Abstract
Voltage-gated Kv1.3 potassium channels control the membrane potential, cellular activation and cell death. Kv1.3 channels have been extensively studied in autoimmune disorders and are promising drug targets for the treatment of solid cancer. However, licensed drugs for specific inhibition of Kv1.3 channels in vivo are not available. Here, we give a short overview of the main characteristics of Kv1.3 channels and then focus on recent advances in the use of Kv1.3 blockers in the treatment of hematological malignancies, proposing memantine as a valuable Kv1.3 channel inhibitor for clinical application.
Keywords
Kv1.3 channel, Lymphoid leukemia, Myeloid leukemia, Memantine, Cell death
Kv1.3 Channels
Voltage-gated potassium channels (Kv) are selectively permeable for potassium ions and are activated by change of the cell membrane voltage [1]. They are grouped into 12 subfamilies (Kv1-Kv12) with the Kv1 subfamily named Shaker type. Kv channels consist of a homotetramer with four α-subunits and a central ion pore. Each α-subunit is composed of six transmembrane domains S1-S6, S4 serving as voltage-gate, S5 and S6 forming the p-loop as selective potassium ion filter. The α-subunits are associated with regulatory subunits such as Kvß, which control channel expression, gating, and potassium current, with integrins [2] and with adapter proteins and protein tyrosine kinase p56lck, which are involved in signal transduction [3]. Initially studied in excitable cells like neurons, expression of Kv channels was detected in different cell types including hematopoietic cells, and Kv channels of the Kv1.3 type were firstly described in human T lymphocytes in 1984 [4,5]. Kv1.3 channels of the plasma membrane prevent accidental depolarization of the T-cell, thereby regulating its resting membrane potential. Upon T-cell receptor engagement, Kv1.3 channels are recruited into the immunological synapse [6] and via potassium efflux prevent continuous depolarization generated by calcium influx through calcium release-activated calcium (CRAC) channels. The permissive hyperpolarization is crucial for sustained calcium influx to allow effective T-cell activation [7]. Elegant studies have shown that Kv1.3 channels are also expressed in the mitochondrial membrane (mitoKv1.3), where they function as mediators of the intrinsic apoptosis pathway [8]. Upon an initial apoptotic stimulus, the pro-apoptotic Bcl-2 family member Bax, which is located in the outer mitochondrial membrane, translocates to the mitoKv1.3 and blocks its central channel pore at Lysin128. Inhibition of mitoKv1.3 channels results in mitochondrial hyperpolarization, enhanced production of reactive oxygen species (ROS) and the release of cytochrome C (CytC), which feeds into the activation of Caspase-9 and Caspase-3 [9]. In addition, Kv1.3 channels have been found to be located in the nuclear membrane of some cancer cell lines, including Jurkat cells [10], but their functional role in the nuclear compartment needs to be unraveled. Kv1.3 channels have been extensively studied in autoimmune disorders [11] and are expected to be promising pharmacological targets for the treatment of solid cancer [12].
Blockade of Kv1.3 Channels
Inhibitors of Kv1.3 channels are classified into peptide toxins and low molecular weight blockers [11]. Peptide toxins comprise scorpion toxins (for instance Vm24 [13] and MgTX [14]) and sea anemone toxins (like ShK [15]) and their derivatives with a size of ~4 kDa. They block the Kv1.3 channel pore with high affinity and selectivity independent of the open probability of the channel. Due to their positive charge, peptide toxins are membrane-impermeable and thus block Kv1.3 channels of the plasma membrane, but not apoptosis-mediating mitoKv1.3 channels. Furthermore, they can cause allergic reactions and need to be applied parenterally. Dalazatide is the first derivative tested in clinical phase I/II trials on healthy volunteers and patients with psoriasis [16]. Other peptide toxins are used for research purposes only. Low molecular weight blockers are characterized by a size below 0.8 kDa and include psoralen derivatives, such as 5-(4-phenylobutoxy) psoralen (Psora-4) and 5-(4-phenoxybutoxy) psoralen (PAP-1, [17]), and clofazimine, a drug applied in the treatment of leprosy [18]. Due to their hydrophobicity, low molecular weight blockers are membrane-permeable and thus are able to block mitoKv1.3 channels to induce apoptosis. Compared to peptide toxins, they have a higher bioavailability, but show a reduced affinity and selectivity [12]. Whereas structural similarities of Kv1.3 channel blockers have not been identified, detailed analyses are available for Kv1.3 channels. Interestingly, the structure of Kv1.3 channels is closely related to ionotropic glutamate receptors (iGluR) showing a significant amino acid homology in the channelforming units. Specifically, the p-loop and S5/S6 domains of Kv1.3 channels structurally resemble the M2 and M1/ M3 domains of iGluR, respectively [19]. Recently, we showed that memantine, a licensed antagonist of iGluRs of the N-methyl-D-aspartate type (NMDARs), also inhibits Kv1.3 channel currents of lymphocytes [20,21].
