Commentary
It is now well accepted that the ionizing radiationgenerated reactive oxygen species (ROS), that constitute ~2/3 of the effects of external beam radiation, do not only produce direct tumor cell death, but also affect the surrounding microenvironment. Moreover, this indirect effect of radiation may result in systemic effects, specifically the initiation of an inflammatory response.
We have previously shown the existence of an endothelial-stem cell linked pathway that is activated by single-high-dose radiotherapy (SDRT) (>8 Gy/fraction). This endothelial-stem cell linkage pathway mediates normal tissue injury after SDRT of the intestines, lung and salivary glands [1-3], suggesting that this represents a generic response mechanism for mammalian tissue damage injury by large single-dose irradiation, i.e. ablative radiation therapy. Prior studies reported by Moeller et al. [4] provided strong evidence that bursts of ROS are generated by waves of hypoxia/reoxygenation that occur after each radiation exposure in response to conventional fractionated radiotherapy in tumors. Recently, Mizrachi et al. [3] demonstrated that SDRTinduced salivary glands (SG) hypofunction was to a large extent mediated by microvascular dysfunction involving ceramide and ROS generation.
ROS generation is important in the maintenance of homeostasis between pro-apoptotic and pro-survival signals [5-7]. However, enhanced accumulation of ROS generates chronic pathological conditions. While a large number of studies on oxidative stress focused on the mitochondria-generated ROS, Wortel et al. [8] in their recent study confirmed that the endothelium is one of the major sources of ROS and identified the different types of ROS generated by the SDRT-induced oxidative stress, both within the plasma membrane and in the cytosol of endothelial cells (Figure 1). Similar findings were previously reported by our collaborators in the same cells in response to Fas ligand, tumor necrosis factor-α (TNF-α), endostatin, and homocysteine [9-15].
