Abstract
The activation of endogenous IFNγ signaling pathway or the administration of recombinant IFNγ increases the expression of MHC-I. MHC-I molecules are core elements for antigen recognition in tumor cells. A better understanding of the regulation of their expression would contribute to counteracting tumor immune escape and enduring permanent tumor rejection. Efficient and functional expression of HLAs dramatically impacts the number of tumor-associated antigens presented to CTL for cell recognition. Many patients diagnosed with various types of cancer have inhibited the IFNγ signaling pathway. This review explores how anomalies associated with IFNγ signaling in tumor cells affect HLA-I expression, current immunotherapies association, and outcome. Globally, MHC-I lesions could be divided into reversible and permanent. Irreversible lesions cannot be recapitulated; hence, the patient will not respond to immunotherapies requiring MHC-I activity. However, gaining precise and systematic molecular knowledge improves tumor stratification, which could help predict which tumors will recover expression of MHC-I. Complementary IFNγ effectors can function as a compensatory mechanism that restores the expression of HLA-I proteins in tumors with deleterious IFNγ pathways. For those non-responsive patients with inactive IFNγ pathways, designing personalized approaches to recover HLA-I expression can make the tumor sensitive to immunotherapy, leading to a better outcome.
Keywords
MHC class I, HLA, IFNγ, Immunotherapy
Highlights
- Inactivation of the IFNγ signaling pathway downregulates MHC-I expression and impairs tumor antigen presentation.
- Recurrent immunotherapy failure observed in patients with IFNγ pathway deficiencies.
- IFNγ signaling pathway and MHC class I regulation require profound molecular understanding to mitigate resistance against cancer treatments.
Abbreviations
CTL: Cytotoxic T Lymphocytes; MHC: Major Histocompatibility Complex; HLA: Human Leukocyte Antigen; IFN: Interferon; JAK: Janus Kinase; STAT: Signal Transducer and Activator of Transcription; ISG: Interferon-Stimulated Genes; IRF: Interferon Regulatory Factor; ISGF: Interferon-Stimulated Gene Factor; GAF: Gamma-Interferon Activation Factor; GAS: Gamma- Interferon Activated Site; ISRE: Interferon- Sensitive Response Element; IRDS: Interferon-Related DNA Damage Resistant Signature; IFITM: Interferon-Induced Transmembrane; NLRC5: NOD-Like Receptor Family CARD Domain Containing 5; PRR: Pattern Recognition Receptor; dsRNA: Double-stranded RNA; LOH: Loss of the Heterozygosity; B2M: Beta-2-Microglobulin; APM: Antigen-Processing Machinery; IFNR: Interferon Receptor; HPV: Human-Papillomavirus; DNMTi: Histone Deacetylase Inhibitors; TAP: Transporter associated with Antigen Processing; LPM: Low-Molecular Mass Polypeptide; MECL: Multi-Catalytic Endopeptidase Complex-Like: DUX4: Double Homeobox 4; LNK: Lymphocyte Adapter Protein; PIN1: Peptidyl-Prolyl Cis-Trans Isomerase NIMA-Interacting 1.
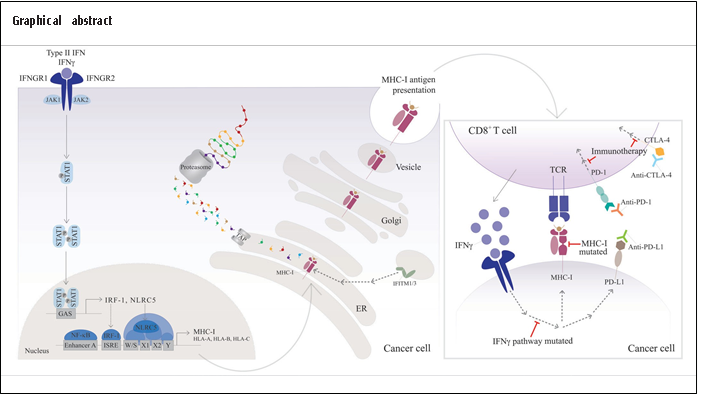
IFNγ Signaling Pathway
Malignant cells are highly adaptive to their microenvironment and develop complex and selective molecular strategies to promote cell survival. Thus, tumor development is a dynamic process sustained during the appearance of the disease, diagnosis, course of treatment, and remission. Furthermore, a decrease in the symptoms and absence of cancer-associated markers may not necessarily translate into the complete disappearance of cancer; dormant cells may remain in the organism causing a future relapse [1]. Hence, the importance of continuous characterization of key molecular abnormalities is desirable to design effective and personalized immunotherapies at all stages [2].
The central paradigm for cancer cell eradication requires effective tumor-derived peptide processing, presentation by MHC-I molecules, and optimal peptide recognition by the cytotoxic T lymphocytes (CTL) [3]. Unfortunately, a comprehensive understanding of the mechanisms behind cancer immunosurveillance is extremely complex. However, this review aims to unravel fundamental features involving the regulation of MHC class I by IFNγ associated with immunotherapy.
The canonical MHC-I (HLA-A, HLA-B, HLA-C) genes are induced by the exceedingly investigated IFN signaling [4]. IFNs are pleiotropic cytokines that rapidly activate the JAK-STAT signaling cascade and mediate the expression of many genes collectively known as interferon-stimulated genes (ISG) [5-11].
In humans, IFNs are classified into three different classes: type I (IFNα, IFNβ, IFNε, IFNκ, and IFNω), type II (IFNγ) and type III (IFNγ) [12-14]. The MHC class I molecules are induced by type I IFN and more efficiently by type II IFN [15-18].
The IFNγ signaling activates the homodimerization of p-STAT1, forming the GAF complex in the cytosol. In addition, p-STAT1 also forms a heterodimer with p-STAT2 and binds to IRF9, constituting the ISGF3 complex, representative of the type I IFN pathway [19,20]. Thus, this illustrates that despite unique receptors for each class of IFN, there is underlying crosstalk between IFNγ and type I IFN pathways. In the primary IFNγ response, the GAF complex translocates to the nucleus. It binds to the GAS sites present in the promoter of ISG genes, such as the transcription factors IRF1 and p48. Sequentially, IRF1 regulates the expression of many genes activated in the secondary response through DNA recognition with ISRE motifs. The p48 enhances the binding specificity between the ISRE boxes to the principal transactivator factor for type I IFN, ISGF3. The induction of MHC class I genes is driven by the binding of IRF1 to the ISRE element presented in the gene's promoter upon IFNγ stimulation [21-23]. More specifically, HLA-A is less responsive to IFNγ than the counterparts HLA-B and HLA-C due to changes in the structure of ISRE sequences [24-28].
IFNγ has numerous protective anti-cancer functions related to the activation of immune cells [29-31], promoting apoptosis [32-34], preventing angiogenesis [35], and inhibiting proliferative processes [36] occurring in cancer cells.
