Abstract
Cancer poses a significant health challenge due to its complex nature and the limitations of current treatments. Over the last decade, emerging research has identified that modulation of immune checkpoint molecules such as CTLA-4, PD-1, and TIM-3 can have potential therapeutic benefits. These molecules are critical regulators of the immune system’s ability to target and eliminate cancer cells. This review explores the roles of these immune checkpoints in cancer and hematological malignancies; examining their mechanisms of action and the potential they hold for enhancing therapeutic outcomes. We highlight recent advancements in immunotherapy that leverage these checkpoints within the tumor microenvironment, demonstrating their potential to improve patient outcomes. Additionally, we discuss therapeutic metrics like objective response rates and treatment related adverse events in therapeutic combinations and in monotherapies; providing a comprehensive overview of the potential benefits, risks, and future directions in immunotherapy.
Keywords
Immune checkpoints, Immunotherapy, PD-1, CTLA-4, TIM-3, Cancer
Introduction
Cancer is the second-leading cause of death in the United States, and with an estimated 2 million new cases along with 600 thousand deaths projected in 2024, the urgency to understand its pathology and find effective treatments for patients has never been more apparent [1,2]. Furthermore, hematological malignancies are a leading cause of global tumor burdens, driven not only by the rapidly aging population but also by advances in innovative technologies, such as next-generation sequencing, which can lead to increased reported incidence. Additionally, over time, the criteria for diagnosing and classifying these malignancies has evolved, contributing to the rising incidence rates [2,3]. For decades, the standard of care was limited to surgery, chemotherapy, and radiation therapy, either as single treatments or in combination [4]. Though in recent history, research on cancer pathways and pathology has led to novel approaches to cancer treatment. A finely tuned immune response is crucial in cancer treatment; an excessively robust response risks autoimmunity, while an inadequate one offers minimal therapeutic benefit against cancer [5]. The mechanisms by which the immune system is stimulated or inhibited involve two kinds of immune checkpoint proteins, commonly known as co-stimulatory and inhibitory molecules. Stimulatory checkpoint molecules facilitate the activation and propagation of T cell mediated immune responses, whereas inhibitory molecules promote the inhibition of that immune response. The inhibition of inhibitory immune checkpoint molecules has shown clinical efficacy in many tumors and autoimmune diseases, including melanoma, non-small cell lung cancer, systemic lupus erythematosus, and type 1 diabetes [5]. In this review, we will outline the role of immune checkpoint molecules in cancer, with a specific focus on their mechanisms and clinical implications.
PD-1/PD-L1
Overview
CD279, also known as Programmed Cell Death 1 (PD-1), is a 55-kDa transmembrane inhibitory receptor primarily expressed on activated T cells across conditions including acute and chronic infection, cancer, and autoimmunity [5-7]. The expression of PD-1 is highly conserved in upstream regulatory regions (CR-B/CR-C) of the PD1 gene, PDCD1; these regions are involved in regulatory functions that influence PD-1 expression levels [6,7,9,10]. The associated proteins with this receptor include Programmed Cell Death Ligand 1 and Ligand 2 (PD-L1, PD-L2) [6-8]. The spatial discrepancy of the expression of these ligands is that PD-L1 is widely expressed in both hematopoietic and non-hematopoietic cells, whereas PD-L2 expression is exclusive to antigen presenting cells (APCs) (Table 1). Furthermore, the expression of both ligands can be induced by inflammatory signals within the tumor microenvironment [5].
|
Immune Checkpoints (IC) |
IC Ligands |
IC Expression |
Ligand Expression |
|
PD-1 |
PD-L1/L2 [5,8,25] |
CD4+/CD8+ T cells [5,8,25] |
PD-L1: hematopoietic and non-hematopoietic cells PD-L2: Antigen Presenting Cells (APCs) [5,8,25] |
|
CTLA-4 |
CD80/CD86 [5,24,25] |
Tregs/Memory T cells [5,24,25] |
APCs CD80: constitutive/ CD86: inducible [5,24,25] |
|
TIM-3 |
Gal-9/Cecam1/HMGB1/PtdSer [5,34-36] |
T cells/NK cells/APCs [5,34-36] |
Gal-9: Tregs/APCs HMGB1: secreted in cellular stress Cecam1: CD4+/CD8+ T cells PtdSer: apoptotic cells [5,34-36] |
Mechanism
To understand the mechanism by which PD-1 modulates immune response, it is first imperative to identify the mechanisms that regulate the expression of the PDCD1 gene [8]. The transcription of PD-1 in CD4+ and CD8+ T cells is regulated by various transcription factors, including NFAT (Nuclear Factor of Activated T cells), which is crucial for T cell activation and IRF9 (Interferon Regulatory Factor 9), which plays a role in cancer by regulating immune responses against tumors [6,8,10,11]. The involvement of these, and other transcription factors varies depending on whether the T cell is in a naïve or an exhausted state. In naïve T cells, which are mature but have not yet encountered their specific antigen, the stimulation of their T cell receptor (TCR) upon antigen recognition, combined with intracellular calcium circulation enhances the activity of NFAT, which then translocates to the nucleus to bind to the conserved promoter region of the PDCD1 gene, ultimately promoting PD-1 transcription (Figure 1) [6,8,11,22]. Similarly, in exhausted T cells, which have undergone chronic stimulation of their TCR due to persistent exposure of their specific antigen, intracellular calcium circulation is necessary, but interferon alpha (IFNα), a cytokine with anti-tumor properties, along with IRF9 binding to the promoter region of the PDCD1 gene promotes PD-1 transcription (Figure 1) [8,11]. Despite the differences in signal transduction molecules associated with PDCD1 gene expression across various T cell states, the core function of PD-1 remains consistent. The overarching function of the PD-1/PD-L1 pathway is to facilitate a balance of immune homeostasis by inhibiting immune responses and thus facilitating peripheral tolerance. The binding of PD-1 to its associated ligands inhibits TCR signaling, while also downregulating the expression of antiapoptotic molecules and proinflammatory cytokines; ultimately, this binding initiates the negative regulation of activated T cells, subsequently, promoting their inactivation [611,22]. To further understand the mechanism, it is critical that we examine the effects of the absence of PD-1 and PD-L1. Genetic ablation studies have shown that PD-1 receptor deficiencies are linked to various autoimmune phenotypes in murine models, highlighting the regulatory role of PD-1 in suppressing T cell response [23]. Similarly, it was illustrated that PD-L1 deficient mice were prone to autoimmune disease, suggesting that PD-L1 is a critical ligand for T cell immunotolerance.