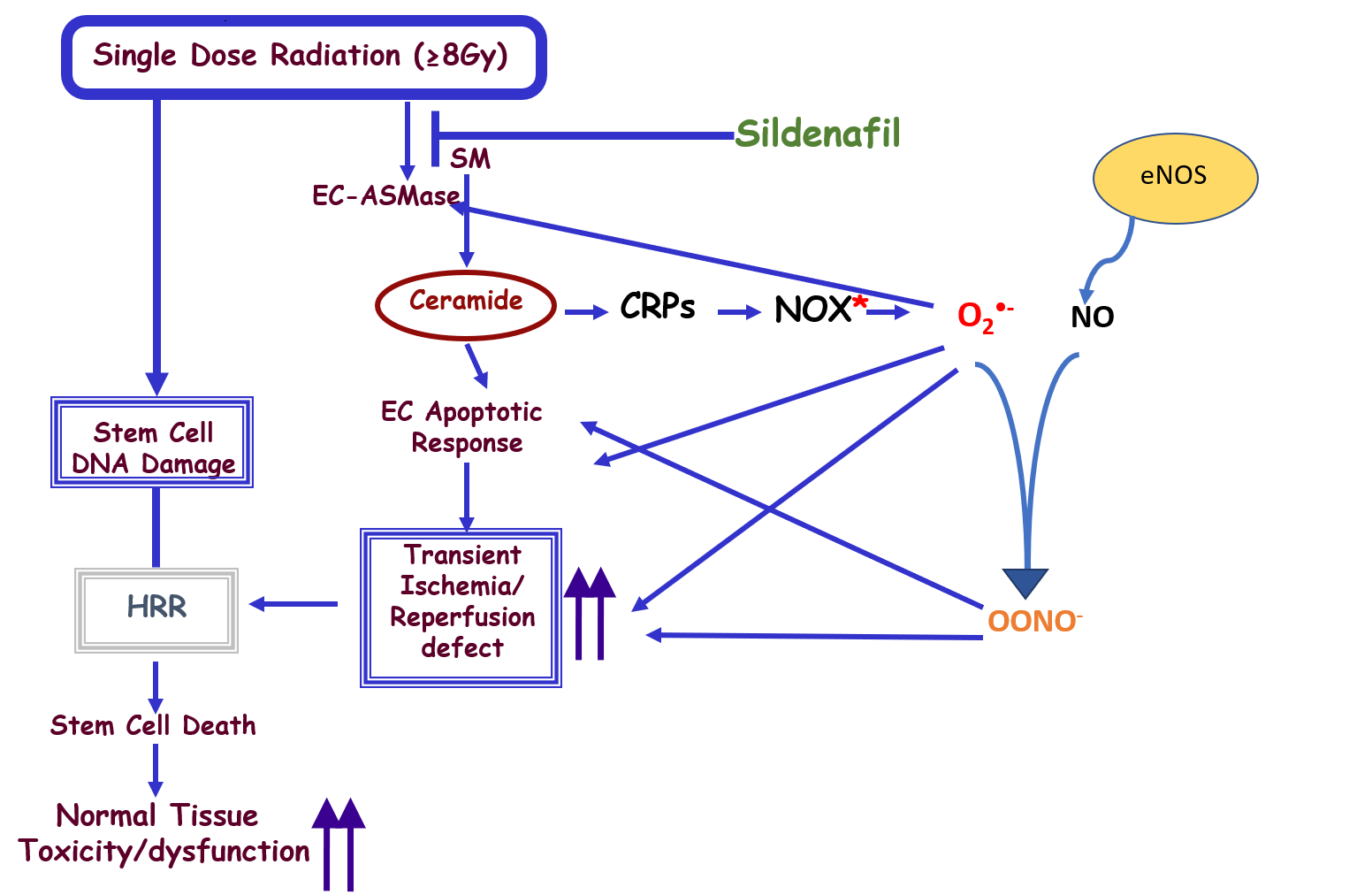
ASMase: Acid Sphingomyelinase; BAECs: Bovine Aortic Endothelial Cells; CRPs: Ceramide-Rich Platforms; NOX: NADPH Oxidase; HRR: Homologous Recombination Repair
Ours as well as others previous studies have shown that the endothelial cells within the different tissues are the most sensitive cells to the effects of ionizing radiation (IR), since they are 20-fold enriched in secretory acid sphingomyelinase (ASMase), as compared to any other cell type in the body [16]. We also showed that ceramide is a sphingolipid messenger capable of initiating apoptotic cascades in response to various stressful stimuli, including IR [17,18]. IR induced alterations in the plasma membrane hydrolyzes sphingomyelin to generate ceramide via sphingomyelinases activation [19,20]. The sphingomyelinases are expressed preferentially in the vascular endothelium [21], suggesting that these mechanisms may be of particular relevance for vascular structure and function. Furthermore, data derived from acid sphingomyelinase-knock-out mice, showed that they have a radioresistant vasculature and are partially protected from end-organ radiation injury [2,22] emphasizing the biological significance of this phenomenon.
Advances in cancer diagnosis and treatment have led to increases in life expectancy, bringing forward the issue of long-term treatment-related morbidity and mortality in these patients. Numerous reports have concluded that these patients should be regarded as long-term cancer survivors rather than as healthy individuals due to their long-term risk of developing treatment-related adverse events, and in particular cardiovascular events. With this clinical background in mind, several groups have embarked in the search for the mechanism(s) by which these effects are inflicted upon the cardiovascular system with special attention to the vascular wall and the vascular endothelium [23-26].