Despite immense progress in the therapy of hematological malignancies, treatment of acute leukemia remains challenging as many patients show relapses or do not tolerate intensive therapeutic regimens. Patients suffering from chronic hematological diseases, such as lymphoma or chronic myeloid leukemia (CML), often require long-term treatment and the reduction of therapy-associated toxicity is of particular challenge. Given that Kv1.3 channels regulate proliferation and cell death of lymphocytes, Kv1.3 channels present attractive oncological targets in fighting hematological neoplasia [22], however, licensed drugs for specific inhibition of Kv1.3 channels in vivo are not available.
Kv1.3 Channels in Lymphocytic Leukemia and Lymphoma
Acute T-lymphoblastic leukemia (T-ALL) and T-cell lymphoma are rare but highly aggressive diseases with restricted therapeutic options. Using memantine to pharmacologically block Kv1.3 channels, we recently demonstrated that inactivation of Kv1.3 channels by memantine potentiates cytarabine (AraC)-induced cell death of T-ALL cell lines (Jurkat and CEM) as well as patients’ primary T leukemic blasts. On the molecular level, memantine co-application promoted a concurrent inhibition of the central AKT, ERK1/2 and c-MYC signaling pathways. In addition, it augmented mitochondrial CytC release and activation of Caspase-9 and Caspase-3. These data propose that in combination with AraC, blockade of Kv1.3 channels by memantine, a licensed and safe drug, may be a therapeutic option for the treatment of T-ALL [23]. Consistent with our data, another report showed that treatment of Jurkat cells with the mitoKv1.3 channel inhibitor PCARBTP, a derivative of the low molecular weight blocker PAP-1, reduced basal and TCR/CD3- induced activation of the signaling molecules ZAP70, PI-3-K, AKT, and JNK. Inhibition of PI-3-K and AKT sensitized Jurkat cells to PCARBTP and allowed induction of cell death at lower drug concentrations [24]. These data are promising, but possible toxic side effects of PCARBTP still need clinical evaluation. Expression of functional Kv1.3 channels was also reported for primary malignant T-cells isolated from patients with Mycosis fungoides (MF), the most common form of cutaneous T-cell lymphoma, and from patients with Sézary syndrome, a leukemic progress of MF. Kv1.3 blockade by the peptide toxins ShK and Vm24 inhibited proliferation, IL-9 production and CD25 induction of the malignant T-cells, but the peptide toxins had no effect on cell death of Sézary cells [25,26]. Thus, Kv1.3 channel inhibition provides potential to combat neoplastic T-cells in Sézary syndrome. However, peptide toxins are not membrane-permeable, and inhibition of apoptosis-regulating mitoKv1.3 channels may be required for killing malignant Sézary cells.