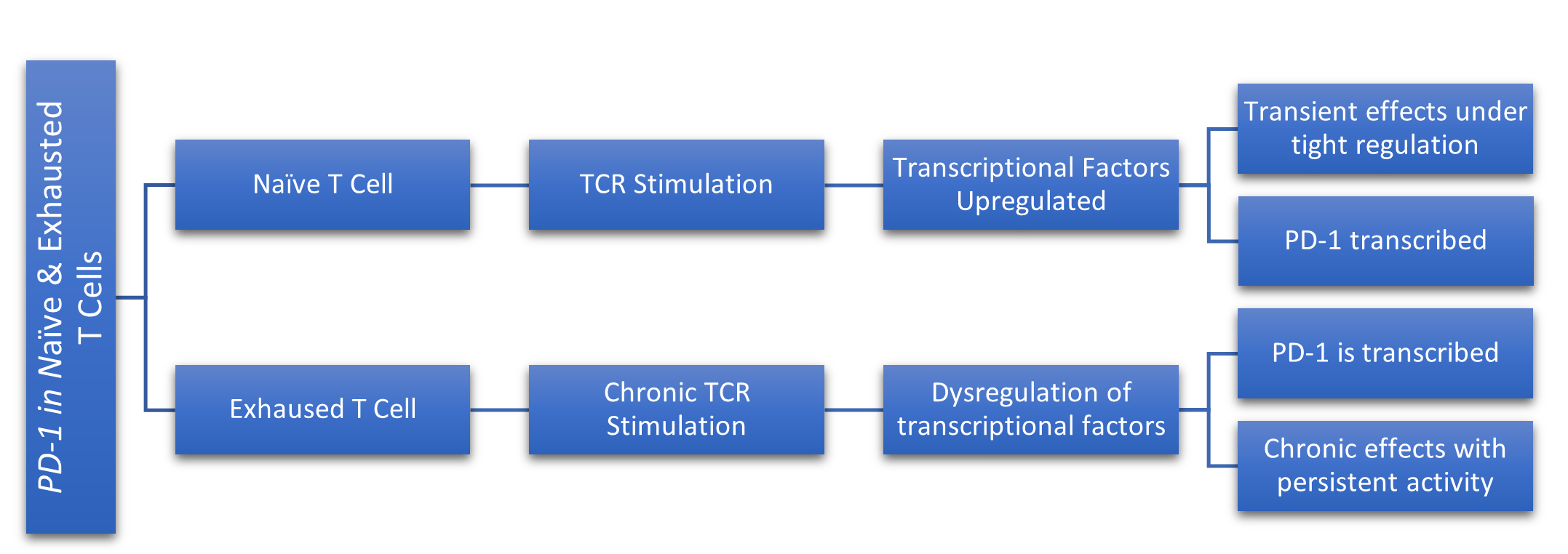
Figure 1. This figure simplifies the differences in PD-1 transcription between naive and exhausted T cells. In naive T cells, TCR stimulation leads to upregulation of transcriptional factors, resulting in transient PD-1 expression under tight regulation. However, in exhausted T cells, chronic TCR stimulation causes dysregulation of transcriptional factors, leading to persistent PD-1 expression and chronic inhibitory effects due to sustained activity.
CTLA-4
Overview
CD152, also known as Cytotoxic T Lymphocyte-associated antigen-4 (CTLA-4), is a transmembrane inhibitory glycoprotein mainly expressed upon activation of regulatory T cells and memory T cells [12,24]. The transcriptional regulation of CTLA-4 remains largely unexplored; however, it is known that CTLA-4 activation relies on NFAT activity. Consequently, modulation of NFAT levels directly impacts CTLA-4 expression [24,25]. CTLA-4 is a co-inhibitory molecule that negatively regulates T cell responses. It is a homolog to CD28, a co-stimulatory molecule that functions to positively regulate T cell responses. Consequently, these two molecules competitively bind the same proteins, CD80 and CD86 [12,25]. The expression patterns of these ligands differ in that CD80 is constitutively expressed on APCs, whereas CD86 exhibits inducible expression on APCs (Table 1 and Figure 2) [13,14,25]. With this in mind, we will examine the mechanism through which CTLA-4 exerts negative regulation on T cells and explore how tumor cells exploit this process to evade immune responses.
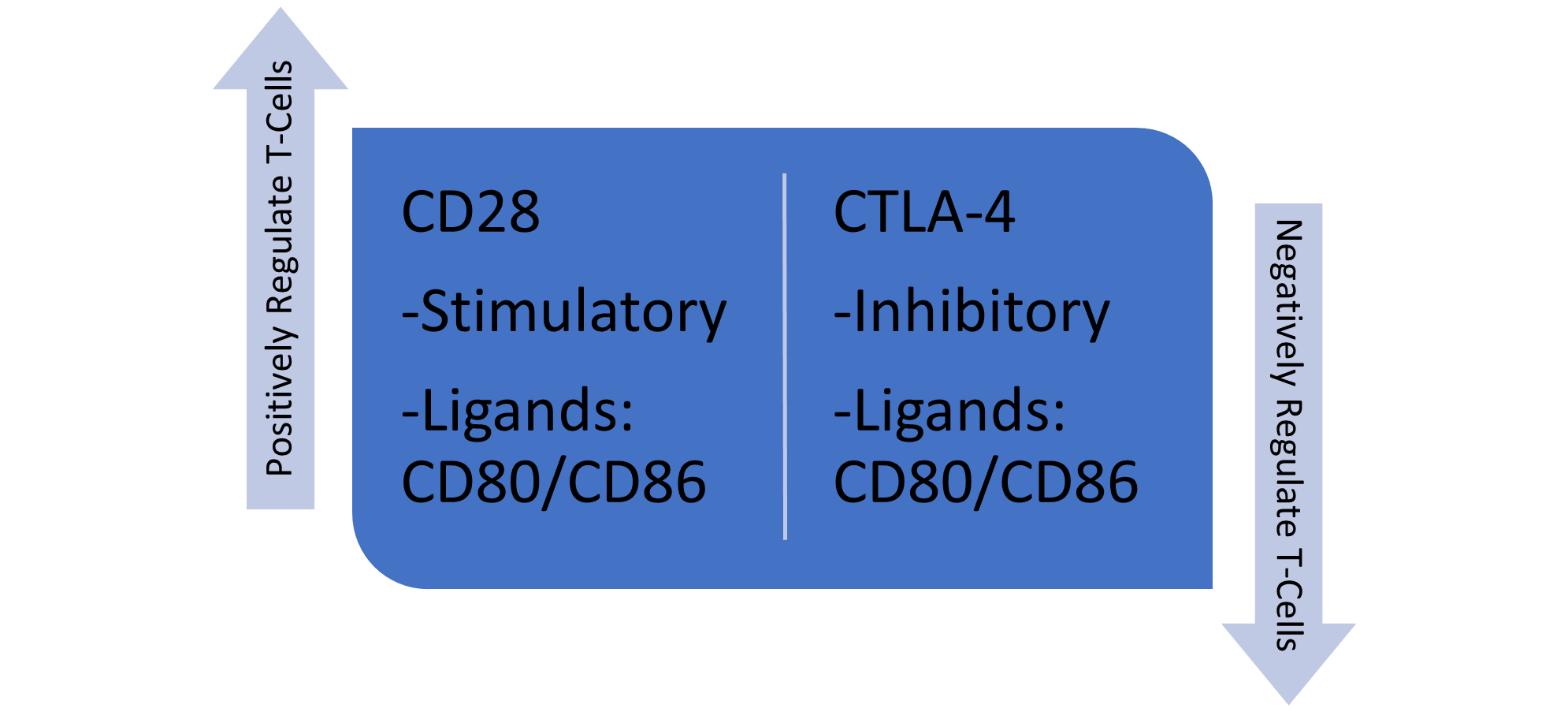
Figure 2. This figure compares the roles of CD28 and CTLA-4 in T cell regulation. CD28 is a stimulatory receptor that binds to CD80/CD86, promoting positive regulation and activation of T cells. In contrast, CTLA-4 is an inhibitory receptor that also binds to CD80/CD86, but it serves to negatively regulate and suppress T cell activity, ultimately, to balance the immune response.