Radiation induces microvascular dysfunction via activation of the acid sphingomyelinase (ASMase)/ ceramide pathway. Microvascular dysfunction is crucial for tumor response to radiation. ASMase activation triggers the generation of ceramide-rich platforms (CRPs), NADPH oxidase (NOX) activation and subsequent production of ROS, resulting in microvascular endothelial dysfunction (Figure 1) [27]. Elevated ROS formation in the vascular wall is a key feature of all cardiovascular diseases and a likely contributor to endothelial cell dysfunction, vascular inflammation and plaque formation. The NOX family of enzymes comprises seven members (NOX1-5, DUOX1-2), with each one of them displaying distinct patterns of expression, intracellular compartmentalization, regulation, and biological function. NOX-derived ROS control multiple aspects of cell physiology via redox-activated signaling pathways. Nevertheless, NOX over-activity, a condition that is typically associated with significant up-regulation of its expression, has been increasingly reported in a variety of cardiovascular diseases [28]. Numerous studies have demonstrated that expression and activity of at least two isoforms of NADPH oxidase - NOX1 and NOX2 - is increased in animal models of hypertension, diabetes and atherosclerosis. Several studies in transgenic mice support the role for NOX1- and/or NOX2-containing oxidases as sources of excessive vascular ROS production and as triggers of endothelial cell dysfunction in hypertension, atherosclerosis and diabetes [29].
The apolipoprotein-E-deficient (ApoE-/-) mouse is the most widely studied animal model of hypercholesterolemia and atherosclerosis [30,31]. It has been reported that ApoE-/-/p47phox-/- double KO are protected, compared to the ApoE-/- single KO mice, from the development of aortic atherosclerotic lesions [32-34]. Moreover, Drummond et al. [24] demonstrated that ApoE-/-/NOX2-/- double KO mice were also protected from endothelial cell dysfunction and development of atherosclerotic lesions, suggesting that at least some of the protective actions of p47phox deletion were likely due to inhibition of NOX2 activity. A major mechanism by which NOX-derived ROS contribute to vascular disease is via superoxide (O2•-)-mediated inactivation of NO, resulting in loss of its vasoprotective actions, and the subsequent generation of the highly reactive ROS, peroxynitrite (OONO-) (Figure 1). Peroxynitrite is a powerful oxidant that causes irreversible damage to macromolecules including proteins, lipids, and DNA, thereby disrupting crucial cell signaling pathways and promoting cell death.
Recent studies have indicated that NOX-derived O2•- production in the extracellular compartment may be markedly increased during vascular disease. Atherogenic stimuli such as tumor necrosis factor-α (TNF-α), endostatin, cholesterol, and homocysteine increase endothelial cell expression of NOX1 and NOX2, cause CRP generation, resulting in NOX2 activation in the plasma membrane (PM) [12,15,35,36]. The translocation of p47phox to the cytosolic face of these CRP-containing NOX2 aggregates is likely to result in ‘hot spots’ of activity in the endothelial cells PM. This, combined with the accumulation of macrophages (which normally express NOX2 oxidase in the PM) in the vessel wall, will result in markedly higher amounts of O2•- being generated in the extracellular space, thereby increasing the likelihood of NO breakdown and production of OONO-. This may explain, at least in part, why the majority of oxidative damage detected in atherosclerotic lesions occurs in the extracellular matrix space [37]. All cardiovascular pathologies have in common excessive NOX-dependent ROS formation associated with upregulation of the various NOX subtypes. In this study, Wortel et al., [8] were able to demonstrate for the first time that IR induces excessive NOX activation and ROS generation. This study and other studies published recently by their collaborators [38], demonstrate that ROS generation is an indispensable mediator of SDRTinduced ischemia/reperfusion pathobiology in tumors. They also demonstrated the transient and immediate O2•- generation and the subsequent accumulation of peroxynitrite (OONO-) after SDRT, which resulted in impaired endothelial function.
Recently, the first class of the dual NOX1 and NOX4 pharmacological inhibitors, GKT137831, received the approval for phase II clinical trial for the treatment of diabetic nephropathy. According to a recent press release of Genkyotex, the leading pharmaceutical company that develops NOX inhibitors, treatment of patients with diabetic nephropathy with GKT137831, significantly reduced liver enzymes and markers of inflammation. Moreover, the beneficial effects of GKT137831 were reported in several experimental models of disease, including atherosclerosis, hypertension, and diabetes [39-41], emphasizing the major role of the endothelial dysfunction in these pathological conditions and the involvement of NOX-mediated ROS.