In contrast, this view is challenged when examining tumor resistance in chemotherapy and radiotherapy patients [37,38]. Throughout cancer treatment and in cancer development, chronic inflammation results in the sustained activation of the IFNγ pathway [39]. Conversely, consistent exposure to IFNγ constitutes an adverse effect promoting cell growth [40,41] and genomic instability [42], depleting immunogenic tumorassociated antigens [43], and exhausting CTL cell recognition [44]. The study of the gene expression in various cancer types has defined a subgroup of ISG genes called IFN-related DNAdamage signature (IRDS), responsible for chemotherapy and radiotherapy resistance [45-47]. Notoriously, HLA-B and HLA-G (non-classical MHC class I molecule) are induced by IRDS genes. However, the current understanding of the cellular mechanism behind the protective phenotype against radiotherapy and chemotherapy conferred by IRDS genes is insufficient. Nonetheless, a steady low dose of IFNγ initiates a turnover from phosphorylated to unphosphorylated STAT1 form that induces the expression of IRDS genes [48,49].
Moreover, multiple factors have been linked with altered IFNγ signaling, including the embryonic transcription factor double homeobox 4 (DUX4), which upregulation in cancer results in a decrease of JAK1/2 and STAT1 and reduces MHC class I and other genes of the antigen-presentation machinery (APM) [50]. In addition, DUX4-mediated suppression of MHC-I and antigen presentation has been associated with resistance to immune checkpoint blockade and promotion of cancer immune escape. The lymphocyte adapter protein (LNK) is another factor that induces the dephosphorylation of STAT1 and, consequently, negatively modulates IFNγ signaling [51]. A further example is a peptidyl-prolyl isomerase (Pin1) that has been shown to play a role in inducing ubiquitination and suppressing the interferon-regulatory factor 3 (IRF3). Increased Pin1 expression has been reported in many types of cancer and has been detected in cancer cells undergoing malignant transformation [52,53].
To increase the complexity of the MHC class I pathway, specific tumor cell subclones can acquire resistance by sabotaging the activation of the IFN pathway, which affects the optimal expression of MHC class I and leads to therapeutic failure. To overcome this event, we have proposed a novel role for the IFN-inducible IFITM1 (which is an IRDS component) and IFITM3 [54,55] mediating protein synthesis in the context of cancer. Our results suggest that IFITM1/3 stimulates the protein synthesis of MHC class I components, most significantly for HLA-B [56]. We hypothesize that the absence of IFITM1/3 expression could decrease MHC class I molecules, resulting in deficient processing of the antigen-presentation pathway and affecting the peptide recognition by CTL. Reduced MHC class I expression followed by malfunctions in recognizing tumorassociated peptides by the CD8+ T cell leads to immune evasion, causing metastasis and lack of response to immunotherapy [57-62]. Our interpretation is consistent with the fact that deficient IFITM1/3 expression observed in cervical cancer specimens is more prone to metastasis [56]. With our work, we would like to expand the knowledge on the HLA regulation and draw interest in non-canonical cellular signaling (s) to activate the expression of MHC class I molecules. Alternative mechanisms are investigated in tumors defective on the IFN pathway to recapitulate the expression of MHC class I in an IFN-independent manner; for example, an increase of the transcriptional activator NLRC5 [63-65] restores the production of MHC class I. In addition to that, an intratumoral injection with the synthetic BO-112 (nanoplexed formulation of poly I:C) restores the pattern recognition receptor (PRR) pathway and activates dsRNA sensing to express MHC class I through the nuclear factor-kB (NF-kB) [66]. All in all, this phenomenon raises the question of whether alternative mechanism(s) responsible for potential tumor sensitivity could be caused (at least partially) by unexplored MHC class I mediators.
Regulation of MHC Class I in Cancer
MHC class I downregulation or total expression loss is frequently observed in malignant tumors [67]. In addition, it is considered a dominant mechanism was facilitating escape from cancer immune surveillance [68]. MHC class I processing and presentation on the cell surface is mediated at the genetic, epigenetic, transcriptional, and posttranscriptional levels. Garrido et al. distinguished two types of alterations in cancers that affect MHC class I expression depending on whether the change is reversible, classified as ‘soft’ mutation, or irreversible, referred to as ‘hard’ mutation [69].
Due to tumor genetic instability, various genetic variations can affect MHC class I expression patterns. These structural irreversible ‘hard’ aberrations can arise through deletions, mutations in chromosome sequences, and somatic recombination [70]. For instance, the loss of the heterozygosity (LOH) by deleting one allele of HLA class I or B2M genes is frequent in numerous types of tumors [71-74]. In particular, loss of MHC class I copy number has been described to occur in 17% out of 59 different cancer types analyzed [75]. Moreover, further studies showed a higher prevalence of LOH events in tumors with a higher mutational burden, thus, more prone to display mutated neoantigens [76]. In cancer immunotherapy, however, targeting irreversible alterations is hindered by technical limitations, whereas reversible events with the possibility of restoring gene expression provide potential target candidates for therapeutic approaches.
Tumor-driven reversible ‘soft’ defects can alter MHC class I expression by targeting components of the APM. For example, numerous studies conducted in several cancer types demonstrated reduced levels of transcription factors and regulators, namely NLRC5 [77-79], that consequently led to attenuated levels of MHC alpha chain (the heavy chain component of the MHC class I complex), B2M (the light chain component of the MHC class I complex). Moreover, other peptide-loading and processing machinery components include the transporter associated with antigen processing (TAP), tapasin, endoplasmic reticulum aminopeptidase 1 (ERAP1) and immunoproteasome subunits.
Deficiencies in the activation of the IFNγ signaling pathway can cause MHC class I inhibition and leads to poor prognosis in patients with cancer. The complex regulatory system of MHC class I transcription consists of multiple components [80] that have been found altered in tumors [81-86]. For instance, loss of the interferon regulatory factor IRF1 in melanoma has been linked with the tumor resistance to immune checkpoint blockade (ICB) therapy [87]. Furthermore, loss of constitutive IRF2 expression is correlated with downregulation of MHC class I, TAP2, and ERAP1; consequently, tumors presenting this phenotype may evade immune detection and elimination [88].
Epigenetic mechanisms silencing MHC class I in tumors have been researched comprehensively as potential therapeutic targets. Hypermethylation of the regulatory elements of MHC-I, B2M, APM components, NLRC5, and elements of the IFN pathway has been described in several types of cancers such as sarcomas, gliomas, breast, lung, colon, thyroid, and human papillomavirus (HPV)-related cancers [89-93]. Nonetheless, suppression of MHC class I by DNA hypermethylation can be recovered with DNA methyltransferase inhibitors (DNMTi). Moreover, the induction of MHC-I genes silenced by histone deacetylation can be restored with histone deacetylase inhibitors (HDACi), leading to increased acetylation levels in melanoma and glioma [94,95]. Currently, there are several epigenetic inhibitors approved by the FDA for treatment or undergoing clinical trials that show an increase in the expression of IFN and genes associated with the MHC class I antigen presentation pathway in cancers both in vitro and in vivo [96-98].