Kv1.3 channels, although most intensely studied in T-cells, are also critical for B-cell function [27]. We previously showed that blockade of Kv1.3 currents by memantine diminishes BCR-induced calcium flux and inhibits B-cell proliferation, antibody production, and migration [21]. Memantine also blocks Kv1.3 channels of Raji Burkitt-lymphoma cells and preliminary results suggest that memantine enhances AraC-induced cell death of primary B acute lymphoblastic leukemia (B-ALL) cells (Figures 1A and 1B) comparable to the effects observed for T-ALL cells [23]. These results indicate that memantine may be suited for Kv1.3 blockade in B-ALL and B-lymphoma treatments, but more detailed studies are required. Chronic lymphocytic leukemia (CLL) is characterized by the accumulation of neoplastic mature B cells, which are resistant to apoptosis. Neoplastic B cells showed higher expression of Kv1.3 channels than healthy B cells, which was linked to oncogenic B-RAF signaling. Inhibition of Kv1.3 channels was proposed as a novel therapeutic strategy to induce cell death of CLL cells while sparing healthy B cells. Whereas the membrane-permeant Kv1.3 channel inhibitors Psora-4, PAP-1, and clofazimine efficiently induced cell death of CLL cells, even in the presence of mesenchymal stem cells, neoplastic B cells were resistant to the membraneimpermeant Kv1.3 channel blocker ShK, indicating that inhibition of mitoKv1.3 is required for apoptosis induction [28,29]. As Kv1.3 channel inhibition effectively killed >98% of primary CLL cells ex vivo independent of the p53 status, it may also be a promising therapeutic option for the group of p53-mutated CLL patients which shows lower response rates and shorter progressionfree survival to established therapies compared to p53- wildtype CLL patients [30]. Fludarabine, a purine analog commonly used in the treatment of CLL, was found to block Kv1.3 channel currents of malignant B-cell lines at concentrations achieved in the plasma of treated patients [31]. Also, rituximab, an anti-CD20 monoclonal antibody regularly used to treat CLL and other B-cell lymphoma, was shown to inhibit Kv1.3 channels in B-lymphoma cell lines. The effect of rituximab on Kv1.3 channels was abolished after selective blockade of FcγRIIB receptors and rituximab-induced apoptosis of neoplastic B cells was attenuated by blockade of FcγRIIB receptors and partially mimicked by inhibition of Kv1.3 channels [32]. Thus, a Kv1.3 channel blockade may in part account for the therapeutic effects of fludarabine and rituximab in CLL treatment.
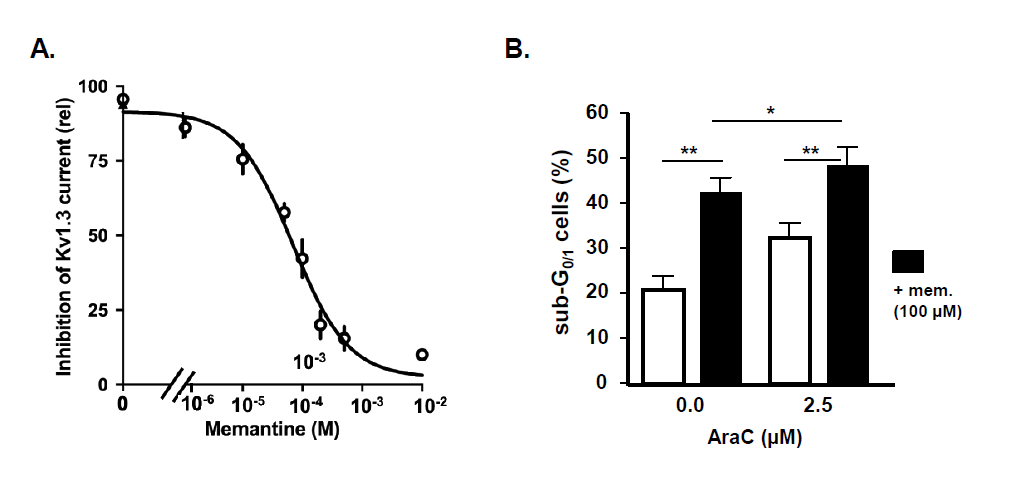
Kv1.3 Channels in Myeloid Leukemia
Detailed studies of Kv1.3 channel expression and function in healthy and malignant T and B lymphoid cells provide an excellent basis for Kv1.3 channel inhibition strategies to treat lymphoid neoplasia. In contrast, less is known about Kv1.3 channels in myeloid cells. Kv1.3 mRNA transcripts were detected in CD34+ hematopoietic stem cells of human blood [33]. We reported functional Kv1.3 channel expression in acute myeloid leukemia (AML) cells using whole-cell patch-clamp technique. Kv1.3 channel currents were inhibited by memantine and AraC-induced cell death of several AML cell lines and primary AML blasts from patients, including FLT3- ITD mutant AML, was potentiated by memantine cotreatment. Inhibition of AKT and ERK1/2 signaling and augmented mitochondrial CytC release was observed upon combined drug treatment, indicating that related cell death mechanisms are initiated in AML and T-ALL cells [23]. Based on the concept that inhibition of mitoKv1.3 induces intrinsic apoptosis [34], in AraC/memantine co-treatment of acute