|
IC Inhibitors (ICI) |
Drug Name |
Type of Antibody (Ab) |
Common Combination Therapy |
Clinical Trial Status |
|
Anti-PD-1/L1 |
Nivolumab/atezolizumab [29,60] |
Monoclonal |
Combined with other ICIs; i.e. CTLA-4/TIM-3 [29,60] |
Phase III Complete; commercialized (NCT01844505) [29,60] |
|
Anti-CTLA-4 |
Ipilimumab [60] |
Monoclonal |
Nivolumab |
Phase III Complete; commercialized (NCT01844505) [60] |
|
Anti-TIM-3 |
Cobolimab (TSR-022) [34,61] |
Monoclonal [34,61] |
Nivolumab |
Phase I - recruiting (NCT02817633) [34,61] |
Mechanism
As previously mentioned, CTLA-4 competes with CD28 for binding of CD80 and CD86. There are two potential mechanisms by which T cells may be inhibited by CTLA-4: through competitive antagonism and direct negative signaling. It is widely accepted that these mechanisms operate in conjunction rather than exclusively [12,13,25]. Competitive antagonism, as it relates to CTLA-4 and CD28, is characterized by the overwhelming avidity that CTLA-4 has for CD80/CD86; this affinity promotes the sequestration of CD80/CD86 molecules to the receptor of CTLA-4, thereby reducing the available co-stimulatory molecules for CD28 [14,15,25]. Inherently, one could understand how this molecular dynamic acts as an evolutionary safeguard against autoimmunity. However, this mechanism is dependent on a higher ratio of expression of CTLA-4:CD28 on T cell surfaces [25]. In the absence of genetic alterations affecting the CTLA-4 phenotype, the second mechanism involves direct negative signaling upon ligand binding through CTLA-4's cytoplasmic tail; ultimately facilitating the transmission of inhibitory signals that lead to T cell inactivation [25]. In the tumor microenvironment, tumor cells manipulate these homeostatic mechanisms of regulatory T cells (Tregs) to suppress immune responses. Evidence from an in vitro study demonstrated that Tregs mediated apoptosis in self-reactive CD8+ T cells upon recognition of self-antigens; researchers also observed the proliferative preference of self-antigen-specific T cells expressing CTLA-4 over self-antigen-specific T cells lacking CTLA-4 expression [26]. Expectedly, this illustrates how tumor cells directly impact tumor-immune interactions and immune regulation by inducing a tumor microenvironment that is conducive to immunosuppression by inhibiting self-reactive T cells and promoting the proliferation of self-tolerant T regulatory cells.
TIM-3
Overview
CD366, commonly known as T cell immunoglobulin domain and mucin domain 3 (TIM-3), is a type 1 transmembrane inhibitory glycoprotein that functions to inhibit both adaptive and innate immune responses; TIM-3 expression is detectable across a variety of immune cell types, including T cells, macrophages, natural killer (NK) cells, dendritic cells (DCs), and, adaptively, on tumor cells [34-36]. Furthermore, substantial evidence suggests that TIM-3 primarily functions as a negative regulator of CD4+ and CD8+ T cells that secrete interferon-gamma (IFN-γ), a pro-inflammatory cytokine critical for immune responses [34-37]. The TIM-3 protein, encoded by the HAVCR2 gene, is transcriptionally upregulated in the presence of pro-proliferative interleukins, particularly IL-12 and IL-27. This induced upregulation in CD4+ and CD8+ T cells is mediated through the STAT1/T-bet and STAT3/NFIL3 pathways, respectively [16,35]. Furthermore, TIM-3 interacts with four ligands: Galectin-9 (Gal-9), Carcinoembryonic antigen cell adhesion molecule 1 (Cecam1), High-mobility group box 1 (HMGB1), and Phosphatidylserine (PtdSer), each utilizing distinct biochemical pathways to exert differential modalities of immunosuppression (Table 1) [34-38]. As we continue to elaborate on TIM-3, we will critically assess the proposed mechanisms by which each of these ligands induces immunosuppression via TIM-3 stimulation, while also elucidating the clinical implications of these pathways.