MHC class I expression is mediated by IFN signaling (primarily by IFNγ) in response to inflammation. Several factors are involved in alterations of the IFNγ signaling, and subsequent MHC class I downregulation; defects in IFNGR1 receptors, mutations in the signaling factors JAK1, JAK2 and modification of downstream components of the IFNγ signaling cascade such as STAT1 and IRF1 have been associated with poor cancer prognosis [99-103]. To emphasize the role that IFNγ plays in regulating antigen presentation pathways, IFNγ also upregulates most components of the peptide-loading complex such as proteases or TAP transporters and three unique components of the immunoproteasome (LPM2, LPM7, and MECL) [104-106]. In addition, IFNγ affects the MHC class I peptidome repertoire presented on the cell surface [107-109].
In this regard, it has been observed an increased presence of mutated neoantigens in the displayed peptidome following MHC class I induction with IFNγ, thus, potentially stimulating T-cell response in the tumor microenvironment [110]. Despite extensive research, IFNγ clinical benefit remains unknown in cancer immunotherapy. In cancers classified with reduced MHC class I, IFNγ was reported to restore MHC class I expression through epigenetic modifications, increased histone acetylation, and DNA demethylation of TAP and immunoproteasome regulatory elements [111,112]. In addition, downregulation of IFNγ signaling has been correlated with resistance to ICB therapy and adoptive cell therapy in lung cancer and melanoma [99,113,114]. However, whether this process involved MHC class I expression remains to be elucidated. Nonetheless, upregulation of IFNγ signaling having a positive outcome on ICB was observed in cancer where IFNγ alterations are less common [115].
Multiple analyses such as immunohistochemistry, microarray, and RNA sequencing showed decreased MHC class I expression from 40% and up to 90% of lesions analyzed across various histological cancers types, including solid tumors [85,116-120] and hematopoietic tumors [121,122], often correlating with worse prognosis [123-125]. In fact, the detection of impaired MHC class I expression during immunotherapy has been identified as a mechanism for acquired resistance to ICB therapy [126-129]. Furthermore, to highlight its role in cancer promotion, the low MHC class I phenotype derived from melanoma metastases in patients undergoing immunotherapy was correlated with cancer progression while metastases regressed in the high MHC class I phenotype [130]. All in all, the complex and polymorphic nature of MHC class I genes coupled with cancer heterogeneity and discrepancies in published clinical data pose a challenge in understanding the precise mechanism behind MHC class I loss.
The Role of IFNγ in Immunotherapy
ICB therapies targeting the CTLA-4 and PD-1 inhibitory pathways are considered a breakthrough therapy for cancer treatment [131,132]. Checkpoint inhibitors have promised positive outcomes in immune responsive tumors, referred to as “hot” tumors, such as in melanoma and lung cancer. In contrast, “cold” tumors that will not activate a strong immune response can resist these immunotherapies by activating multiple mechanisms [133]. For instance, low expression of MHC-I molecules and a lack of tumor infiltrating lymphocytes (TIL). In addition, MHC-I molecules are very poorly present in various cancer lines; however, IFNγ can stimulate their expression [134,135]. Conversely, “cold” sarcomas treated with IFNγ enhance the expression of MHC-I and increase the infiltration and activation of cytotoxic T cells [136]. Furthermore, treatment with IFNγ upregulates the expression of PD-L1 [137-139], CTLA-4 [140], and IDO1[139,141] in tumor cells which will benefit ICB therapy.
Several clinical trials are ongoing with only IFNγ or combined with other drugs for various types of cancer. For example, patients with HER-2 positive breast cancer are treated with IFNγ, Paclitaxel, Trastuzumab, and Pertuzumab in a phase I-II study (NCT03112590). Another clinical trial exclusively uses the recombinant IFNγ as an immunotherapy agent to treat soft tissue sarcoma (NCT01957709). A growing body of evidence shows that IFNγ interferes with the growth of cancer cells [142,143]. Hence, it is considered for a clinical trial in phase I to treat patients with recurrent metastatic melanoma or other solid tumors (NCT00004016). In addition, the recombinant IFNγ-1b form has also been considered alone or combined with Lexatumumab to treat children with various solids tumors (NCT00428272). Moreover, the chemotherapy drugs cyclophosphamide and cisplatin combined with IFNγ yielded a significant benefit in prolonging progression-free survival in patients with ovarian cancer [144].
Ionizing radiation and chemotherapeutical agents act more effectively with recombinant IFNγ [144-147]. Radiation in a murine colon adenocarcinoma model increases the peptide repertoire, the MHC-I presentation, and CTL recognition dosedependent [148]. However, radiation has proven insufficient to cure some human cancers. The combination of IFNγ with radiation therapy has improved T cells activation and the immune system for a more effective response against colon cancer [146].
Furthermore, functional IFNγ signaling improves the response to PD-L1 inhibitor treatment. It increases the survival of patients affected with non-small cell lung cancer and urothelial cancer [149-151]. The combination of PD-1 and CTLA-4 blockers increases the infiltration and the production of IFNγ in effector T cells [152]. In fact, clinical trials conducted on patients with bladder cancer revealed that the treatment with anti-CTLA-4 antibody was directly associated with increased IFNγ-producing CD4+ICOShi (inducible co-stimulator) cells ratio effector to regulatory T cells [153].
Murine models have shown that inactivation of the IFNpathway is a prevalent cause for resistance to ICB therapy failure [154,155]. Moreover, deficiencies in the IFN signaling inactivate MHC class I expression, and consequently, antigens are presented on the cell surface in a deficient manner [83,155]. Comparably, mutated B2M is recurrent in patients nonresponsive to ICB therapy [129]. In addition, tumor resistance to ICB targeting PD-L1 and CTLA-4 co-receptors sustain the inflammatory response of the IFN signaling modifying STAT1, increasing ISG gene expression and activating T cell inhibitory receptors [156]. However, this adverse effect is mitigated by inhibiting the type I and type II IFN receptors in cancer cells.
Lack of response to the cancer treatment or uncomplete clearance of tumor cells is originated by factors intrinsic to the immune infiltrated cells and/or tumor cells [157]; the immunerelated functions of IFNγ present a stimulatory and suppressive role in cancer [158]. How these dual antagonistic activities of IFNγ coexist is still under debate. Nonetheless, the implications of IFNγ in the cancer cell may diverge from the one exerted by the neighboring immune cell. Thus, the IFN regulation between cancer cells and immune cells is fundamental to define resistance or response to immunotherapy. In this regard, suppressing the IFNγ pathway diminishes the expression of ISG genes in tumor cells. However, higher production of IFNγ in exhausted T cells induces ISG genes [159]. Therefore, the homeostatic balance of the IFNγ is crucial for a favorable immunotherapeutic outcome. Thus, there is an urgent need to improve the predictive biomarkers for immune checkpoint inhibitors prior to treatment. Besides the various biomarkers based on gene expression profiling and RNA analysis, the ratio of the IFNγ signature to the immunosuppression signature better predicts the response to anti-PD-1 therapy in patients with melanoma [160].