Obtaining profound knowledge on the pathogenesis of IR-induced vascular injury could help stratify patients who might be at risk for cardiovascular events and potentially identify specific biomarkers that would warrant close surveillance and indicate specific diagnostic interventions. We postulate that acute IR-induced microvascular dysfunction of normal tissue vasculature may contribute to and increase the risk for long-term cardiovascular morbidity and other vascular related diseases.
Wortel’s [8] finding that sildenafil protected endothelial cells from RT-induced oxidative stress in the endothelial cells via reduction of NOX-mediated ROS formation and, thus inhibited the pro-apoptotic ASMase/ceramide pathway, is of paramount importance. The endothelium plays a role in the initiation of pulmonary oxidative injury induced by Ischemia/Reperfusion (I/R), and endothelial cells could be either a source or a target of oxidants. Oxidative injury to endothelium has been found to set the stage for secondary pulmonary injury by leukocytes [42,43].
While it is known that sildenafil affects NO levels in the blood vessels, Wortel et al., [8] elucidated the cellular mechanism by which sildenafil, attenuates endothelial dysfunction and protects against radiation-induced erectile dysfunction (ED). This study showed that sildenafil inhibited RT-induced NOX generation of ROS in endothelial cells and protected them from apoptotic death via the acid sphingomyelinase (ASMase)/ceramide pathway. Specifically, by inhibiting ASMase and ceramide generation, sildenafil significantly inhibited O2•- generation, and subsequently NO was not used to generate ONOO- [44]. ONOO- can disrupt crucial cell signaling pathways and initiate cell death by causing damage to macromolecules, including proteins, lipids, and more critically DNA [3,44]. In addition, to reducing the toxicity of ONOO-, sildenafil maintained the bioavailability of NO in the endothelial cells, and therefore preserved its vasoprotective properties. Reduced NO bioavailability is considered one of the main characteristics of endothelial dysfunction, which is also linked to erectile dysfunction [45]. In addition, since endothelial cells are a major target for RT-induced pneumonitis, GI syndrome, kidney damage, and xerostomia and because it is known that sildenafil is well tolerated, repurposing this drug should be considered for the treatment and prevention of RTinduced side effects, including cardiotoxicity for which it was initially successfully introduced.
Many cancer survivors find themselves struggling with health issues related to prior cancer treatment many years after they are declared cancer-free. These problems include chronic pain, neuropathy, infertility, recurrent infections, memory problems, sexual health issues, cognitive impairments and more, including increased risk of secondary malignancy. For many cancer survivors, these health issues last a lifetime, and some might even be life threatening. Mitigating treatment side effects by protecting the vasculature, in particular the generation of ROS, may result in a significant improvement of patient’s quality of life by reducing morbidity and mortality.
Funding
This study was funded by the NIH Grant 5R01 DK114321-02.
References
2. Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, Haimovitz-Friedman A, Cordon-Cardo C, Kolesnick R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001 Jul 13;293(5528):293-7.
3. Mizrachi A, Cotrim AP, Katabi N, Mitchell JB, Verheij M, Haimovitz-Friedman A. Radiation-induced microvascular injury as a mechanism of salivary gland hypofunction and potential target for radioprotectors. Radiation Research. 2016 Aug;186(2):189-95.
4. Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004 May 1;5(5):429-41.
5. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita KI, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Reports. 2001 Mar 1;2(3):222-8.
6. Choi TG, Lee J, Ha J, Kim SS. Apoptosis signalregulating kinase 1 is an intracellular inducer of p38 MAPK-mediated myogenic signalling in cardiac myoblasts. Biochimica et Biophysica Acta (BBA)- Molecular Cell Research. 2011 Aug 1;1813(8):1412-21.
7. Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. ROS-dependent activation of the TRAF6- ASK1-p38 pathway is selectively required for TLR4- mediated innate immunity. Nature Immunology. 2005 Jun;6(6):587-92.
8. Wortel RC, Mizrachi A, Li H, Markovsky E, Enyedi B, Jacobi J, Brodsky O, Cao J, Lippert AR, Incrocci L, Mulhall JP. Sildenafil Protects Endothelial Cells From Radiation-Induced Oxidative Stress. The Journal of Sexual Medicine. 2019 Nov 1;16(11):1721-33.