Mechanism
To understand the receptor-ligand interaction of these distinct ligands, it is imperative to delineate the structural arrangement of the TIM-3 protein and its influence on the signaling pathway. The TIM-3 transmembrane inhibitory protein consists of four signaling domains: an extracellular variable immunoglobulin domain (IgV), a mucin-like domain, a transmembrane domain, and an intracellular cytoplasmic tail [34-37]. The IgV domain serves as the principal site for receptor-ligand interactions. This domain is composed of beta strands that form loops, which are stabilized by disulfide bonds, thereby resulting in a pocket-like structure consisting of the functional CC’ and FG loops [34,36,37]. When TIM-3 is engaged by ligands, the tyrosine residues within its cytoplasmic tail become phosphorylated via an interleukin-inducible kinase. This state facilitates the release of HLA-B-associated transcript 3 (BAT3) from the cytoplasmic tail of TIM-3; which initiates FYN, another tyrosine kinase, ultimately, negatively regulating T cell activation [34,38]. As aforementioned, the associated ligands of TIM-3 induce an immunosuppressive affect in T cells upon TIM-3 stimulation; the mechanisms by which this occurs differ in each ligand. Galectin-9, or Gal-9, is an approximate 36 kDa soluble protein that binds to the IgV domain of TIM-3 resulting in cell death of T cells, specifically Th1 cells; thereby, suppressing pro-inflammatory cytokine secretion and tissue inflammation (Figure 3) [17,36,37]. Moreover, it has been illustrated that Gal-9 induces programmed cell death in TIM-3+ CD8+ tumor-infiltrating-lymphocytes (TILs) in colon cancer; conversely, Gal-9 exhibits contrasting effects in NK cells, where it has been observed to enhance IFN-γ secretion; thereby promoting a cell-mediated immune response [18,37]. These opposing immune responses at the Gal-9-TIM-3 immunological synapse suggests that the effects of Gal-9 binding to TIM-3 are dependent on the cell of which TIM-3 is expressed. The second TIM-3 ligand, Cecam1, has unique properties and expression that allow for cis and trans type binding at the TIM-3-Cecam1 immunological synapse (Figure 3) [34,36,37]. Interestingly, TIM-3's inhibitory function is contingent upon co-expression with Ceacam1. This dependence on Ceacam1 expression suggests that Ceacam1 acts as a requisite ligand facilitating the intrinsic cis-binding mechanism necessary for TIM-3 to exert its inhibitory effects. Moreover, trans-binding of Ceacam1 facilitates similar downstream effects to that of cis-binding Ceacam1, ultimately leading to the suppression of effector T cell responses [37]. Despite these unique binding properties, CD8+ T cells that co-express Cecam1 and TIM-3 are subjected to T cell exhaustion and thus have dampened IFN-γ secretion, resulting in suppressed antitumor immunity [34]. The third associated ligand of TIM-3, high-mobility group box 1 (HMGB1), is a damage-associated molecular pattern (DAMP) protein that is secreted into the extracellular space by various cells in response to cellular stress [34,36,37,39]. While HMGB1 is recognized by TIM-3, it is also recognized by toll-like receptors (TLRs) and pattern-recognition receptors (PRRs), where it exerts effects counteractive to those mediated by TIM-3. Specifically, HMGB1 interaction with TLRs, such as TLR4, induces immune cell recruitment and pro-inflammatory cytokine secretion, thereby promoting anti-tumor immunity (Figure 3) [39-42]. Furthermore, TIM-3 competes with nucleic acids for HMGB1 binding; successful binding of HMGB1 by TIM-3 effectively inhibits nucleic acid interaction with HMGB1, thereby attenuating immune cell recruitment and activation. In contrast, formation of an HMGB1-nucleic acid-PRR complex triggers PRR-mediated immune responses, which have effects opposite to those of TIM-3 [39-42]. Lastly, the phospholipid, Phosphatidylserine (PtdSer), is one of the four ligands related to TIM-3. PtdSer is expressed on the cell surface of apoptotic cells. Biochemical studies have proposed that a metal cation, specifically calcium, is necessary for PtdSer to bind to the IgV domain of TIM-3 [34-37,43]. Once bound, the TIM-3-PtdSer complex in DCs promotes and initiates phagocytosis of apoptotic cells, while also inducing a cross-talking mechanism to present specific antigens, including PtdSer to T cells (Figure 3), who cannot directly phagocytize apoptotic cells, but instead employ a programmed cell death pathway directed at antigen-specific cells, similar effects to that of the Gal-9-TIM-3 complex [34-37,43,44]. Thus, in the PtdSer-TIM-3 interaction, TIM-3 functions as an immunomodulator that enhances the effects of immune tolerance and facilitates apoptotic cell clearance [36,43,44]. In conclusion, genetic mutations impacting the TIM-3 phenotype or sustained expression or activation by ligands and cytokines, commonly observed in infectious diseases and prominently in cancer, drive the protein into a dysfunctional state, leading to the indication of T cell exhaustion [34,45-47]. Given TIM-3’s regulatory role, we will further examine its clinical implications as it relates to immunotherapy, by highlighting preclinical and clinical studies.
