Commentary
Tissues are composed of various types of interacting cells [1]. To understand the cellular organization and function in tissues, it is necessary to identify all of the different cell types and the locations of these different cell types within tissue structures. The transformative advances in experimental and computational methods will help us to build the complex map of the tissues and study how tissue organization influences the cell’s molecular state and interactions in healthy and diseased tissue [1-6].
Over the past few years, the development and application of single-cell sequencing methods have revolutionized the entire field of biology thus enabling us to study the cellular heterogeneity of the cancer tissues [7]. Understanding the clonal architecture of the tumor and the interplay between the malignant and non-malignant cells within the tumor ecosystem provides significant insights into the tumor recurrence, treatment, initiation, progression, and metastasis [8-10]. For example, the ratios of specific immune cell types in the tumor predict overall survival and response to different immunotherapies [11]. Researchers used single-cell RNA sequencing (scRNA-seq) to examine heterogeneities in malignant and nonmalignant cell types and states in various cancer types such as melanoma and glioma [7,12,13]. With the advent of scRNA-Seq, it is now possible to identify tumor-infiltrating immune cell types and tumor-associated malignant/non-malignant cell types such as endothelial cells (ECs) and cancer-associated fibroblasts (CAFs) in the tumor and identify transcriptional alterations within these cells groups [7,12-17]. Single-cell DNA sequencing is another new approach for elucidating the genomic diversity of tumor clonal architecture [2,10]. However, we still do not have the technological capabilities to simultaneously probe the genome and transcriptome at the single cell level and a large scale.
Many algorithms have been developed for detecting copy number variations (CNV) events from DNA sequencing data using depth of coverage analysis [18,19]. These tools rely on uniform coverage of genome by DNA-sequencing reads. However, statistical approaches for CNV detection using RNA-sequencing data is very limited since it is very hard to discriminate between differential expression and an underlying copy number variation using only RNA-Seq data. Another challenge is that RNA-Seq signal is generally concentrated on the exonic regions and most of the genome is not covered. Thus, the identified CNVs will reflect the copy number states of the genes and the copy number of intergenic regions may not be represented well. Regardless, the copy number of genes is extremely useful information for characterizing CNV architecture of, for example, the copy number of oncogenes and tumor suppressor genes. It is worth noting that this issue is similar to the whole exome-sequencing based CNV detection because the whole exome-sequencing covers only the targeted exonic regions in the genome.
Although many tools identify CNVs from exome sequencing data, there is a lack of methods for detecting CNVs solely from RNA sequencing data [7,20]. We developed one of the first methods that identifies, visualizes and integrates CNV events using scRNA-Seq data [21]. In addition to CNVs, we also need to estimate SNPs and indels from scRNA-Seq data for understanding the clonal architecture of the tumor. Identification of SNPs and indels from scRNA-Seq data, however, is also very challenging because of allelic dropouts, non-uniform and low coverage. This makes it very hard to distinguish between real variations and technical artifacts.
Inference of CNV, SNP, and indel from RNA-Seq data is essential for understanding the correlation between the genomic and the transcriptomic properties of different cell types and clones within the tumor ecosystem [22]. These correlations will provide significant insight into tumor initiation, progression, and metastasis. Since, it remains technically challenging to assay both the genome and transcriptome from the same cell, until now gene signatures of different tumor clones have not been studied very well. There is an increasing need for developing CNV, SNP and indel inference algorithms from scRNA-Seq data especially with the growing number of scRNA-Seq studies.
Another important motivation in calling CNV, SNP, and indels from scRNA-Seq datasets is the capability of detecting low-allele frequency variants. Somatic variants in even a small minority of cells can have large phenotypic effects. Detecting somatic mutations with a low allele frequency is especially important in cancer. It is very challenging to detect low allele frequency mutations using bulk DNA sequencing. Even though several methods have been developed to detect low allele frequency mutations such as MuTect and Strelka from bulk DNA sequencing [23,24], these methods still cannot reliably detect mutations with lower than 0.1 allele frequency rate for the samples with an average sequencing depth. Unfortunately, currently, there is a lack of cancer single-cell DNA sequencing data but instead, there is a growing number of cancer scRNA-Seq data. Therefore, there is an increasing need for developing CNV, SNP and indel inference algorithm from scRNA-Seq data.
