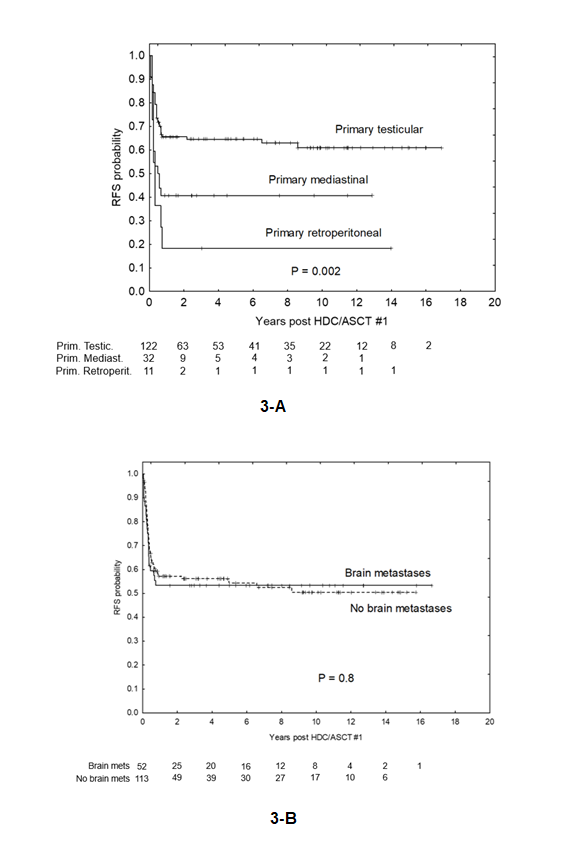
Figure 3. Outcomes of special populations. 3-A: Relapse-free survival (RFS) stratified by primary tumor site: testicular (N=122), mediastinal (N=32), or retroperitoneal (N=11). 3-B: RFS of patients with (N=52) and without (N=113) brain metastases. Statistical comparisons used the log-rank test.
Our multivariable analyses, limited by the relatively small size of 165 patients, identified achievement of CR or PRm- at the time of the first HDC cycle as the strongest favorable prognostic factor. Embryonal carcinoma histology was also associated with improved outcomes. In contrast, adverse prognostic factors included primary extragonadal tumors, very elevated AFP (>1,000 ng/mL) and HCG (>1,000 IU/mL) levels at the time of relapse, extensive metastatic involvement including liver, bone, or brain, and a history of multiple prior relapses.
In summary, our study showed that sequential HDC with GemDMC followed by CE can achieve durable remissions in patients with multiply relapsed or highly refractory GCT, a population historically associated with extremely poor outcomes. Long-term follow-up confirmed that survival plateaus are reached early, indicating that a substantial proportion of patients are likely cured rather than experiencing delayed relapse. Importantly, the outcomes we observed in our trial patients were reproduced in an independent, prospectively followed validation cohort, supporting the robustness and generalizability of the approach beyond the clinical trial setting.
The survival rates we observed compare favorably with outcomes reported from other HDC series, despite our cohort having more advanced disease, greater treatment resistance, and a higher frequency of second or later relapses (Table 2). Established prognostic models for patients in first relapse (Beyer, Indiana, and IPFSG) retained discriminatory power in this population, validating their relevance beyond first relapse and reinforcing the unusually strong performance of this regimen relative to predicted expectations. Notably, we showed for the first time that the IPFSG model remains informative in patients treated beyond initial relapse.