leukemia cells AraC may act as an initial apoptotic stimulus inducing translocation of Bax to mitoKv1.3 channels and memantine may enhance mitochondrial CytC release through further inhibition of mitoKv1.3 channels. This hypothesis assumes that memantine is membrane-permeable and enters the cell, which has not been investigated though. However, memantine is a hydrophobic molecule and crosses the blood-brain barrier, suggesting that memantine can act on intracellular Kv1.3 channels (mitoKv1.3 and possibly nuclear Kv1.3). Janus-kinase 2 (JAK2) is a tyrosine kinase involved in cytokine and growth factor signaling and plays a central role in proliferation and differentiation of immune cells. The frequently occurring gain-of-function JAK2 V617F mutation gives rise to a constitutively active JAK2 kinase and can drive the pathogenesis of myeloproliferative neoplasms (MPN) [35]. Since JAK2 regulates Kv1.3 channel activity and protein expression [36], Kv1.3 channels may be promising drug targets for treating MPN. Recently, it was shown that clofazimine induces apoptosis of blood mononuclear cells and CD34+ progenitor cells of CML patients and synergizes with imatinib, a protein tyrosine kinase inhibitor commonly used in CML therapy. Hematopoietic progenitor cells from healthy donors were not affected. The authors demonstrated that clofazimine induces peroxisome proliferator-activated receptor-γ (PPARγ)-mediated apoptosis in CML cells via induction of ROS and activation of Caspase-9 and Caspase-3 [37]. Since clofazimine inhibits Kv1.3 channels and induces similar cell death mechanisms in other neoplastic cells [28], Kv1.3 channel inhibition may contribute to the clofazimine-mediated cell death in CML cells.
Kv1.3 Channels and Memantine
Kv1.3 channels are not only expressed in immune cells, but also in fibroblasts, osteoclasts, brain, lung, testis, kidney, and islets [2]. Thus, specific blockade of these channels by high affinity inhibitors may cause severe side effects and Kv1.3 channel blockers with sufficient hydrophobicity to induce apoptosis, oral applicability and good clinical tolerability are in demand. Memantine’s inhibitory effect on neuronal NMDARs was elucidated in 1989 and memantine is clinically applied in therapy of moderate to severe Alzheimer disease [38]. It is a well tolerated drug and even accidental over-dosing (until 10- fold of standard dose, i.e. 200 mg/d) showed no or only limited toxic side effects [38]. We reported that memantine blocks Kv1.3 channels on lymphocytes and leukemic cells in vitro and ex vivo and promotes concurrent inhibition of AKT/mTOR/S6, ERK1/2, and c-MYC [23], i.e. signaling pathways that are often deregulated in leukemia and are linked with particular aggressive disease and therapy resistance [39]. Furthermore, standard memantine treatment of Alzheimer patients inhibits Kv1.3 currents of lymphocytes in vivo [40], suggesting that memantine could be re-purposed for in vivo blockade of Kv1.3 channels, for instance in AraC/memantine combinatory therapies of acute leukemia. Since memantine crosses the blood-brain barrier, it could also be effective in combination with AraC in the prophylaxis or treatment of meningeosis leukemia. Besides Kv1.3 channels, memantine blocks calcium-regulated KCa3.1 potassium channels of lymphocytes [20], which could be relevant with regard to induction of resistance to Kv1.3 channel inhibitors. Given that Kv1.3 and ORAI1 subunits of CRAC channels show some structural similarities [1,7] and memantine reduces antigen receptor-induced calcium flux in lymphocytes [20,21], it can be hypothesized that memantine might also directly interfere with CRAC channel opening.