In addition to the above points, the analysis framework that CaSpER utilizes can be extended to other functional genomics datasets such as ChIP-Sequencing, epigenomics, and spatial transcriptomics datasets, which are currently not performed as often as RNA-sequencing. Spatial approaches aid us to build spatially resolved gene expression patterns of different cell types within a tissue. The inference of CNV events from spatial transcriptomics datasets provides us the clonal architecture of the tumor together with the spatial information.
Single-cell RNA-seq is powerful in detecting different cell types or states but requires tissue dissociation, thus losing the information about the original location of the cells. On the other hand, spatial approaches provide spatial information but measures expression levels of only a small number of transcripts (in situ hybridization) or lack singlecell resolution (spatial transcriptomics assays) [3-6,25]. We can infer the spatial location of cells by integrating scRNA-Seq with spatial transcriptomics. In the future, we believe that there will be advances in approaches that integrate spatially transcriptomics data with scRNA-Seq data. These approaches will enable us to generate the complex map of the tumor revealing the interactions of different cell types within the tumor ecosystem (Figure 1).
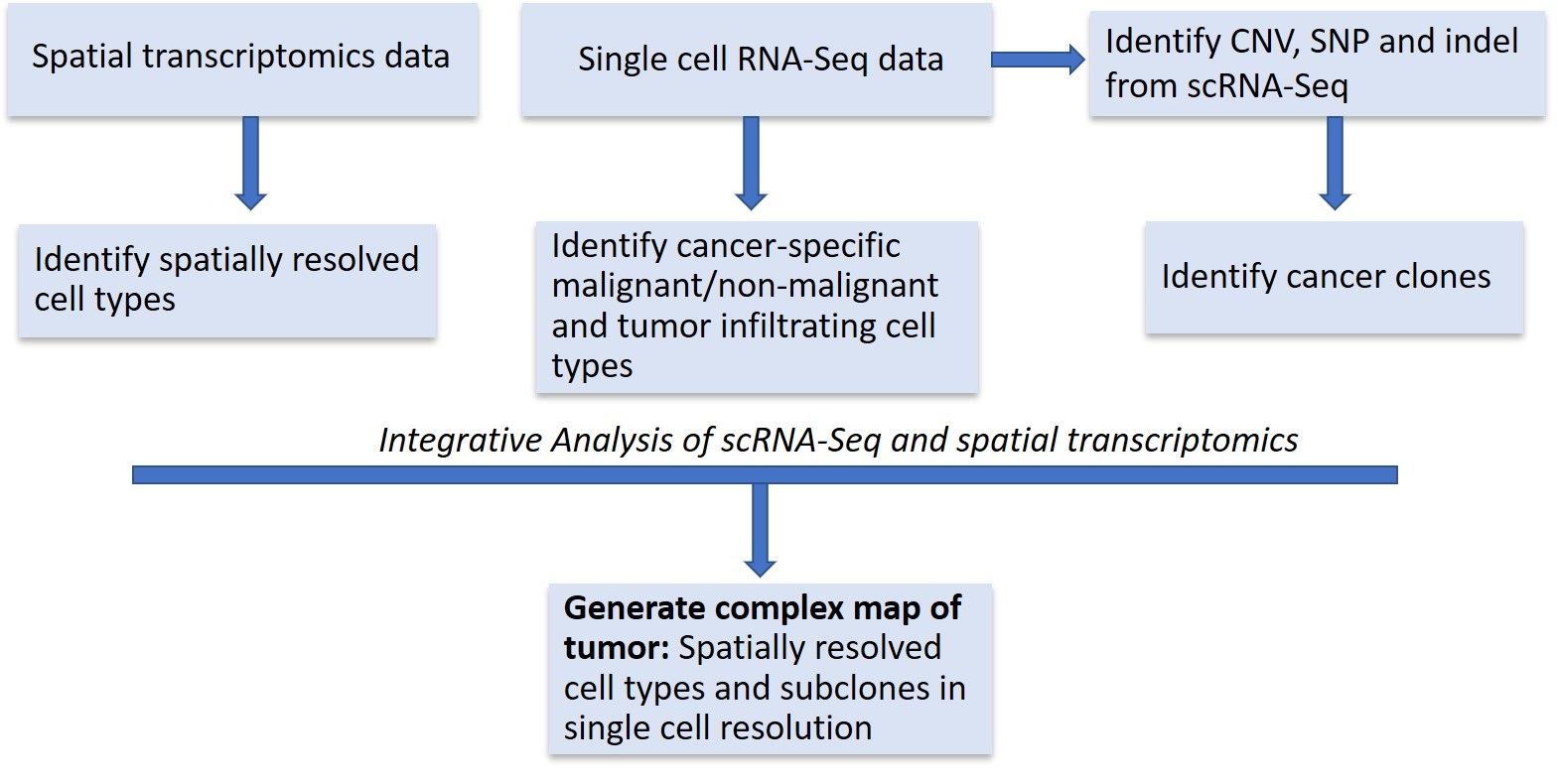
Figure 1: Approaches that integrate spatially transcriptomics data with scRNA-Seq data.
References
2. Pan X. Single cell analysis: from technology to biology and medicine. Single Cell Biology. 2014;3(1).
3. Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015 Apr 24;348(6233):aaa6090.
4. Wang X, Allen WE, Wright MA, Sylwestrak EL, Samusik N, Vesuna S, et al. Three-dimensional intacttissue sequencing of single-cell transcriptional states. Science. 2018 Jul 27;361(6400):eaat5691.
5. Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016 Jul 1;353(6294):78-82.
6. Crosetto N, Bienko M, Van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nature Reviews Genetics. 2015 Jan;16(1):57-66.
7. Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016 Apr 8;352(6282):189-96.
8. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74.
9. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013 Sep;501(7467):328-37.
10. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011 Apr;472(7341):90.
11. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Scientific Reports. 2015 Oct 14;5:15179.
12. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20;344(6190):1396-401.
13. Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016 Nov;539(7628):309-13.
14. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nature Communications. 2017 May 5;8(1):1-2.
15. Ledergor G, Weiner A, Zada M, Wang SY, Cohen YC, Gatt ME, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nature Medicine. 2018 Dec;24(12):1867-76.
16. MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nature Communications. 2018 Oct 22;9(1):1-21.
17. Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, et al. Neuronal subtypes and diversity revealed by singlenucleus RNA sequencing of the human brain. Science. 2016 Jun 24;352(6293):1586-90.
18. Sathirapongsasuti JF, Lee H, Horst BA, Brunner G, Cochran AJ, Binder S, et al. Exome sequencing-based copynumber variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011 Oct 1;27(19):2648-54.
19. Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, et al. Copy number variation detection and genotyping from exome sequence data. Genome Research. 2012 Aug 1;22(8):1525-32.
20. Fan J, Lee HO, Lee S, Ryu DE, Lee S, Xue C, Kim SJ, Kim K, Barkas N, Park PJ, Park WY. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome research. 2018 Aug 1;28(8):1217-27.
21. Harmanci AS, Harmanci AO, Zhou X. CaSpER: Identification, visualization and integrative analysis of CNV events in multiscale resolution using single-cell or bulk RNA sequencing data. bioRxiv. 2018 Jan 1:426122.
22. Müller S, Liu SJ, Di Lullo E, Malatesta M, Pollen AA, Nowakowski TJ, et al. Single-cell sequencing maps gene expression to mutational phylogenies in PDGF-and EGFdriven gliomas. Molecular Systems Biology. 2016 Nov 1;12(11).
23. Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Källberg M, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nature Methods. 2018 Aug;15(8):591-4.
24. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology. 2013 Mar;31(3):213.
25. Shah S, Lubeck E, Zhou W, Cai L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron. 2016 Oct 19;92(2):342-57.