|
|
Indiana University [2] |
Indiana University [3] |
MSKCC [5] |
MDACC [18] |
P value |
|
|
N |
184 |
364 |
107 |
165 |
||
|
Time period |
1996–2004 |
2004–2014 |
1993–2006 |
2008–2017 |
||
|
HDC regimens |
CE x 2 |
CE x 2 |
TI-CE x 3 |
GemDMC – (I)CE |
||
|
Primary tumor |
Testicular Mediastinal Retroperitoneal |
100% 0% 0% |
87% 5% 8% |
67% 20% 6% |
74% 19% 7% |
<0.0001 (<0.0001*) |
|
Cisplatin sensitivity |
Sensitive Refractory |
78% 22% |
66% 34% |
26% 74% |
32% 68% |
<0.0001 |
|
No. prior lines of therapy |
1 2 3+ |
73% 24% 2% |
83% 15% 2% |
83% 15% 2% |
36% 31% 33% |
<0.0001 |
|
IPFSG |
Very low Low Intermediate High Very high |
N/A N/A N/A N/A N/A |
11% 18% 25% 25% 20% |
N/A N/A N/A N/A N/A |
0% 0% 7% 14% 79% |
<0.0001 |
|
Long-term EFS rate |
63% |
60% |
47% |
57% |
||
|
(*) The 2nd P value is calculated after omitting the 1st series from Indiana, which only included primary testicular cancers. |
||||||
Outcomes in patients with extragonadal primaries, particularly PMNSGCT, were especially noteworthy. Historically, this subgroup has shown limited benefit from HDC, leading to uncertainty about the value of aggressive therapy in such cases [24,25]. In contrast, a meaningful fraction (34%) of patients with multiply relapsed mediastinal disease in our study achieved long-term remission, even when treated in the setting of active progression. These findings suggest that carefully selected patients with extragonadal disease may still derive substantial benefit from intensified multimodal therapy.
Similarly, patients with brain metastases—often excluded from aggressive protocols—experienced outcomes comparable to those without central nervous system involvement. This supports the concept that effective local control prior to HDC, particularly SRS, is a critical component of successful treatment and that brain involvement should not automatically preclude consideration of curative-intent HDC.
From a biological standpoint, the GemDMC regimen represents a mechanistically distinct strategy compared with traditional CE-based HDC. We combined gemcitabine and carboplatin—two commonly used drugs for GCT— with docetaxel and melphalan, which are uncommonly used but have intrinsic activity in GCT [26–30]. Most importantly, this combination leverages the inhibition of DNA damage repair—particularly nucleotide excision repair—allowing the regimen to overcome key mechanisms of platinum resistance (Figure 4) [31]. The use of high-dose, prolonged-infusion gemcitabine is central to this strategy, avoiding saturation of its intracellular activating enzyme and increasing the availability of its active triphosphate metabolite for incorporation into the DNA [32,33]. This, in turn, potentiates the cytotoxic effects of the other three agents, which constitutes the mechanism of their synergy [34].
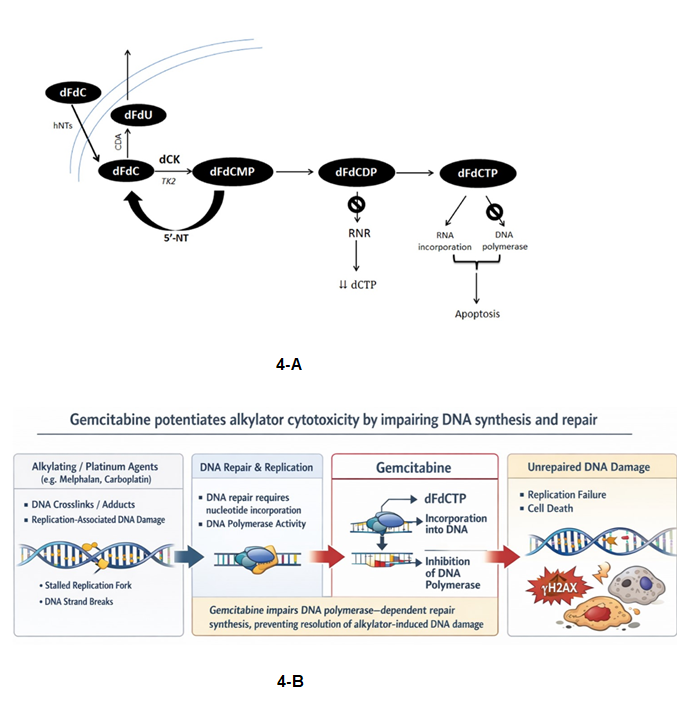
Figure 4. Synergistic interaction between gemcitabine and alkylating agents. 4-A: Intracellular activation of gemcitabine (dFdC, difluorodeoxycytidine) through sequential phosphorylation steps by the enzyme dCK (deoxycytidine kinase) with final production of its active metabolite dFdCTP (dFdC triphosphate), responsible for the inhibition of DNA polymerase. In this process, the intermediary metabolite dFdCDP (dFdC diphosphate) inhibits ribonucleotide reductase (RNR), which results in depletion of cellular pools of nucleotides. In our trial, the intracellular activation of gemcitabine was optimized by its continuous infusion at a fixed dose rate, which avoids saturation of dCK. 4-B: Graphic representation of the mechanism of synergy between gemcitabine and alkylators via inhibition of DNA damage repair.