Personalized cancer immunotherapies based on therapeutic vaccination and the transfer of TILs targeting tumor-specific neoantigens have shown promising clinical outcomes [161-163]. IFNγ has the potential to remodel the antigen presentation and MHC expressions in the tumor cells [107]. The effective activation of antigen-presenting cells (APCs) and its machinery by IFNγ signaling pathways promote the T cell functioning against the tumor cells. However, the loss of IFNγ inhibits the activation of T cells and allows tumor development and immunoediting [164]. Therefore, it is crucial to consider the role of IFNγ while developing the approach to identify tumor neoantigens. The identification of neoantigens is further limited by the low number of T cells recognizing a specific tumor antigen. Therefore, the development of NeoScreen as a neoantigen screening pipeline is valuable for selecting relevant and personalized targeted tumor antigens for further cancer vaccine and T cell therapy development [165].
Taking everything together, the complexity of the IFNγ reflects the need to study the state of the canonical and alternative IFNγ signaling pathways to design, understand and better predict the outcomes of IFNγ based therapy [166-168]. We would like to remark the need for more advanced analysis of the IFN pathway, novel non-canonical molecules, and the study of individual patient-associated immunopeptides presented in tumor cells to design combined personalized (immuno)therapies and predict with more confidence the responsiveness to anti-cancer treatment [127].
Funding
This research was funded by POWROTY/Reintegration program of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund, project number POIR.04.04.00-00- 3E52/17-00, and The International Centre for Cancer Vaccine Science, a project carried out within the International Research Agendas program of the Foundation for Polish Science cofinanced by the European Union under the European Regional Development Fund, project number MAB/2017/3.
Author Contributions
MGH: Conceptualization, original draft preparation, writing, reviewing, editing, funding acquisition. MP: conceptualization, original draft preparation, writing, reviewing. SK: conceptualization, original draft preparation, writing. TH: supervision.
Conflicts of Interest
The authors declare no conflicts of interest.
References
2. Villanueva MT. Unmasking cancer cell character. Nature Reviews Cancer. 2019 Jan 31;19(3):130–130.
3. Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation. International Review of Cell and Molecular Biology. 2019 Jan 1;348:69–121.
4. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proceedings of the Royal Society of London Series B - Biological Sciences. 1957 Sep 12;147(927):258–67.
5. Shuai K, Schindler C, Prezioso V, Darnell J. Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science. 1992;258(5089):1808–12.
6. Schindler C, Darnell JE. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annual Review of Biochemistry. 1995;64:621–51.
7. Fu X, Schindler C, Improta T, Aebersold R, Darnell J. The proteins of ISGF-3, the interferon alpha-induced transcriptional activator, define a gene family involved in signal transduction. PNAS. 1992;89(16):7840–3.
8. Darnell J, Kerr I, Stark G. Jak-STAT pathways and transcriptional activation in response. Science. 1994;264(5164):1415–21.
9. Silvennoinen O, Ihle J, Schlessinger J, Levy D. Interferoninduced nuclear signaling by Jak protein tyrosine kinases. Nature. 1993;366(6455):583–5.
10. Williams B. Transcriptional regulation of interferon-stimulated genes. European Journal of Biochemistry. 1991;200(1):1–11.
11. Hubel P, Urban C, Bergant V, Schneider WM, Knauer B, Stukalov A, et al. A protein-interaction network of interferonstimulated genes extends the innate immune system landscape. Nature Immunology. 2019 Apr 1;20(4):493–502.
12. Bekisz J, Schmeisser H, Hernandez J, Goldman ND, Zoon KC. Human Interferons Alpha, Beta and Omega. Growth Factors. 2004;22(4):243–51.
13. Borden EC;, Sen GC;, Uze G;, Silverman RH;, Ransohoff RM, Foster GR;, et al. Interferons at age 50: past, current and future impact on biomedicine. Nature Reviews: Drug Discovery. 2007;6:975–90.
14. Platanias LC. Mechanisms of type-I- and type-II-interferonmediated signaling. Nature Reviews Immunology. 2005;5(5):375–86.
15. King DP, Jones PP. Induction of Ia and H-2 antigens on a macrophage cell line by immune interferon. The Journal of Immunology. 1983;131(1).
16. Trinchieri G, Perussia B. Immune interferon: a pleiotropic lymphokine with multiple effects. Immunology Today. 1985 Apr 1;6(4):131–6.
17. Chang C, Furue M, Tamaki K. Selective regulation of ICAM- 1 and major histocompatibility complex class I and II molecule expression on epidermal Langerhans cells by some of the cytokines released by keratinocytes and T cells. European Journal of Immunology. 1994;24(11):2889–95.
18. Gupta SL. Regulation of cellular gene expression by interferongamma: Involvement of multiple pathways. The International Journal of Cell Cloning. 1990 Jan 1;8(S1):92–102.
19. Ihle J. STATs: signal transducers and activators of transcription. Cell. 1996 Feb 9;84(3):331–4.
20. Platanitis E, Demiroz D, Schneller A, Fischer K, Capelle C, Hartl M, et al. A molecular switch from STAT2-IRF9 to ISGF3 underlies interferon-induced gene transcription. Nature Communications. 2019 Dec 1;10(1).
21. Gobin S, van Zutphen M, Woltman A, van den Elsen P. Transactivation of classical and nonclassical HLA class I genes through the IFN-stimulated response element. Journal of Immunology. 1999. p. 163, 1428–1434.
22. Chang C, Hammer J, Loh J, Fodor W, Flavell R. The activation of major histocompatibility complex class I genes by interferon regulatory factor-1 (IRF-1). Immunogenetics. 1992 Apr;35(6):378– 84.
23. Hobart M, Ramassar V, Goes N, Urmson J, Halloran P. IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. Journal of Immunology. 1997. p. 158(9):4260-9.
24. Schmidt H, Gekeler V, Haas H, Engler-Blum G, Steiert I, Probst H, et al. Differential regulation of HLA class I genes by interferon. Immunogenetics. 1990 Apr;31(4):245–52.
25. Girdlestone J, Isamat M, Gewert D, Milstein C. Transcriptional regulation of HLA-A and -B: differential binding of members of the Rel and IRF families of transcription factors. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(24):11568–72.
26. Girdlestone J, Milstein C. Differential expression and interferon response of HLA class I genes in thymocyte lines and response variants. European Journal of Immunology. 1988;18(1):139–43.
27. Hakem R, Jezo-Brémond A, le Bouteiller P, Harper K, Lemonnier FA. Differential transcription inducibility by interferon of the HLA-A3 and HLA-B7 class-I genes. International Journal of Cancer. 1991;47(S6):2–9.
28. van den Elsen P. Expression regulation of major histocompatibility complex class I and class II encoding genes. Frontiers in Immunology. 2011;2(48).