9. Zhang C, Hu JJ, Xia M, Boini KM, Brimson C, Li PL. Redox signaling via lipid raft clustering in homocysteineinduced injury of podocytes. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2010 Apr 1;1803(4):482-91.
10. Yi F, Jin S, Li PL. Lipid raft-redox signaling platforms in plasma membrane. Methods in Molecular Biology. 2009;580:93-107.
11. Jin S, Yi F, Zhang F, Poklis JL, Li PL. Lysosomal targeting and trafficking of acid sphingomyelinase to lipid raft platforms in coronary endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008 Nov 1;28(11):2056-62.
12. Jin S, Zhang Y, Yi F, Li PL. Critical role of lipid raft redox signaling platforms in endostatin-induced coronary endothelial dysfunction. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008 Mar 1;28(3):485-90.
13. Jin S, Yi F, Li PL. Contribution of lysosomal vesicles to the formation of lipid raft redox signaling platforms in endothelial cells. Antioxidants & Redox Signaling. 2007 Sep 1;9(9):1417-26.
14. Zhang C, Li PL. Membrane raft redox signalosomes in endothelial cells. Free Radical Research. 2010 Aug 1;44(8):831-42.
15. Zhang AY, Yi F, Zhang G, Gulbins E, Li PL. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension. 2006 Jan 1;47(1):74-80.
16. Tabas I. Secretory sphingomyelinase. Chemistry and Physics of Lipids. 1999 Nov 1;102(1-2):123-30.
17. Schenck M, Carpinteiro A, Grassmé H, Lang F, Gulbins E. Ceramide: physiological and pathophysiological aspects. Archives of Biochemistry and Biophysics. 2007 Jun 15;462(2):171-5.
18. Liu H, Geng S, Feng C, Xie X, Wu B, Chen X, et al. A DNA vaccine targeting p42. 3 induces protective antitumor immunity via eliciting cytotoxic CD8+ T lymphocytes in a murine melanoma model. Human vaccines & immunotherapeutics. 2013 Oct 4; 9(10):2196- 202.
19. Haimovitz-Friedman A, Kan CC, Ehleiter D, Persaud RS, Mcloughlin M, Fuks Z, Kolesnick RN. Ionizing radiation acts on cellular membranes to generate ceramide and initiate apoptosis. The Journal of Experimental Medicine. 1994 Aug 1;180(2):525-35.
20. Chmura SJ, Nodzenski E, Beckett MA, Kufe DW, Quintans J, Weichselbaum RR. Loss of ceramide production confers resistance to radiation-induced apoptosis. Cancer Research. 1997 Apr 1;57(7):1270-5.
21. Marathe S, Schissel SL, Yellin MJ, Beatini N, Mintzer R, Williams KJ, Tabas I. Human vascular endothelial cells are a rich and regulatable source of secretory sphingomyelinase implications for early atherogenesis and ceramide-mediated cell signaling. Journal of Biological Chemistry. 1998 Feb 13;273(7):4081-8.
22. American Cancer Society Facts and Figures. 2008.
23. Takac I, Schröder K, Brandes RP. The Nox family of NADPH oxidases: friend or foe of the vascular system?. Current Hypertension Reports. 2012 Feb 1;14(1):70-8.
24. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease?. Trends in Endocrinology & Metabolism. 2014 Sep 1;25(9):452-63.
25. Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxidants & Redox Signaling. 2014 Jun 10;20(17):2794-814.
26. Kero AE, Järvelä LS, Arola M, Malila N, Madanat- Harjuoja LM, Matomäki J, Lähteenmäki PM. Cardiovascular morbidity in long-term survivors of earlyonset cancer: a population-based study. International Journal of Cancer. 2014 Feb 1;134(3):664-73.
27. Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005 Jan 1;25(1):29-38.
28. Manea A. NADPH oxidase-derived reactive oxygen species: involvement in vascular physiology and pathology. Cell and Tissue Research. 2010 Dec 1;342(3):325-39.
29. Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxidants & Redox Signaling. 2014 Jan 1;20(1):164-82.
30. Jawień J, Nastałek P, Korbut R. Mouse models of experimental atherosclerosis. Journal of Physiology and Pharmacology. 2004 Sep;55(3):503-17.
31. Getz GS, Reardon CA. Animal models of atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012 May;32(5):1104-15.
32. Barry-Lane PA, Patterson C, Van Der Merwe M, Hu Z, Holland SM, Yeh ET, Runge MS. p47phox is required for atherosclerotic lesion progression in ApoE–/– mice. The Journal of Clinical Investigation. 2001 Nov 15;108(10):1513-22.
33. Vendrov AE, Hakim ZS, Madamanchi NR, Rojas M, Madamanchi C, Runge MS. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arteriosclerosis, thrombosis, and vascular biology. 2007 Dec 1;27(12):2714-21.
34. Judkins CP, Diep H, Broughton BR, Mast AE, Hooker EU, Miller AA, Selemidis S, Dusting GJ, Sobey CG, Drummond GR. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. American Journal of Physiology-Heart and Circulatory Physiology. 2010 Jan;298(1):H24-32.
35. Wei YM, Li X, Xiong J, Abais JM, Xia M, Boini KM, Zhang Y, Li PL. Attenuation by statins of membrane raftredox signaling in coronary arterial endothelium. Journal of Pharmacology and Experimental Therapeutics. 2013 May 1;345(2):170-9.
36. Boini KM, Zhang C, Xia M, Han WQ, Brimson C, Poklis JL, Li PL. Visfatin-induced lipid raft redox signaling platforms and dysfunction in glomerular endothelial cells. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2010 Dec 1;1801(12):1294-304.
37. Woods AA, Linton SM, Davies MJ. Detection of HOCl-mediated protein oxidation products in the extracellular matrix of human atherosclerotic plaques. Biochemical Journal. 2003 Mar 1;370(2):729-35.
38. Bodo S, Campagne C, Thin TH, Higginson DS, Vargas HA, Hua G, Fuller JD, Ackerstaff E, Russell J, Zhang Z, Klingler S. Single-dose radiotherapy disables tumor cell homologous recombination via ischemia/reperfusion injury. The Journal of Clinical Investigation. 2019 Jan 14;129(2).
39. Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP. NADPH oxidase 1 plays a key role in diabetes mellitus–accelerated atherosclerosis. Circulation. 2013 May 7;127(18):1888-902.
40. Gorin Y, Cavaglieri RC, Khazim K, Lee DY, Bruno F, Thakur S, Fanti P, Szyndralewiez C, Barnes JL, Block K, Abboud HE. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. American Journal of Physiology-Renal Physiology. 2015 Jun 1;308(11):F1276-87.
41. Somanna NK, Valente AJ, Krenz M, Fay WP, Delafontaine P, Chandrasekar B. The Nox1/4 dual inhibitor GKT137831 or Nox4 knockdown inhibits angiotensin-IIinduced adult mouse cardiac fibroblast proliferation and migration. AT1 physically associates with Nox4. Journal of Cellular Physiology. 2016 May;231(5):1130-41.
42. Seibert AF, Haynes J, Taylor A. Ischemia-reperfusion injury in the isolated rat lung: role of flow and endogenous leukocytes. American Review of Respiratory Disease. 1993 Feb;147(2):270-5.
43. Lubna NJ, Nakamura Y, Cao X, Wada T, Izumi- Nakaseko H, Ando K, Sugiyama A. Cardiac safety profile of sildenafil: chronotropic, inotropic and coronary vasodilator effects in the canine isolated, blood-perfused heart preparations. The Journal of Toxicological Sciences. 2016 Dec 1;41(6):739-44.
44. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease?. Trends in Endocrinology & Metabolism. 2014 Sep 1;25(9):452-63.
45. Castela Â, Costa C. Molecular mechanisms associated with diabetic endothelial–erectile dysfunction. Nature Reviews Urology. 2016 May;13(5):266.