The addition of bevacizumab, while theoretically attractive due to its potential effects on tumor vasculature and drug delivery, did not translate into improved efficacy and was associated with increased toxicity. Thus, our findings argue against incorporating anti-VEGF therapy into this high-dose platform and underscore the importance of balancing biologic rationale with tolerability in heavily pretreated patients. It also highlights the important point that a strong biological rationale does not always translate into clinical benefit, key lesson for future trial design.
While the non-randomized design and protocol modifications represent limitations, most patients ultimately received a refined version of the regimen with reduced toxicity and preserved efficacy. Given the rarity and clinical complexity of this population, randomized trials are unlikely to be feasible. In this context, the consistency of outcomes across cohorts and extended follow-up strengthens the credibility of the findings.
Commentary to Our Study by Kollmansberger et al.
In an accompanying editorial, Kollmansberger et al. argue against our conclusions [35]. These authors consider our outcomes to be promising but difficult to interpret, and that they ultimately do not improve on established HDC approaches.
In their opinion, CE-based HDC remains the standard salvage strategy, supported by decades of clinical experience. Prior evidence also indicates that adding additional drugs with limited single-agent activity mainly increases toxicity without improving survival. Along this line, they consider the biologic rationale for GemDMC weak, as its components have limited effectiveness in germ cell tumors and uncertain synergy.
They argue that the trial’s design further complicates interpretation: it was conducted at a single institution, involved heavily pretreated patients, and relied on prognostic models (Beyer and IPFSG) that had not been validated for multiply relapsed disease. Their main point is that outcomes appear similar to modern tandem CE results, most notably those reported by the Indiana group [3], but with substantially higher acute and long-term toxicity, including significant kidney disease, neuropathy, and treatment-related mortality.
Overall, they conclude that sequential GemDMC-based therapy is not ready to replace established HDC with tandem cycles of CE, which—ideally at first or second relapse—remains the recommended standard while awaiting results from ongoing randomized trials.
Rebuttal to Kollmansberger et al.
The arguments advanced by these authors in their commentary to our article rely heavily on comparisons with historical HDC series—most notably the Indiana University experience and IPFSG-stratified cohorts—that are not representative of the population treated in our study. This fundamental mismatch in baseline risk severely limits the validity of the conclusions they draw.
First, the majority of patients in the Indiana dataset were treated in first relapse (82%) and almost all of them received only one or two prior chemotherapy regimens (98%) [3], a group well known to have markedly better outcomes with CE-based HDC (Table 2). In contrast, our cohort consisted predominantly of multiply relapsed (median 2, range, 1–6 relapses; ≥ 3 relapses in 47% of patients), refractory (40% refractory and 34% absolutely refractory to cisplatin), and poor-risk patients (12% patients with a high and 81% with a very high risk IPFSG score, respectively), including individuals who had failed multiple (up to eight) salvage regimens—features consistently associated with dramatically inferior survival. Outcomes observed in earlier-relapse populations therefore cannot be used as a benchmark for efficacy or toxicity in a more advanced setting.
Second, we agree that the Beyer or IPFSG prognostic models had not been validated in multiply relapsed disease, at least not until our study, where we first showed their applicability beyond first relapse. Despite this, their critique repeatedly extrapolates survival expectations from these models to our cohort. This creates an internal inconsistency: if indirect comparisons are “impossible”, then claims of comparable or superior outcomes with tandem CE are likewise unsupported. The apparent similarity in survival outcomes between our study and earlier-line HDC series may, in fact, suggest meaningful activity of the GemDMC-based approach given the substantially worse baseline prognosis of our patients.