Conclusion
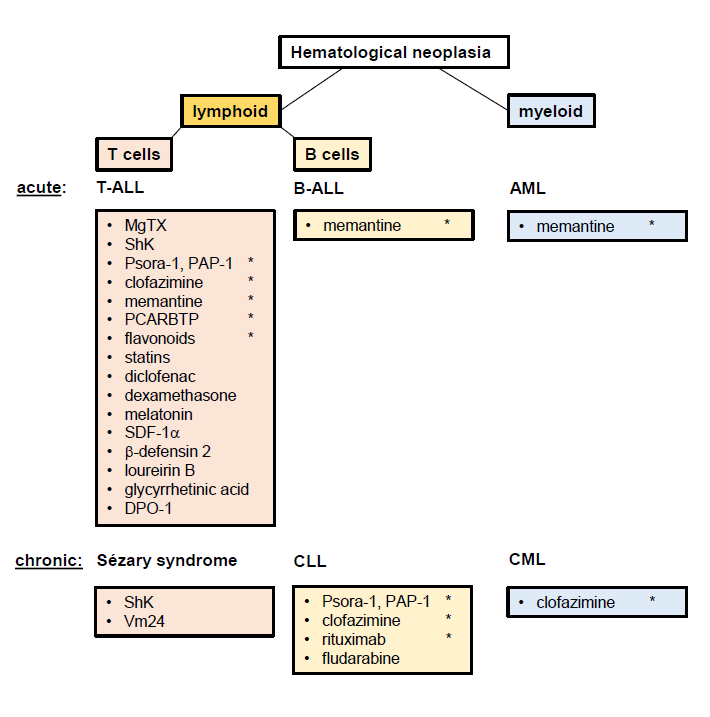
It has become evident that Kv1.3 channels are potent oncological targets for therapy of hematological neoplasia such as leukemia and lymphoma. The most promising Kv1.3 channel inhibitors have been psoralen derivatives like Psora-4, PAP-1 and newer generation derivatives, but clinical trials of these inhibitors are not concluded. Besides highly specific inhibitors, a number of unrelated substances have been described to inhibit Kv1.3 channels, including dexamethasone [41], diclofenac [42], statins [43-45], flavonoids [12,45], melatonin [46], SDF1α [47], human defensins [48], 18beta-glycyrrhetinic acid [49], loureirin B [50], and DPO-1 [51] (Figure 2). Some of these Kv1.3 inhibitors, like memantine, have the advantage that they are already applied in clinical therapies and thus could be tested in various drug combinations to improve treatments of hematological neoplasia. Altogether, more detailed studies are required to define the molecular mechanisms regulating Kv1.3 expression and function, for instance how Kv1.3 channels interfere with intracellular signaling pathways, like AKT or ERK1/2 activation, to modulate cellular responses and, vice versa, how different receptors and signaling molecules influence Kv1.3 channel activity. Future studies also have to verify whether re-purposed drugs like clofazimine or memantine are indeed exploitable as a potential lead compound to improve treatments in leukemia and possibly solid cancers.
Conflict of Interest
The authors declare no conflict of interest.
Funding
This work was supported by DFG grant SFB854.
References
2. Levite M, Cahalon L, Peretz A, Hershkoviz R, Sobko A, Ariel A, et al. Extracellular K(+) and opening of voltagegated potassium channels activate T cell integrin function: physical and functional association between Kv1.3 channels and beta1 integrins. The Journal of Experimental Medicine. 2000 Apr 3;191(7):1167-76.