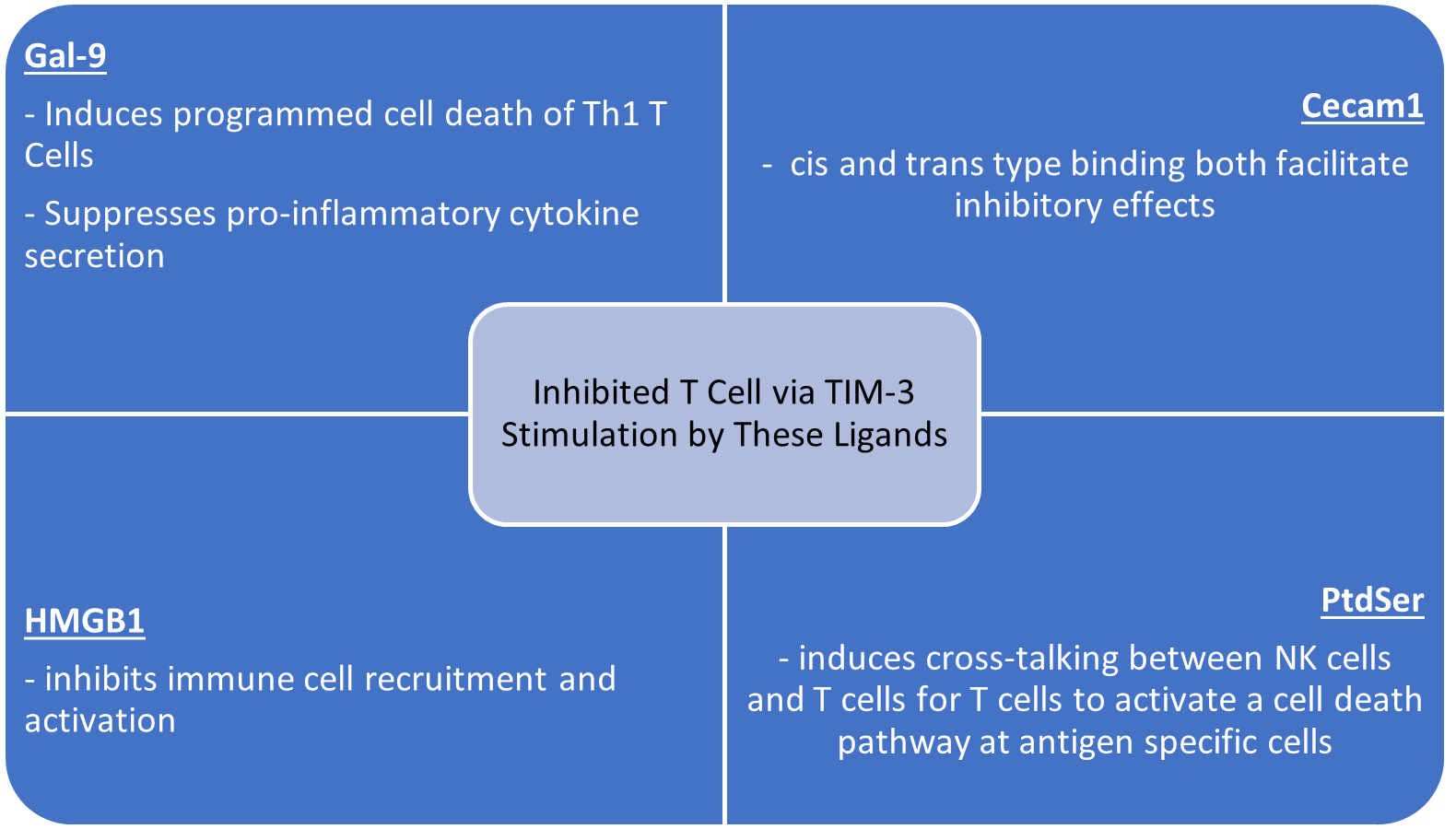
Figure 3. The figure illustrates how TIM-3 stimulation by its respective ligands inhibits T cell activity.
Applications
Overview
For well over two decades, cancer researchers have aimed to utilize the inherent mechanisms of the immune system to improve patient outcomes. The traditional standard of care, including surgery, chemotherapy, and radiation therapy, has achieved a measurable amount of success. These modalities, however, have a natural ceiling effect. As a result, scientists began exploring modulation of the immune system through immunotherapy in the 1990s and subsequently subjected it to clinical investigation in the 2010s [27]. These investigative efforts have led to the discovery of immune checkpoints that can be targeted with engineered antibodies (Ab). The rationale behind immune checkpoint inhibitors (ICIs) is based on blocking the molecular interactions between inhibitory molecules, such as PD-1 and CTLA-4, and their respective ligands, PD-L1/PD-L2 and CD80/CD86. Herein, we will evaluate the preclinical and clinical efficacy of ICIs as both monotherapy and in-combination, with a particular emphasis on their mechanism of action and effectiveness.
Monotherapy (Anti-PD-1/Anti-PD-L1)
Preclinical monotherapy: As aforementioned, the binding of PD-L1 to PD-1 promotes the negative regulation of T cell activation; moreover, this molecular interaction also contributes to the downregulation of regulatory T cell proliferation [19,22,28]. Within the tumor microenvironment, this attenuated immune response contributes to the increased PD-1 expression on regulatory T cells, thereby enhancing the immunosuppressive environment [28]. Investigators have identified two potential molecular targets for disrupting these interactions and signaling pathways. The first target, Anti-PD-1 blockade, has demonstrated in in vitro studies on non-small cell lung carcinoma (NSCLC) and ovarian tumor cells an increase in antigen-specific CD8+ T cells, leading to elevated cytokine production [28]. In the same study, the second target, Anti-PD-L1 blockade, also demonstrated a significant augmentation in CD8+ T cells [28].
Clinical trial monotherapy: Since the latter half of the 2010s, the FDA has sanctioned various PD-1/PD-L1 inhibitors, including atezolizumab. Phase I clinical trials of atezolizumab have demonstrated favorable tolerability and efficacy in patients with NSCLC, with a minimal incidence of treatment-related adverse events (10%) and an objective response rate of 23% across the cohort. Additionally, the one-year survival rate reached 63% for all participants [29]. Furthermore, in a phase II clinical trial involving a sub-cohort of 27 patients, from a larger cohort, with stage III or IV cervical cancer, 44% demonstrated no progression of their cancer after 6 weeks of treatment with atezolizumab (Table 2) [62]. Despite the promising outcomes observed in preclinical and clinical studies, further research is necessary to optimize pharmacodynamics and enhance the objective response rate.
Monotherapy (Anti-CTLA-4)
Preclinical monotherapy (Anti-CTLA-4): As previously noted, CTLA-4 competes with the co-stimulatory molecule CD28 for the binding of CD80/CD86. Consequently, antibodies targeting these ligands would be counterproductive, hence the investigation into Anti-CTLA-4. Inherently, blocking CTLA-4 increases the available ligands, CD80/CD86, for CD28 to bind and disrupt the immunosuppressive environment [30]. In preclinical murine models, researchers illustrated that CD28 blockade suppressed antitumor immunity. In contrast, they observed that CTLA-4 blockade led to increased cytokine production via CD28 stimulation, including IL-2, which in turn upregulated anti-apoptotic genes and disrupted the immunosuppressive tumor microenvironment [31].
Clinical trial monotherapy (Anti-CTLA-4): The FDA approval of the ICI, ipilimumab, demonstrated an improved overall survival (OS) in stage IV melanoma patients in its phase 3 trial [30]. The hypothesized mechanism underlying these patient outcomes suggests that anti-CTLA-4-mediated tumor eradication correlates with an elevated ratio of CD4+ effector cells to Tregs, as well as a similar effect on CD8+ effector cells [30]. Moreover, the preponderance of preclinical and clinical studies, suggests that CTLA-4 blockade induces a diffusive immunity, rather than a tumor-specific immunity; evident by trials in advanced melanoma and renal cell carcinoma, showing minimal objective responses [31]. While this drug proves effective, the consequent immune-related adverse events (irAEs) possibly stem from the elevated infiltration of T cells, most commonly manifesting as a rash [32].