29. Swann J, Hayakawa Y, Zerafa N, Sheehan K, Scott B, Schreiber R, et al. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. Journal of Immunology (Baltimore, Md : 1950). 2007 Jun 15;178(12):7540–9.
30. Strehl B, Seifert U, Krüger E, Heink S, Kuckelkorn U, Kloetzel P. Interferon-gamma, the functional plasticity of the ubiquitinproteasome system, and MHC class I antigen processing. Immunological Reviews. 2005 Oct;207:19–30.
31. Greiner J, Guadagni F, Goldstein D, Smalley R, Borden E, Simpson J, et al. Intraperitoneal administration of interferongamma to carcinoma patients enhances expression of tumorassociated glycoprotein-72 and carcinoembryonic antigen on malignant ascites cells. Journal of Clinical Oncology: official journal of the American Society of Clinical Oncology. 1992;10(5):735–46.
32. Cao Z, Zheng Q, Li G, Hu X, Feng S, Xu G, et al. STAT1-mediated down-regulation of Bcl-2 expression is involved in IFN-γ/TNF-α- induced apoptosis in NIT-1 cells. PloS One. 2015 Mar 26;10(3).
33. Bello C, Vazquez-Blomquist D, Miranda J, Garcia Y, Novoa L, Palenzuela D, et al. Regulation by IFN-α/IFN-γ co-formulation (HerberPAG®) of genes involved in interferon-STAT-pathways and apoptosis in U87MG. Current Topics in Medicinal Chemistry. 2014 Jan 27;14(3):351–8.
34. Wall L, Burke F, Barton C, Smyth J, Balkwill F. IFN-gamma induces apoptosis in ovarian cancer cells in vivo and in vitro. Clinical Cancer Research: an official journal of the American Association for Cancer Research. 2003 Jul 1;9(7):2487–96.
35. Battle T, Lynch R, Frank D. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Research. 2006 Apr 1;66(7):3649–57.
36. Yang G, Xu Y, Chen X, Hu G. IFITM1 plays an essential role in the antiproliferative action of interferon-gamma. Oncogene. 2007 Jan 25;26(4):594–603.
37. Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proceedings of the National Academy of Sciences. 2004;101(6):1714–9.
38. Khodarev NN, Minn AJ, Efimova E v., Darga TE, Labay E, Beckett M, et al. Signal transducer and activator of transcription 1 regulates both cytotoxic and prosurvival functions in tumor cells. Cancer Research. 2007;67(19):9214–20.
39. Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Seminars in Oncology.2014;.
40. Hanada T, Kobayashi T, Chinen T, Saeki K, Takaki H, Koga K, et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. The Journal of Experimental Medicine. 2006 Jun 12;203(6):1391–7.
41. Bernabei P, Coccia EM, Rigamonti L, Bosticardo M, Forni G, Pestka S, et al. Interferon-γ receptor 2 expression as the deciding factor in human T, B, and myeloid cell proliferation or death. Journal of Leukocyte Biology. 2001;70(6):950–60.
42. Takeda K, Nakayama M, Hayakawa Y, Kojima Y, Ikeda H, Imai N, et al. IFN-γ is required for cytotoxic T cell-dependent cancer genome immunoediting. Nature Communications. 2017 Feb 24;8:14607.
43. McFadden D, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter S, Cibulskis K, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014 Mar 13;156(6):1298–311.
44. Morel S, Lévy F, Burlet-Schiltz O, Brasseur F, Probst-Kepper M, Peitrequin A, et al. Processing of some antigens by the standard proteasome but not by the immunoproteasome results in poor presentation by dendritic cells. Immunity. 2000;12(1):107–17.
45. Shiffman D, Pare G, Oberbauer R, Louie JZ, Rowland CM, Devlin JJ, et al. A gene variant in CERS2 is associated with rate of increase in albuminuria in patients with diabetes from ONTARGET and TRANSCEND. PLoS One. 2014 Sep 19;9(9):e106631.
46. Duarte CW, Willey CD, Zhi D, Cui X, Harris JJ, Vaughan LK, et al. Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtypespecific manner. PLoS ONE. 2012;7(1):1–8.
47. Tsai M-H, Cook JA, Chandramouli GVR, DeGraff W, Yan H, Zhao S, et al. Gene Expression Profiling of Breast, Prostate, and Glioma Cells following Single versus Fractionated Doses of Radiation. Cancer Research. 2007;67(8):3845–52.
48. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell. 2010;140(6):805–20.
49. Cheon H, Stark GR. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proceedings of the National Academy of Sciences. 2009;106(23):9373–8.
50. Chew GL, Campbell AE, de Neef E, Sutliff NA, Shadle SC, Tapscott SJ, et al. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Developmental Cell. 2019 Sep 9;50(5):658-671.e7.
51. Ding LW, Sun QY, Edwards JJ, Fernández LT, Ran X bin, Zhou SQ, et al. LNK suppresses interferon signaling in melanoma. Nature Communications. 2019 Dec 1;10(1).
52. Chen Y, Wu Y ran, Yang H ying, Li X zhe, Jie M meng, Hu C jiang, et al. Prolyl isomerase Pin1: a promoter of cancer and a target for therapy. Cell Death & Disease. 2018 Sep 1;9(9).
53. Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, et al. Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nature Immunology. 2006 Jun;7(6):598–605.
54. Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR. Transcriptional and posttranscriptional regulation of interferoninduced gene expression in human cells. Cell. 1984;38(3):745–55.
55. Ackrill AM, Reid LE, Gilbert CS, Gewert DR, Porter AC, Lewin AR, et al. Differential response of the human 6-16 and 9-27 genes to alpha and gamma interferons. Nucleic Acids Research. 1991;19(3):591–8.
56. Gómez-Herranz M, Nekulova M, Faktor J, Hernychova L, Kote S, Sinclair E, et al. The effects of IFITM1 and IFITM3 gene deletion on IFNγ stimulated protein synthesis. Cellular Signaling. 2019 Aug 1;60:39–56.
57. Ferns D, Heeren A, Samuels S, Bleeker M, de Gruijl T, Kenter G, et al. Classical and non-classical HLA class I aberrations in primary cervical squamous- and adenocarcinomas and paired lymph node metastases. Journal for Immunotherapy of Cancer. 2016 Nov 15;4(1).
58. Connor M, Stern P. Loss of MHC class-I expression in cervical carcinomas. International Journal of Cancer. 1990;46(6):1029–34.
59. Vermeulen C, Jordanova E, Zomerdijk-Nooijen Y, ter Haar N, Peters A, Fleuren G. Frequent HLA class I loss is an early event in cervical carcinogenesis. Human Immunology. 2005 Nov;66(11):1167–73.
60. Hilders C, Munoz I, Nooyen Y, Fleuren G. Altered HLA expression by metastatic cervical carcinoma cells as a factor in impaired immune surveillance. Gynecologic Oncology. 1995;57(3):366–75.
61. Garrido F, Ruiz-Cabello F, Cabrera T, Pérez-Villar J, López-Botet M, Duggan-Keen M, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunology Today. 1997 Feb;18(2):89–95.