3. Hanada T, Lin L, Chandy KG, Oh SS, Chishti AH. Human homologue of the Drosophila discs large tumor suppressor binds to p56lck tyrosine kinase and Shaker type Kv1.3 potassium channel in T lymphocytes. Journal of Biological Chemistry. 1997 Oct 24;272(43):26899-904.
4. Matteson DR, Deutsch C. K channels in T lymphocytes: a patch clamp study using monoclonal antibody adhesion. Nature. 1984 Feb 2;307(5950):468-71.
5. Cahalan MD, Chandy KG, DeCoursey TE, Gupta S. A voltage-gated potassium channel in human T lymphocytes. The Journal of Physiology. 1985 Jan 1;358(1):197-237.
6. Panyi G, Vamosi G, Bacso Z, Bagdany M, Bodnar A, Varga Z, et al. Kv1.3 potassium channels are localized in the immunological synapse formed between cytotoxic and target cells. Proceedings of the National Academy of Sciences. 2004 Feb 3;101(5):1285-90.
7. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annual Review of Immunology. 2015 Mar 21;33:291-353.
8. Szabo I, Bock J, Grassme H, Soddemann M, Wilker B, Lang F, et al. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proceedings of the National Academy of Sciences. 2008 Sep 30;105(39):14861-6.
9. Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E, et al. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Molecular Medicine. 2012 Jul;4(7):577-93.
10. Jang SH, Byun JK, Jeon WI, Choi SY, Park J, Lee BH, et al. Nuclear localization and functional characteristics of voltage-gated potassium channel Kv1.3. Journal of Biological Chemistry. 2015 May 15;290(20):12547-57.
11. Chandy KG, Norton RS. Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease. Current Opinion in Chemical Biology. 2017 Jun 1;38:97- 107.
12. Teisseyre A, Palko-Labuz A, Sroda-Pomianek K, Michalak K. Voltage-Gated Potassium Channel Kv1.3 as a Target in Therapy of Cancer. Frontiers in Oncology. 2019;9:933.
13. Varga Z, Gurrola-Briones G, Papp F, Rodriguez de la Vega RC, Pedraza-Alva G, Tajhya RB, et al. Vm24, a natural immunosuppressive peptide, potently and selectively blocks Kv1.3 potassium channels of human T cells. Molecular Pharmacology. 2012 Sep 1;82(3):372-82.
14. Garcia-Calvo M, Leonard RJ, Novick J, Stevens SP, Schmalhofer W, Kaczorowski GJ, et al. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. Journal of Biological Chemistry. 1993 Sep 5;268(25):18866-74.
15. Pennington MW, Byrnes ME, Zaydenberg I, Khaytin I, de Chastonay J, Krafte DS, et al. Chemical synthesis and characterization of ShK toxin: a potent potassium channel inhibitor from a sea anemone. International Journal of Peptide and Protein Research. 1995 Nov;46(5):354-8.
16. Tarcha EJ, Olsen CM, Probst P, Peckham D, Munoz- Elias EJ, Kruger JG, et al. Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS One. 2017 Jul 19;12(7):e0180762.
17. Schmitz A, Sankaranarayanan A, Azam P, Schmidt- Lassen K, Homerick D, Hansel W, et al. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Molecular Pharmacology. 2005 Nov 1;68(5):1254-70.
18. Ren YR, Pan F, Parvez S, Fleig A, Chong CR, Xu J, et al. Clofazimine inhibits human Kv1.3 potassium channel by perturbing calcium oscillation in T lymphocytes. PloS one. 2008 Dec 23;3(12):e4009.
19. Kuner T, Seeburg PH, Guy HR. A common architecture for K+ channels and ionotropic glutamate receptors? Trends in Neuroscience. 2003 Jan 1;26(1):27-32.
20. Kahlfuss S, Simma N, Mankiewicz J, Bose T, Lowinus T, Klein-Hessling S, et al. Immunosuppression by N-methyl-D-aspartate receptor antagonists is mediated through inhibition of Kv1.3 and KCa3.1 channels in T cells. Molecular and Cellular Biology. 2014 Mar 1;34(5):820-31.