Third, the regimen GemDMC is criticized by the “limited single-agent activity” of two of its components, docetaxel and melphalan. While these drugs are uncommon in GCT management and the reports of their activity are scarce, this should not be considered tantamount to limited activity. Indeed, intrinsic activity in GCT has been reported both for docetaxel [26,27] and melphalan [28–30]. More importantly, the efficacy of GemDMC is not based on the added activity of each component, but rather, on its supra-additive synergistic interaction based on gemcitabine-mediated DDR inhibition, which potentiates the other agents (Figure 1).
Fourth, toxicity comparisons must be interpreted in light of cumulative treatment burden. Our patients were heavily pretreated with platinum, ifosfamide, taxanes, and in some cases HDC—factors known to independently increase the risk of renal, neurologic, and mucosal toxicity. Attributing long-term toxicities solely to the GemDMC regimen without adequately accounting for prior therapy exposure risks overstating regimen-specific harm and understating the contribution of disease biology and treatment history.
Fifth, the critique assumes that CE remains the optimal backbone across all relapse settings. While this seems well supported in earlier relapse, there is no high-level evidence, from a randomized trial or even large prospective data, demonstrating superiority of CE-based HDC in multiply relapsed or HDCT-refractory patients, precisely the population addressed in our trial. Therefore, in this multiply relapsed scenario, the default to CE as a comparator benchmark becomes itself an assumption, not an evidence-based standard. In such patients, exploring regimens incorporating non–cross-resistant agents with documented activity in platinum-refractory GCT represents a rational and necessary therapeutic strategy, rather than an unjustified deviation from standard practice.
Finally, the assertion that the regimen is “not ready for prime time” implicitly applies a standard derived from earlier-line salvage therapy to a population with few effective options and historically dismal outcomes. In this context, our phase II data, which demonstrates durable survival in subsets of patients—despite heavy pretreatment and adverse prognostic features—should be interpreted as clinically meaningful, not dismissed on the basis of inappropriate cross-trial comparisons.
In summary, their critique largely rests on comparisons to patient populations with fundamentally different prognostic profiles, treatment histories, and expected outcomes. When viewed within the appropriate clinical context—multiply relapsed, refractory, poor-risk GCT—the results of our study address an unmet need and cannot be invalidated by data derived from earlier-line, more favorable cohorts. Moving forward, it would seem reasonable to adopt a risk-adapted strategy, using tandem or triple CE for patients in first relapse (if corroborated by the randomized TIGER trial) and GemDMC-CE for patients with very advanced disease beyond first relapse, particularly with a very high-risk IPFSG score.
Conclusions
Our work supports sequential HDC with GemDMC followed by CE as a highly effective salvage strategy for patients with unfavorable, multiply relapsed, or refractory GCT, including PMNSGCT. Our results challenge historical pessimism surrounding this advanced poor-prognosis scenario and provide a strong rationale for biologically driven intensification strategies in selected high-risk patients.
References
2. Einhorn LH, Williams SD, Chamness A, Brames MJ, Perkins SM, Abonour R. High-dose chemotherapy and stem-cell rescue for metastatic germ-cell tumors. N Engl J Med. 2007 Jul 26;357(4):340–8.
3. Adra N, Abonour R, Althouse SK, Albany C, Hanna NH, Einhorn LH. High-Dose Chemotherapy and Autologous Peripheral-Blood Stem-Cell Transplantation for Relapsed Metastatic Germ Cell Tumors: The Indiana University Experience. J Clin Oncol. 2017 Apr 1;35(10):1096–02.
4. Gleeson JP, Knezevic A, Bromberg M, Patil S, Sheinfeld J, Carver BS, et al. Paclitaxel, Ifosfamide, and Cisplatin as Initial Salvage Chemotherapy for Germ Cell Tumors: Long-Term Follow-Up and Outcomes for Favorable- and Unfavorable-Risk Disease. J Clin Oncol. 2024 Sep 10;42(26):3130–39.