62. Seliger B, Cabrera T, Garrido F, Ferrone S. HLA class I antigen abnormalities and immune escape by malignant cells. Seminars in Cancer Biology. 2002;12(1):3–13.
63. Meissner T, Li A, Biswas A, Lee K, Liu Y, Bayir E, et al. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proceedings of the National Academy of Sciences of the United States of America. 2010 Aug 3;107(31):13794–9.
64. Biswas A, Meissner T, Kawai T, Kobayashi K. Cutting edge: impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. Journal of Immunology (Baltimore, Md : 1950). 2012 Jul 15;189(2):516–20.
65. Neerincx A, Rodriguez G, Steimle V, Kufer T. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosomedependent manner. Journal of Immunology (Baltimore, Md : 1950). 2012 May 15;188(10):4940–50.
66. Kalbasi A, Tariveranmoshabad M, Hakimi K, Kremer S, Campbell K, Funes J, et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Science Translational Medicine. 2020 Oct 14;12(565).
67. Cornel AM, Mimpen IL, Nierkens S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers [Internet]. 2020 Jul 1;12(7):1–33.
68. Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Molecular Medicine Today. 1999 Apr 1;5(4):178–86.
69. Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. International Journal of Cancer. 2010 Jul 15;127(2):249–56.
70. Garrido F. MHC Class-I Loss and Cancer Immune Escape. 2019;1151.
71. Feenstra M, Veltkamp M, van Kuik J, Wiertsema S, Slootweg P, van den Tweel J, et al. HLA class I expression and chromosomal deletions at 6p and 15q in head and neck squamous cell carcinomas. Tissue Antigens. 1999;54(3):235–45.
72. Maleno I, López-Nevot M, Cabrera T, Salinero J, Garrido F. Multiple mechanisms generate HLA class I altered phenotypes in laryngeal carcinomas: high frequency of HLA haplotype loss associated with loss of heterozygosity in chromosome region 6p21. Cancer Immunology, Immunotherapy: CII. 2002;51(7):389– 96.
73. Maleno I, Romero JM, Cabrera T, Paco L, Aptsiauri N, Cozar JM, et al. LOH at 6p21.3 region and HLA class I altered phenotypes in bladder carcinomas. Immunogenetics. 2006 Jul;58(7):503–10.
74. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell. 2017;171(6):1259-1271.e11.
75. Montesion M, Murugesan K, Jin DX, Sharaf R, Sanchez N, Guria A, et al. Somatic HLA Class I Loss Is a Widespread Mechanism of Immune Evasion Which Refines the Use of Tumor Mutational Burden as a Biomarker of Checkpoint Inhibitor Response. Cancer Discovery. 2021 Feb 1;11(2):282–92.
76. Zhao J, Xiao X, Li Y, Gao X, Zhang X, Liu Z, et al. The prevalence of HLA-I LOH in Chinese pan-cancer patients and genomic features of patients harboring HLA-I LOH. Human Mutation. 2021 Oct 1;42(10):1254–64.
77. Vijayan S, Sidiq T, Yousuf S, van den Elsen PJ, Kobayashi KS. Class I transactivator, NLRC5: a central player in the MHC class I pathway and cancer immune surveillance. Immunogenetics. 2019 Mar 1;71(3):273–82.
78. Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proceedings of the National Academy of Sciences of the United States of America. 2016 May 24;113(21):5999–6004.
79. Shukla A, Cloutier M, Santharam MA, Ramanathan S, Ilangumaran S. The MHC Class-I Transactivator NLRC5: Implications to Cancer Immunology and Potential Applications to Cancer Immunotherapy. International Journal of Molecular Sciences. 2021 Feb 2;22(4):1–36.
80. Jongsma MLM, Guarda G, Spaapen RM. The regulatory network behind MHC class I expression. Molecular Immunology. 2019 Sep 1;113:16–21.
81. Kasajima A, Sers C, Sasano H, Jöhrens K, Stenzinger A, Noske A, et al. Down-regulation of the antigen processing machinery is linked to a loss of inflammatory response in colorectal cancer. Human Pathology. 2010 Dec;41(12):1758–69.
82. Mehta AM, Jordanova ES, Kenter GG, Ferrone S, Fleuren GJ. Association of antigen processing machinery and HLA class I defects with clinicopathological outcome in cervical carcinoma. Cancer Immunology, Immunotherapy: CII. 2008 Feb;57(2):197–206.
83. Seliger B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunology, Immunotherapy: CII. 2008 Nov;57(11):1719–26.
84. Delp K, Momburg F, Hilmes C, Huber C, Seliger B. Functional deficiencies of components of the MHC class I antigen pathway in human tumors of epithelial origin. Bone Marrow Transplantation. 2000;25 Suppl 2:S88–95.
85. Liu Q, Hao C, Su P, Shi J. Down-regulation of HLA class I antigenprocessing machinery components in esophageal squamous cell carcinomas: association with disease progression. Scandinavian Journal of Gastroenterology. 2009;44(8):960-9.
86. Seliger B, Stoehr R, Handke D, Mueller A, Ferrone S, Wullich B, et al. Association of HLA class I antigen abnormalities with disease progression and early recurrence in prostate cancer. Cancer Immunology, Immunotherapy : CII. 2010 Apr;59(4):529–40.
87. Smithy JW, Moore LM, Pelekanou V, Rehman J, Gaule P, Wong PF, et al. Nuclear IRF-1 expression as a mechanism to assess “Capability” to express PD-L1 and response to PD-1 therapy in metastatic melanoma. Journal for Immunotherapy of Cancer. 2017 Mar 21;5(1).
88. Kriegsman BA, Vangala P, Chen BJ, Meraner P, Brass AL, Garber M, et al. Frequent Loss of IRF2 in Cancers Leads to Immune Evasion through Decreased MHC Class I Antigen Presentation and Increased PD-L1 Expression. Journal of Immunology (Baltimore, Md : 1950). 2019 Oct 1;203(7):1999–2010.
89. Gunda V, Frederick DT, Bernasconi MJ, Wargo JA, Parangi S. A potential role for immunotherapy in thyroid cancer by enhancing NY-ESO-1 cancer antigen expression. Thyroid: official journal of the American Thyroid Association. 2014 Aug 1;24(8):1241–50.
90. Krishnadas DK, Bao L, Bai F, Chencheri SC, Lucas K. Decitabine facilitates immune recognition of sarcoma cells by upregulating CT antigens, MHC molecules, and ICAM-1. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35(6):5753–62.
91. Natsume A, Wakabayashi T, Tsujimura K, Shimato S, Ito M, Kuzushima K, et al. The DNA demethylating agent 5-aza-2’- deoxycytidine activates NY-ESO-1 antigenicity in orthotopic human glioma. International Journal of Cancer. 2008 Jun 1;122(11):2542–53.
92. Šímová J, Polláková V, Indrová M, Mikyšková R, Bieblová J, Stěpánek I, et al. Immunotherapy augments the effect of 5-azacytidine on HPV16-associated tumours with different MHC class I-expression status. British Journal of Cancer. 2011 Nov 8;105(10):1533–41.