Combination therapy (Anti-PD-1/Anti-PD-L1 + Anti-CTLA-4)
Preclinical combination: Both Anti-PD-1 and Anti-CTLA-4 individually confer therapeutic benefits. As a result, researchers have endeavored to combine the two treatments to investigate potential synergistic or additive effects. Investigators have shown in preclinical models that simultaneous blockade of PD-1 and CTLA-4 enhances OS and reduces tumor growth in both small and large established flank melanoma tumors [33]. Furthermore, they postulated that the simultaneous blockade significantly increased the ratios of both CD4+ and CD8+ effector T cells to Tregs [33]. In a separate preclinical investigation, scientists illustrated that inhibiting both immune checkpoint molecules, PD-1 and CTLA-4, during T cell priming led to increased T cell proliferation and cytokine secretion [28].
Clinical trial combination: Moreover, in a longitudinal clinical trial monitoring patients with metastatic melanoma receiving both nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4), the overall response rate was 40%, and 65% of the cohort exhibited varying degrees of tumor regression [28]. These studies indicate that, although ICIs act to modulate the immune response, they do so through distinct mechanisms. Future research will focus on elucidating the specific mechanisms of PD-1 and CTLA-4. Furthermore, advancements in combination therapy will address the unmet clinical needs in immunotherapy, which will be pivotal in counteracting the diverse strategies tumor cells employ to manipulate the immune system.
Combination therapy (Anti-TIM-3/Anti-PD-1)
Clinical trial combination: Over years of preclinical and clinical investigations, researchers have identified a correlation between elevated TIM-3 expression on exhausted CD8+ T cells in chronic viral infections [20,21,37]. Subsequent studies have further characterized TIM-3 expression in oncology, recognizing TIM-3 as a checkpoint inhibitor that contributes to the exhausted T cell phenotype within the tumor microenvironment, thereby suppressing antitumor immunity [21,37]. This has led to the immunotherapeutic exploration of drug candidates that target TIM-3; despite these investigative endeavors, monotherapy with anti-TIM-3 Ab has shown tolerability, but little clinical efficacy in overall response rates (ORR) [52-54]. In a Novartis-sponsored Phase I/II multicenter, open-label, double-arm clinical trial, investigators enrolled 219 patients with various metastatic solid tumors, predominantly ovarian cancer, to assess the clinical implications of sabatolimab, an anti-TIM-3 Ab, and spartalizumab, an anti-PD-1 Ab, either as monotherapy or in-combination. The study found no objective response rate with sabatolimab alone [49,52]. Moreover, in a phase I clinical trial funded by Tesaro, 173 patients were assigned randomly to three treatment cohorts assessing the ORR of TSR-022 (anti-TIM-3 Ab), TSR-022 + TSR-042 (anti-PD-1 Ab), and TSR-022 + docetaxel (chemotherapy) in the context of advanced solid tumor progression. The study reported an overall response rate (ORR) across all groups, with notable efficacy observed in the TSR-022 + TSR-042 arm. Importantly, grade ≥ 3 TRAEs were limited to 7.5% of participants in this cohort [34]. This study elucidates the specificity of anti-TIM-3 Abs, which contributed to the observed ORR, potentially through the induction of IFN-γ secretion in CD8+ T cells, while mitigating treatment-related adverse events [51].
Preclinical combination: Furthermore, in a preclinical study evaluating checkpoint immunotherapy, investigators inoculated murine glioma into C57BL/6 mice and assessed tumor progression under eight different randomized treatment arms: control, stereotactic radiosurgery (SRS), anti-TIM-3 Ab, anti-PD-1 Ab, anti-TIM-3 Ab + SRS, anti-PD-1 Ab + SRS, anti-TIM-3 Ab + anti-PD-1 Ab, and anti-TIM-3 Ab + anti-PD-1 Ab + SRS. In alignment with the previously referenced clinical study, it was observed that monotherapy with the anti-TIM-3 Ab did not yield a significant therapeutic effect [35]. Additionally, elevated TIM-3 expression, alongside chronic PD-1 expression, characterizes the exhausted T cell phenotype [21,37,52]. Based on this observation, the proposed rationale for employing a dual checkpoint blockade targeting TIM-3 and PD-1 is rooted in the concept of adaptive resistance. Tumor cells that exhibit refractoriness to anti-PD-1 treatment likely employ adaptive resistance mechanisms [37,52]. Given that TIM-3 expression is correlated with PD-1 expression in exhausted T cells, TIM-3 is upregulated, thus contributing to the immunosuppressive milieu [52]. TIM-3 represents a promising secondary target in conjunction with anti-PD-1 therapy; given that TIM-3 is predominantly expressed on IFN-γ-secreting T cells, targeting TIM-3 with specific antibodies can localize the immune response to tumor cells. This specificity has the potential to reduce treatment-related adverse events (TRAEs) [34,37]. In anti-TIM-3 immunotherapy, significant advancements have been achieved through combination approaches, particularly with anti-PD-1, as opposed to anti-TIM-3 monotherapy [36,37,52-54]. Through several preclinical and clinical trials, it is evident that TIM-3 immunotherapy is emerging as a promising immunotherapeutic target capable of reversing the exhausted T cell phenotype in the tumor microenvironment through multiple mechanisms, of particular interest, by inducing IFN-γ secretion through CD8+ T cells. Despite these significant advancements, the limitations of anti-TIM-3 Abs are evident due to the complex interactions with TIM-3 ligands. Future research should focus on augmenting bispecific antibodies that can bind multiple antigens, given the multifaceted ligand interactions of TIM-3. Additionally, considering the observed efficacy of anti-TIM-3/anti-PD-1 combination regimens, further optimization of these therapies could be achieved through in-depth investigations into the molecular interactions of TIM-3, its ligands, and the downstream signaling pathways within the tumor microenvironment.
Limitations
While immune checkpoint inhibitors have revolutionized cancer therapy, several limitations must be considered to fully understand their impact and applicability. These limitations encompass variability in patient responses, adverse events, and challenges in biomarker identification, all of which highlight the need for ongoing research and personalized treatment strategies. Inherent to the field of immunotherapy, inducing an immune response is often accompanied with general treatment-related adverse events, which can affect the therapeutic efficacy of the immune check point inhibitor. Moreover, the unpredictability of response rates adds to the inconsistencies seen in objective response rates among patients, and this uncertainty can be amplified in combination therapies. In relation to Figures 4 and 5, consolidating data from various clinical trials may introduce biases into the analysis; however, we maintained consistent variables across disease status and treatment type. Though, it is important to note that treatment duration and dosage varied across these trials.