5. Feldman DR, Sheinfeld J, Bajorin DF, Fischer P, Turkula S, Ishill N, et al. TI-CE high-dose chemotherapy for patients with previously treated germ cell tumors: results and prognostic factor analysis. J Clin Oncol. 2010 Apr 1;28(10):1706–13.
6. Beyer J, Stenning S, Gerl A, Fossa S, Siegert W. High-dose versus conventional-dose chemotherapy as first-salvage treatment in patients with non-seminomatous germ-cell tumors: a matched-pair analysis. Ann Oncol. 2002 Apr;13(4):599–605.
7. Lorch A, Bascoul-Mollevi C, Kramar A, Einhorn L, Necchi A, Massard C, et al. Conventional-dose versus high-dose chemotherapy as first salvage treatment in male patients with metastatic germ cell tumors: evidence from a large international database. J Clin Oncol. 2011 Jun 1;29(16):2178–84.
8. Gilligan T, Lin DW, Aggarwal R, Chism D, Cost N, Derweesh IH, et al. Testicular Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2019 Dec;17(12):1529–54.
9. Oldenburg J, Fosså SD, Nuver J, Heidenreich A, Schmoll HJ, Bokemeyer C, et al. Testicular seminoma and non-seminoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013 Oct;24 Suppl 6:vi125-32.
10. Chevreau C, Droz JP, Pico JL, Biron P, Kerbrat P, Cure H, et al. Early intensified chemotherapy with autologous bone marrow transplantation in first line treatment of poor risk non-seminomatous germ cell tumours. Preliminary results of a French randomized trial. Eur Urol. 1993;23(1):213–7.
11. Daugaard G, Skoneczna I, Aass N, De Wit R, De Santis M, Dumez H, et al. A randomized phase III study comparing standard dose BEP with sequential high-dose cisplatin, etoposide, and ifosfamide (VIP) plus stem-cell support in males with poor-prognosis germ-cell cancer. An intergroup study of EORTC, GTCSG, and Grupo Germinal (EORTC 30974). Ann Oncol. 2011 May;22(5):1054–61.
12. Motzer RJ, Nichols CJ, Margolin KA, Bacik J, Richardson PG, Vogelzang NJ, et al. Phase III randomized trial of conventional-dose chemotherapy with or without high-dose chemotherapy and autologous hematopoietic stem-cell rescue as first-line treatment for patients with poor-prognosis metastatic germ cell tumors. J Clin Oncol. 2007 Jan 20;25(3):247–56.
13. Feldman DR, Huddart R, Hall E, Beyer J, Powles T. Is high dose therapy superior to conventional dose therapy as initial treatment for relapsed germ cell tumors? The TIGER Trial. J Cancer. 2011;2:374-7.
14. Broun ER, Nichols CR, Tricot G, Loehrer PJ, Williams SD, Einhorn LH. High dose carboplatin/VP-16 plus ifosfamide with autologous bone marrow support in the treatment of refractory germ cell tumors. Bone Marrow Transplant. 1991 Jan;7(1):53–6.
15. Cavallo F, Feldman DR, Barchi M. Revisiting DNA damage repair, p53-mediated apoptosis and cisplatin sensitivity in germ cell tumors. Int J Dev Biol. 2013;57(2-4):273–80.
16. Cavallo F, Graziani G, Antinozzi C, Feldman DR, Houldsworth J, Bosl GJ, et al. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS One. 2012;7(12):e51563.
17. Caggiano C, Cavallo F, Giannattasio T, Cappelletti G, Rossi P, Grimaldi P, et al. Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways. Cancers (Basel). 2021 Feb 13;13(4):787.
18. Nieto Y, Ward JF, Hofstetter W, Rice D, Karam JA, Pisters L, et al. High-Dose Chemotherapy for Multiply or Poor-Risk Relapsed Germ Cell Tumors. Clin Cancer Res. 2026 Jan 16;32(2):300–11.
19. Nieto Y, Shpall EJ, Bearman SI, McSweeney PA, Cagnoni PJ, Matthes S, et al. Phase I and pharmacokinetic study of docetaxel combined with melphalan and carboplatin, with autologous hematopoietic progenitor cell support, in patients with advanced refractory malignancies. Biol Blood Marrow Transplant. 2005 Apr;11(4):297–306.