93. Ye Q, Shen Y, Wang X, Yang J, Miao F, Shen C, et al. Hypermethylation of HLA class I gene is associated with HLA class I down-regulation in human gastric cancer. Tissue Antigens. 2010 Jan;75(1):30–9.
94. Khan ANH, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunology, Immunotherapy : CII. 2008 May;57(5):647–54.
95. Sun T, Li Y, Yang W, Wu H, Li X, Huang Y, et al. Histone deacetylase inhibition up-regulates MHC class I to facilitate cytotoxic T lymphocyte-mediated tumor cell killing in glioma cells. Journal of Cancer. 2019;10(23):5638–45.
96. Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nature Communications. 2018 Dec 1;9(1).
97. Wang L, Amoozgar Z, Huang J, Saleh MH, Xing D, Orsulic S, et al. Decitabine Enhances Lymphocyte Migration and Function and Synergizes with CTLA-4 Blockade in a Murine Ovarian Cancer Model. Cancer Immunology Research. 2015 Sep 1;3(9):1030–41.
98. Ritter C, Fan K, Paschen A, Reker Hardrup S, Ferrone S, Nghiem P, et al. Epigenetic priming restores the HLA class-I antigen processing machinery expression in Merkel cell carcinoma. Scientific Reports. 2017 Dec 1;7(1).
99. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016 Oct 6;167(2):397-404.e9.
100. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Reports. 2017 May 9;19(6):1189–201.
101. Groner B, von Manstein V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Molecular and Cellular Endocrinology. 2017 Aug 15;451:1–14.
102. Meissl K, Macho-Maschler S, Müller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine. 2017 Jan 1;89:12–20.
103. Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNγ resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nature Communications. 2017 May 31;8.
104. Kloetzel PM. Antigen processing by the proteasome. Nature Reviews Molecular Cell Biology. 2001 Mar;2(3):179–87.
105. Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nature Immunology. 2002 Dec 1;3(12):1169–76.
106. Zhou F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. International Reviews of Immunology. 2009;28(3–4):239–60.
107. Chong C, Marino F, Pak H, Racle J, Daniel RT, Mü Ller M, et al. High-throughput and Sensitive Immunopeptidomics Platform Reveals Profound Interferonγ-Mediated Remodeling of the Human Leukocyte Antigen (HLA) Ligandome. Molecular & Cellular Proteomics : MCP. 2018 Mar 1;17(3):533–48.
108. Goncalves G, Mullan KA, Duscharla D, Ayala R, Croft NP, Faridi P, et al. IFNγ Modulates the Immunopeptidome of Triple Negative Breast Cancer Cells by Enhancing and Diversifying Antigen Processing and Presentation. Frontiers in Immunology. 2021 Apr 22;12.
109. Javitt A, Barnea E, Kramer MP, Wolf-Levy H, Levin Y, Admon A, et al. Pro-inflammatory Cytokines Alter the Immunopeptidome Landscape by Modulation of HLA-B Expression. Frontiers in Immunology. 2019;10(FEB).
110. Olsson N, Heberling ML, Zhang L, Jhunjhunwala S, Phung QT, Lin S, et al. An Integrated Genomic, Proteomic, and Immunopeptidomic Approach to Discover Treatment-Induced Neoantigens. Frontiers in Immunology. 2021 Apr 15;12.
111. Angell TE, Lechner MG, Jang JK, LoPresti JS, Epstein AL. MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in vitro. Clinical Cancer Research: an official journal of the American Association for Cancer Research. 2014 Dec 1;20(23):6034–44.
112. Li H, Chiappinelli KB, Guzzetta AA, Easwaran H, Yen RWC, Vatapalli R, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancers. Oncotarget. 2014;5(3):587–98.
113. Walsh RJ, Soo RA. Resistance to immune checkpoint inhibitors in non-small cell lung cancer: biomarkers and therapeutic strategies. Therapeutic Advances in Medical Oncology. 2020;12.
114. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. The New England Journal of Medicine. 2016 Sep;375(9):819–29.
115. Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-γ inducible chemokines. Cancer Research. 2012 Oct 15;72(20):5209–18.
116. Kageshita T, Hirai S, Ono T, Hicklin DJ, Ferrone S. Down regulation of HLA class I antigen-processing molecules in malignant melanoma: association with disease progression. The American Journal of Pathology. 1999;154(3):745–54.
117. Kloor M, Becker C, Benner A, Woerner SM, Gebert J, Ferrone S, et al. Immunoselective pressure and human leukocyte antigen class I antigen machinery defects in microsatellite unstable colorectal cancers. Cancer Research. 2005 Jul 15;65(14):6418–24.
118. Meissner M, Reichert TE, Kunkel M, Gooding W, Whiteside TL, Ferrone S, et al. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clinical Cancer Research : an official journal of the American Association for Cancer Research. 2005 Apr 1;11(7):2552–60.
119. Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clinical and Experimental Immunology. 2007 Apr;148(1):127–35.
120. Raffaghello L, Prigione I, Bocca P, Morandi F, Camoriano M, Gambini C, et al. Multiple defects of the antigenprocessing machinery components in human neuroblastoma: immunotherapeutic implications. Oncogene. 2005 Jul 7;24(29):4634–44.
121. Hoves S, Aigner M, Pfeiffer C, Laumer M, Obermann EC, Mackensen A. In situ analysis of the antigen-processing machinery in acute myeloid leukaemic blasts by tissue microarray. Leukemia. 2009;23(5):877–85.
122. Racanelli V, Leone P, Frassanito MA, Brunetti C, Perosa F, Ferrone S, et al. Alterations in the antigen processing-presenting machinery of transformed plasma cells are associated with reduced recognition by CD8+ T cells and characterize the progression of MGUS to multiple myeloma. Blood. 2010 Feb 11;115(6):1185–93.
123. Bandoh N, Ogino T, Katayama A, Takahara M, Katada A, Hayashi T, et al. HLA class I antigen and transporter associated with antigen processing downregulation in metastatic lesions of head and neck squamous cell carcinoma as a marker of poor prognosis. Oncology Reports. 2010;23(4):933–9.
124. Kikuchi E, Yamazaki K, Torigoe T, Cho Y, Miyamoto M, Oizumi S, et al. HLA class I antigen expression is associated with a favorable prognosis in early stage non-small cell lung cancer. Cancer Science. 2007 Sep;98(9):1424–30.
125. Mizukami Y, Kono K, Maruyama T, Watanabe M, Kawaguchi Y, Kamimura K, et al. Downregulation of HLA Class I molecules in the tumour is associated with a poor prognosis in patients with oesophageal squamous cell carcinoma. British Journal of Cancer. 2008 Oct 4;99(9):1462–7.
126. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discovery. 2017 Dec 1;7(12):1420–35.