21. Simma N, Bose T, Kahlfuss S, Mankiewicz J, Lowinus T, Luhder F, et al. NMDA-receptor antagonists block B-cell function but foster IL-10 production in BCR/CD40- activated B cells. Cell Communication and Signaling. 2014 Dec 1;12(1):75.
22. Leanza L, Venturini E, Kadow S, Carpinteiro A, Gulbins E, Becker KA. Targeting a mitochondrial potassium channel to fight cancer. Cell calcium. 2015 Jul 1;58(1):131-8.
23. Lowinus T, Heidel FH, Bose T, Nimmagadda SC, Schnoder T, Cammann C, et al. Memantine potentiates cytarabine-induced cell death of acute leukemia correlating with inhibition of Kv1.3 potassium channels, AKT and ERK1/2 signaling. Cell Communication and Signaling. 2019 Dec;17(1):1-3.
24. Bergermann T, Born L, Ferguson F, Latkovic P, Scheul A, Sonnenschein N, et al. Inhibition of PI-3-K and AKT Amplifies Kv1.3 Inhibitor-Induced Death of Human T Leukemia Cells. Cellular Physiology & Biochemistry. 2019 Jan 1;53(S1):1-0.
25. Hu T, Buus TB, Krejsgaard T, Nansen A, Lundholt BK, Spee P, et al. Expression and function of Kv1.3 channel in malignant T cells in Sezary syndrome. Oncotarget. 2019 Aug 6;10(47):4894.
26. Hu T, Krejsgaard T, Nastasi C, Buus TB, Nansen A, Hald A, et al. Expression of the Voltage-Gated Potassium Channel Kv1.3 in Lesional Skin from Patients with Cutaneous T-Cell Lymphoma and Benign Dermatitis. Dermatology. 2020;236(2):123-32.
27. Wulff H, Knaus HG, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity.The Journal of Immunology. 2004 Jul 15;173(2):776-86.
28. Leanza L, Trentin L, Becker KA, Frezzato F, Zoratti M, Semenzato G, et al. Clofazimine, Psora-4 and PAP-1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia. 2013 Aug;27(8):1782-5.
29. Szabo I, Trentin L, Trimarco V, Semenzato G, Leanza L. Biophysical characterization and expression analysis of Kv1.3 potassium channel in primary human leukemic B cells. Cellular physiology and biochemistry. 2015;37(3):965-78.
30. Leanza L, Romio M, Becker KA, Azzolini M, Trentin L, Manago A, et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell. 2017 Apr 10;31(4):516-31.
31. de la Cruz A, Vera-Zambrano A, Peraza DA, Valenzuela C, Zapata JM, Perez-Chacon G, et al. Fludarabine Inhibits KV1.3 Currents in Human B Lymphocytes. Frontiers in Pharmacology. 2017 Mar 31;8:177.
32. Wang LH, Wang N, Lu XY, Liu BC, Yanda MK, Song JZ, et al. Rituximab inhibits Kv1.3 channels in human B lymphoma cells via activation of FcgammaRIIB receptors. Biochimica et Biophysica Acta. 2012 Feb 1;1823(2):505- 13.
33. Park KS, Pang B, Park SJ, Lee YG, Bae JY, Park S, et al. Identification and functional characterization of ion channels in CD34(+) hematopoietic stem cells from human peripheral blood. Molecules and Cells. 2011 Aug 1;32(2):181-8.
34. Checchetto V, Prosdocimi E, Leanza L. Mitochondrial Kv1.3: a New Target in Cancer Biology? Cellular Physiology & Biochemistry. 2019;53(S1):52-62.
35. Lanikova L, Babosova O, Prchal JT. Experimental Modeling of Myeloproliferative Neoplasms. Genes. 2019 Oct;10(10):813.
36. Hosseinzadeh Z, Warsi J, Elvira B, Almilaji A, Shumilina E, Lang F. Up-regulation of Kv1.3 channels by janus kinase 2. The Journal of Membrane Biology. 2015 Apr 1;248(2):309-17.