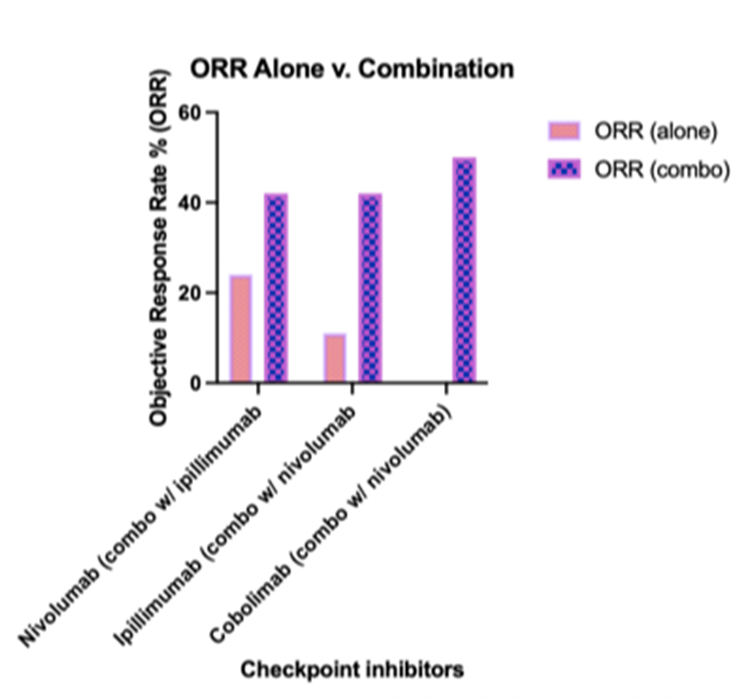
Figure 4. It is important to note that significant limitations exist, as the data has been consolidated from various clinical studies with differing trial designs. Nevertheless, the graph illustrates the relationship between monotherapy and combination therapy on overall response rate (ORR). Despite said limitations, the graph provides a clear comparative illustration of monotherapy versus combination therapy on ORR.
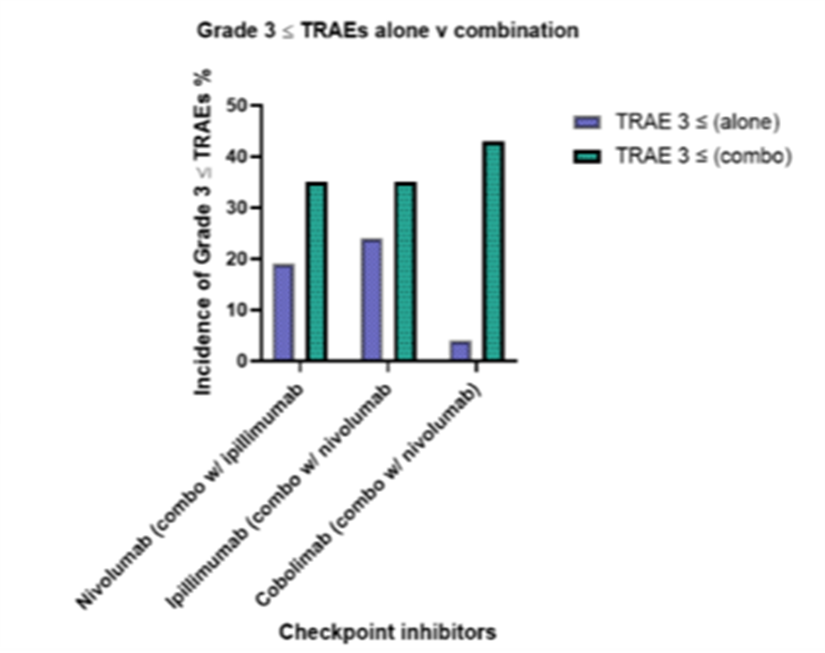
Figure 5. It is important to acknowledge that the data has been derived and consolidated from various clinical studies that differ in trial designs, which introduce significant limitations. Despite this, the graph depicts the comparative impact of monotherapy versus combination therapy on treatment-related adverse events (TRAEs).
Conclusion
The intricate molecular dynamics of immune checkpoint molecules such as PD-1, CTLA-4, and TIM-3 are pivotal in the regulation of immune responses and have profound implications for cancer therapy. The PD-1 and TIM-3 signaling pathways are crucial for maintaining immune homeostasis and peripheral tolerance; their dysregulation can result in autoimmunity, while in the context of cancer, it often enhances anti-tumor immunity. Likewise, CTLA-4 functions as a pivotal regulator by competing with CD28 for ligand binding, thereby modulating T cell activation. The therapeutic targeting of these checkpoints has ushered in a new modality of cancer treatment, with ICIs demonstrating promising clinical efficacy. Monotherapy with agents such as anti-PD-1 and anti-CTLA-4 has shown promising results, leading to improved patient outcomes and tumor regression. The success of agents like atezolizumab and ipilimumab in clinical trials highlights the potential of these therapies. On the contrary, the associated TRAEs highlight the need for careful management and further optimization of these treatments. Furthermore, combination therapies targeting multiple immune checkpoints offer a compelling strategy, potentially amplifying therapeutic benefits through synergistic or additive effects. The enhanced proliferation of effector cells observed in preclinical and clinical studies provides a strong rationale for continued exploration of combination regimens. As we advance, researchers must remain focused on unraveling the precise mechanisms of these immune checkpoints and refining therapeutic approaches to balance efficacy and safety. By harnessing the body's immune system, we edge closer to more effective and personalized cancer treatments, addressing the urgent need to improve outcomes for the millions affected by cancer and hematological malignancies globally.
The Author Disclosure Statement
The authors have nothing to disclose.
Acknowledgments
This study was supported by a grant from Des Moines University for Dr. Yujiang Fang (IOER 112-3119).
References
2. Zhang N, Wu J, Wang Q, Liang Y, Li X, Chen G, et al. Global burden of hematologic malignancies and evolution patterns over the past 30 years. Blood Cancer J. 2023 May 17;13(1):82.
3. Global Burden of Disease 2019 Cancer Collaboration; Kocarnik JM, Compton K, Dean FE, Fu W, Gaw BL, Harvey JD, et al. Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life Years for 29 Cancer Groups From 2010 to 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. JAMA Oncol. 2022 Mar 1;8(3):420-44.
4. Debela DT, Muzazu SG, Heraro KD, Ndalama MT, Mesele BW, Haile DC, et al. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med. 2021 Aug 12;9:20503121211034366.
5. Huang C, Zhu HX, Yao Y, Bian ZH, Zheng YJ, Li L, et al. Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J Autoimmun. 2019 Nov;104:102333.