127. Lee M, Jeon J, Sievers C, Allen C. Antigen processing and presentation in cancer immunotherapy. Journal for Immunotherapy of Cancer. 2020 Aug 1;8(2).
128. Paulson KG, Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, et al. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nature Communications. 2018 Dec 1;9(1).
129. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily- Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nature Communications. 2017 Dec 1;8(1).
130. Carretero R, Romero JM, Ruiz-Cabello F, Maleno I, Rodriguez F, Camacho FM, et al. Analysis of HLA class I expression in progressing and regressing metastatic melanoma lesions after immunotherapy. Immunogenetics. 2008 Aug;60(8):439–47.
131. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of Experimental Medicine. 2000 Oct 2;192(7):1027–34.
132. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (New York, NY). 1996 Mar 22;271(5256):1734–6.
133. Bonaventura P, Shekarian T, Alcazer V, Valladeau- Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Frontiers in Immunology. 2019;10:168.
134. Maschek U, Pulm W, Hammerling GJ. Altered regulation of MHC class I genes in different tumor cell lines is reflected by distinct sets of DNase I hypersensitive sites. The EMBO Journal. 1989;8(8):2297.
135. Alavi S, Stewart AJ, Kefford RF, Lim SY, Shklovskaya E, Rizos H. Interferon Signaling Is Frequently Downregulated in Melanoma. Frontiers in Immunology. 2018 Jun 21;9(JUN).
136. Zhang S, Kohli K, Graeme Black R, Yao L, Spadinger SM, He Q, et al. Systemic Interferon-γ Increases MHC Class I Expression and T-cell Infiltration in Cold Tumors: Results of a Phase 0 Clinical Trial. Cancer Immunology Research. 2019;7(8):1237–43.
137. Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, et al. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. British journal of cancer. 2015 Apr 28;112(9):1501–9.
138. Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, et al. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015 Jan 1;4(6).
139. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Science Translational Medicine. 2013 Aug 28;5(200).
140. Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villarreal R, et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. Journal of the American Society of Nephrology. 2015 Jan 1;26(1):133-47.
141. Brody JR, Costantino CL, Berger AC, Sato T, Lisanti MP, Yeo CJ, et al. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle (Georgetown, Tex). 2009 Jun 15;8(12):1930–4.
142. Lu C, Klement JD, Ibrahim ML, Xiao W, Redd PS, Nayak- Kapoor A, et al. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. Journal for Immunotherapy of Cancer. 2019 Jun 22;7(1).
143. Kortylewski M, Komyod W, Kauffmann ME, Bosserhoff A, Heinrich PC, Behrmann I. Interferon-gamma-mediated growth regulation of melanoma cells: involvement of STAT1-dependent and STAT1-independent signals. The Journal of Investigative Dermatology. 2004;122(2):414–22.
144. Windbichler GH, Hausmaninger H, Stummvoll W, Graf AH, Kainz C, Lahodny J, et al. Interferon-gamma in the first-line therapy of ovarian cancer: a randomized phase III trial. British Journal of Cancer. 2000;82(6):1138–44.
145. Miller CHT, Maher SG, Young HA. Clinical Use of Interferongamma. Annals of the New York Academy of Sciences. 2009;1182:69–79.
146. Gerber SA, Sedlacek AL, Cron KR, Murphy SP, Frelinger JG, Lord EM. IFN-γ Mediates the Antitumor Effects of Radiation Therapy in a Murine Colon Tumor. The American Journal of Pathology [Internet]. 2013;182(6):2345–54.
147. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature Medicine. 2007 Sep;13(9):1050–9.
148. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, K.Wansley E, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. The Journal of Experimental Medicine [Internet]. 2006;203(5):1259-71.
149. Higgs BW, Morehouse CA, Streicher K, Brohawn PZ, Pilataxi F, Gupta A, et al. Interferon Gamma Messenger RNA Signature in Tumor Biopsies Predicts Outcomes in Patients with Non-Small Cell Lung Carcinoma or Urothelial Cancer Treated with Durvalumab. Clinical Cancer Research : an official journal of the American Association for Cancer Research. 2018 Aug 15;24(16):3857–66.
150. Karachaliou N, Gonzalez-Cao M, Crespo G, Drozdowskyj A, Aldeguer E, Gimenez-Capitan A, et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Therapeutic Advances in Medical Oncology. 2018 Jan 1;10.
151. Gao Y, Yang J, Cai Y, Fu S, Zhang N, Fu X, et al. IFN-γ-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. International Journal of Cancer. 2018 Aug 15;143(4):931–43.
152. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA- 4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences of the United States of America. 2010 Mar 2;107(9):4275–80.
153. Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proceedings of the National Academy of Sciences of the United States of America. 2008 Sep 30;105(39):14987–92.
154. Seliger B, Ruiz-Cabello F, Garrido F. IFN inducibility of major histocompatibility antigens in tumors. Advances in Cancer Research. 2008;101:249–76.
155. Rodríguez T, Méndez R, del Campo A, Jiménez P, Aptsiauri N, Garrido F, et al. Distinct mechanisms of loss of IFN-gamma mediated HLA class I inducibility in two melanoma cell lines. BMC Cancer. 2007;7.
156. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016 Dec 1;167(6):1540-1554.e12.
157. Patel SA, Minn AJ. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity. 2018 Mar 20;48(3):417–33.
158. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of IFN-γ in tumor progression and regression: a review. Biomarker Research. 2020 Sep 29;8(1).
159. Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell. 2019 Aug 8;178(4):933-948.e14.
160. Cui C, Xu C, Yang W, Chi Z, Sheng X, Si L, et al. Ratio of the interferon-γ signature to the immunosuppression signature predicts anti-PD-1 therapy response in melanoma. NPJ Genomic Medicine. 2021 Dec 1;6(1).
161. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (New York, NY). 2014;344(6184):641–5.
162. Chen F, Zou Z, Du J, Su S, Shao J, Meng F, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. The Journal of Clinical Investigation. 2019 May 1;129(5):2056–70.
163. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (New York, NY). 2015 May 15;348(6236):803–8.
164. Ikeda H, Old LJ, Schreiber RD. The roles of IFNy in protection against tumor development and cancer immunoediting. Cytokine and Growth Factor Reviews. 2002;13(2):95–109.
165. Arnaud M, Chiffelle J, Genolet R, Navarro Rodrigo B, Perez MAS, Huber F, et al. Sensitive identification of neoantigens and cognate TCRs in human solid tumors. Nature Biotechnology. 2021 Nov 15;
166. Ni L, Lu J. Interferon gamma in cancer immunotherapy. Cancer Medicine . 2018 Sep 1;7(9):4509–16.
167. Marth C, Windbichler GH, Hausmaninger H, Petru E, Estermann K, Pelzer A, et al. Interferon-gamma in combination with carboplatin and paclitaxel as a safe and effective first-line treatment option for advanced ovarian cancer: results of a phase I/II study. International Journal of Gynecological Cancer. 2006 Jul;16(4):1522–8.
168. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Frontiers in Immunology. 2018 May 4;9(847).