37. Kumar H, Chattopadhyay S, Das N, Shree S, Patel D, Mohapatra J, et al. Leprosy drug clofazimine activates peroxisome proliferator-activated receptor-gamma and synergizes with imatinib to inhibit chronic myeloid leukemia cells. Haematologica. 2020 Apr 1;105(4):971-86.
38. Rammes G, Danysz W, Parsons CG. Pharmacodynamics of memantine: an update. Current Neuropharmacology. 2008 Mar 1;6(1):55-78.
39. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012 Oct;3(10):1068- 111.
40. Lowinus T, Bose T, Busse S, Busse M, Reinhold D, Schraven B, et al. Immunomodulation by memantine in therapy of Alzheimer’s disease is mediated through inhibition of Kv1.3 channels and T cell responsiveness. Oncotarget. 2016 Aug 16;7(33):53797.
41. Lampert A, Muller MM, Berchtold S, Lang KS, Palmada M, Dobrovinskaya O, et al. Effect of dexamethasone on voltage-gated K+ channels in Jurkat T-lymphocytes. Pflugers Archives. 2003 Nov 1;447(2):168-74.
42. Villalonga N, David M, Bielanska J, Gonzalez T, Parra D, Soler C, et al. Immunomodulatory effects of diclofenac in leukocytes through the targeting of Kv1.3 voltage-dependent potassium channels. Biochemical Pharmacology. 2010 Sep 15;80(6):858-66.
43. Kazama I, Baba A, Maruyama Y. HMG-CoA reductase inhibitors pravastatin, lovastatin and simvastatin suppress delayed rectifier K(+)-channel currents in murine thymocytes. Pharmacological Reports. 2014 Jul;66(4):712- 7.
44. Zhao N, Dong Q, Qian C, Li S, Wu QF, Ding D, et al. Lovastatin blocks Kv1.3 channel in human T cells: a new mechanism to explain its immunomodulatory properties. Scientific Reports. 2015 Nov 30;5:17381.
45. Zhao N, Dong Q, Fu XX, Du LL, Cheng X, Du YM, et al. Acacetin blocks kv1.3 channels and inhibits human T cell activation. Cellular Physiology and Biochemistry. 2014;34(4):1359-72.
46. Varga Z, Panyi G, Peter M, Jr., Pieri C, Csecsei G, Damjanovich S, et al. Multiple binding sites for melatonin on Kv1.3. Biophysical Journal. 2001 Mar 1;80(3):1280-97.
47. Matsushita Y, Ohya S, Suzuki Y, Itoda H, Kimura T, Yamamura H, et al. Inhibition of Kv1.3 potassium current by phosphoinositides and stromal-derived factor-1alpha in Jurkat T cells. American Journal of Physiology-Cell Physiology. 2009 May;296(5):C1079-85.
48. Yang W, Feng J, Xiang F, Xie Z, Zhang G, Sabatier JM,et al. Endogenous animal toxin-like human beta-defensin 2 inhibits own K(+) channels through interaction with channel extracellular pore region. Cellular and molecular life sciences. 2015 Feb 1;72(4):845-53.
49. Fu XX, Du LL, Zhao N, Dong Q, Liao YH, Du YM. 18beta-Glycyrrhetinic acid potently inhibits Kv1.3 potassium channels and T cell activation in human Jurkat T cells. Journal of ethnopharmacology. 2013 Jul 9;148(2):647-54.
50. Yin S, Hu Q, Luo J, Li Y, Lu C, Chen X, et al. Loureirin B, an essential component of Sanguis Draxonis, inhibits Kv1.3 channel and suppresses cytokine release from Jurkat T cells. Cell & Bioscience. 2014 Dec;4(1):1-8.
51. Zhao N, Dong Q, Du LL, Fu XX, Du YM, Liao YH. Potent suppression of Kv1. 3 potassium channel and IL-2 secretion by diphenyl phosphine oxide-1 in human T cells. PLoS One. 2013 May 22;8(5):e64629.