6. Salmaninejad A, Khoramshahi V, Azani A, Soltaninejad E, Aslani S, Zamani MR, et al. PD-1 and cancer: molecular mechanisms and polymorphisms. Immunogenetics. 2018 Feb;70(2):73-86.
7. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020 Mar 1;10(3):727-42.
8. Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, et al. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J Cell Physiol. 2019 Aug;234(10):16824-37.
9. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity. 2011 Sep 23;35(3):400-12.
10. Chi Z, Lu Y, Yang Y, Li B, Lu P. Transcriptional and epigenetic regulation of PD-1 expression. Cell Mol Life Sci. 2021 Apr;78(7):3239-46.
11. Bally AP, Austin JW, Boss JM. Genetic and Epigenetic Regulation of PD-1 Expression. J Immunol. 2016 Mar 15;196(6):2431-7.
12. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. 2018 Jan 4;131(1):58-67.
13. Jaffar ZH, Stanciu L, Pandit A, Lordan J, Holgate ST, Roberts K. Essential role for both CD80 and CD86 costimulation, but not CD40 interactions, in allergen-induced Th2 cytokine production from asthmatic bronchial tissue: role for alphabeta, but not gammadelta, T cells. J Immunol. 1999 Dec 1;163(11):6283-91.
14. Thompson CB, Allison JP. The emerging role of CTLA-4 as an immune attenuator. Immunity. 1997 Oct;7(4):445-50.
15. Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016 Feb;39(1):98-106.
16. Banerjee H, Kane LP. Immune regulation by Tim-3. F1000Res. 2018 Mar 14;7:316.
17. Yang R, Sun L, Li CF, Wang YH, Yao J, Li H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021 Feb 5;12(1):832.
18. Rahmati A, Bigam S, Elahi S. Galectin-9 promotes natural killer cells activity via interaction with CD44. Front Immunol. 2023 Mar 16;14:1131379.
19. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013 Apr;13(4):227-42.
20. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010 Aug 17;107(33):14733-8.
21. Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol. 2014 Aug 15;193(4):1525-30.
22. McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med. 2013 Oct;2(5):662-73.
23. Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015 Sep;125(9):3384-91.
24. Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol. 2006;24:65-97.
25. Zhang H, Dai Z, Wu W, Wang Z, Zhang N, Zhang L, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res. 2021 Jun 4;40(1):184.
26. Sobhani N, Tardiel-Cyril DR, Davtyan A, Generali D, Roudi R, Li Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers (Basel). 2021 Mar 22;13(6):1440.
27. Chamoto K, Hatae R, Honjo T. Current issues and perspectives in PD-1 blockade cancer immunotherapy. Int J Clin Oncol. 2020 May;25(5):790-800.
28. Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol Immunol. 2015 Oct;67(2 Pt A):4-17.
29. Horn L, Gettinger SN, Gordon MS, Herbst RS, Gandhi L, Felip E, et al. Safety and clinical activity of atezolizumab monotherapy in metastatic non-small-cell lung cancer: final results from a phase I study. Eur J Cancer. 2018 Sep;101:201-9.
30. Blank CU, Enk A. Therapeutic use of anti-CTLA-4 antibodies. Int Immunol. 2015 Jan;27(1):3-10.
31. Wolchok JD, Saenger Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist. 2008;13 Suppl 4:2-9.
32. Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006 Apr;18(2):206-13.
33. Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013 Sep 15;19(18):4917-24.
34. Zeidan AM, Komrokji RS, Brunner AM. TIM-3 pathway dysregulation and targeting in cancer. Expert Rev Anticancer Ther. 2021 May;21(5):523-34.
35. Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol. 2018 Oct 11;11(1):126.
36. Zhao L, Cheng S, Fan L, Zhang B, Xu S. TIM-3: An update on immunotherapy. Int Immunopharmacol. 2021 Oct;99:107933.
37. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017 Mar;276(1):97-111.
38. Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl Oncol. 2020 Mar;13(3):100738.
39. Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. 2022 Feb;54(2):91-102.
40. Li G, Liang X, Lotze MT. HMGB1: The Central Cytokine for All Lymphoid Cells. Front Immunol. 2013 Mar 20;4:68.
41. Srikrishna G, Freeze HH. Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia. 2009 Jul;11(7):615-28.
42. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007 Sep;13(9):1050-9.
43. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010 Feb 15;184(4):1918-30.
44. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood. 2009 Apr 16;113(16):3821-30.
45. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010 Sep 27;207(10):2187-94.
46. Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest. 2012 Apr;122(4):1271-82.
47. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011 Apr 28;117(17):4501-10.
48. Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. 2020 Jun;8(1):e000911.
49. Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin Cancer Res. 2021 Jul 1;27(13):3620-9.
50. Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin Cancer Res. 2017 Jan 1;23(1):124-36.
51. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011 May 15;71(10):3540-51.
52. Sauer N, Janicka N, Szlasa W, Skinderowicz B, Kołodzińska K, Dwernicka W, et al. TIM-3 as a promising target for cancer immunotherapy in a wide range of tumors. Cancer Immunol Immunother. 2023 Nov;72(11):3405-25.
53. Shaffer A. Novel Immunotherapy Combos Target TIM-3 and PD-1/PD-L1 Networks. Oncology Live. 2022;22(24).
54. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010 Sep 27;207(10):2175-86.
55. Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017 Jan;18(1):31-41.
56. Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, et al. Nivolumab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016 Sep 1;34(25):2980-7.
57. Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, et al. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2015 Jun 20;33(18):2004-12.
58. Farzeen Z, Khan RRM, Chaudhry AR, Pervaiz M, Saeed Z, Rasheed S, et al. Dostarlimab: A promising new PD-1 inhibitor for cancer immunotherapy. J Oncol Pharm Pract. 2024 Jul 26:10781552241265058.
59. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015 May 21;372(21):2006-17.
60. Phase 3 Study of Nivolumab or Nivolumab Plus Ipilimumab Versus Ipilimumab Alone in Previously Untreated Advanced Melanoma (CheckMate 067). (2024, July 3). Clinicaltrials.gov. https://www.clinicaltrials.gov/study/NCT01844505
61. A Study of TSR-022 in Participants With Advanced Solid Tumors (AMBER). (2024, June 20). Clinicaltrials.gov. https://clinicaltrials.gov/study/NCT02817633
62. Tabernero J, Andre F, Blay JY, Bustillos A, Fear S, Ganta S, et al. Phase II multicohort study of atezolizumab monotherapy in multiple advanced solid cancers. ESMO Open. 2022 Apr;7(2):100419.