20. Nieto Y, Aldaz A, Rifón J, Pérez-Calvo J, Zafra A, Zufia L, et al. Phase I and pharmacokinetic study of gemcitabine administered at fixed-dose rate, combined with docetaxel/melphalan/carboplatin, with autologous hematopoietic progenitor-cell support, in patients with advanced refractory tumors. Biol Blood Marrow Transplant. 2007 Nov;13(11):1324–37.
21. Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004 Feb;10(2):145–7.
22. Nieto Y, Tu SM, Bassett R, Jones RB, Gulbis AM, Tannir N, et al. Bevacizumab/high-dose chemotherapy with autologous stem-cell transplant for poor-risk relapsed or refractory germ-cell tumors. Ann Oncol. 2015 Oct;26(10):2125–32.
23. Feldman DR, Glezerman I, Patil S, Van Alstine L, Bajorin DF, Fischer P, et al. Phase I/II Trial of Paclitaxel With Ifosfamide Followed by High-Dose Paclitaxel, Ifosfamide, and Carboplatin (TI-TIC) With Autologous Stem Cell Reinfusion for Salvage Treatment of Germ Cell Tumors. Clin Genitourin Cancer. 2015 Oct;13(5):453–60.
24. Saxman SB, Nichols CR, Einhorn LH. Salvage chemotherapy in patients with extragonadal nonseminomatous germ cell tumors: the Indiana University experience. J Clin Oncol. 1994 Jul;12(7):1390–3.
25. Hartmann JT, Einhorn L, Nichols CR, Droz JP, Horwich A, Gerl A, et al. Second-line chemotherapy in patients with relapsed extragonadal nonseminomatous germ cell tumors: results of an international multicenter analysis. J Clin Oncol. 2001 Mar 15;19(6):1641–8.
26. Berruti A, Saini A, Gorzegno G, Tampellini M, Borasio P, Dogliotti L. Durable complete remission after weekly docetaxel administration in a patient with mediastinal non-seminomatous germ-cell tumor refractory to cisplatin-based chemotherapy. Ann Oncol. 2003 Oct;14(10):1589-90.
27. Yamada S, Saito H, Ohara S, Yamashita S, Mitsuzuka K, Namiki S, et al. Salvage chemotherapy with docetaxel, ifosfamide and nedaplatin (DIN) for patients with advanced germ cell tumors: a preliminary report. Jpn J Clin Oncol. 2013 Jul;43(7):734–9.
28. Blokhin N, Larionov L, Perevodchikova N, Chebotareva L, Merkulova N. [Clinical experiences with sarcolysin in neoplastic diseases]. Ann N Y Acad Sci. 1958 Apr 24;68(3):1128–32.
29. Li MC, Whitmore WF Jr, Golbey R, Grabstald H. Effects of combined drug therapy on metastatic cancer of the testis. JAMA. 1960 Nov 5;174:1291-9.
30. Solomon J, Steinfeld JL, Bateman JR. Chemotherapy of germinal tumors. Cancer. 1967;20:747–50.
31. Cavallo F, Graziani G, Antinozzi C, Feldman DR, Houldsworth J, Bosl GJ, et al. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS One. 2012;7(12):e51563.
32. Grunewald R, Kantarjian H, Du M, Faucher K, Tarassoff P, Plunkett W. Gemcitabine in leukemia: a phase I clinical, plasma, and cellular pharmacology study. J Clin Oncol. 1992 Mar;10(3):406–13.
33. Gandhi V, Plunkett W, Du M, Ayres M, Estey EH. Prolonged infusion of gemcitabine: clinical and pharmacodynamic studies during a phase I trial in relapsed acute myelogenous leukemia. J Clin Oncol. 2002 Feb 1;20(3):665–73.
34. Plunkett W, Huang P, Searcy CE, Gandhi V. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol. 1996 Oct;23(5 Suppl 10):3–15.
35. Kollmannsberger C, Nappi L, Nichols CR. Novel High-Dose Chemotherapy Regimens in Relapsed/Refractory Germ Cell Cancer: Have We Made Progress? Clin Cancer Res. 2026 Jan 16;32(2):257–59.