Abstract
Neuroblastoma (NB) is one of the most common causes of pediatric cancer mortality. Cancer is well known for its complexity and wide variety of presentations. At its worst, NB presents with early metastasis and resistance to therapies. It contains complex molecular and genetic features and has an immunosuppressive microenvironment that forms a complex case that is fatal in around 50% of affected patients, even with the current multimodal treatment standard for high-risk neuroblastoma (HR-NB). This has highlighted a need for comprehensive molecular understanding of the tumor, as well as multi-targeted therapeutic approaches. Current targets that remain a focus for NB include GD2 Disialoganglioside (GD2), v-myc avian myelocytomatosis viral oncogene neuroblastoma-derived homolog (MYCN), glypican 2 (GPC2), B7 Homolog 3 (B7-H3) also known as CD276, and Anaplastic Lymphoma Kinase (ALK). Recent advances covered in this review include further developments in anti-GD2 immunotherapy, targeted therapy for ALK-driven tumors, Chimeric Antigen Receptor T-cell therapy (CAR-T), combination therapies, and other immunotherapies. This review aims to compile the most up-to-date information on the recent advances in the molecular landscape of immunotherapy in a comprehensive manner to understand and identify the shortcomings and future directions for treatment targets of HR-NB.
Keywords
Immunotherapy, Neuroblastoma, Molecular pathogenesis, Precision oncology
Introduction
NBs are part of a spectrum of neuroblastic pediatric tumors that originate from primitive sympathetic ganglion cells, also known as neural crest cells [1–3]. It is the most common extracranial solid tumor in children and is responsible for approximately 11–15% of all pediatric cancer deaths [4–6]. NBs are well known for their diversity in progression and presentation, whether it is on a clinical or cellular level [3,7,8]. Clinical presentation varies widely with tumor location, although the most common clinical presentation of neuroblastoma is an abdominal mass [5,9]. Other possible presentations include hypertension, opsoclonus-myoclonus syndrome, and symptoms based on areas of compression [5,9]. Due to their complex makeup, NBs are classified based on multiple prognostic factors including age at diagnosis, tumor staging, histopathology, MYCN amplification status, segmental chromosomal aberrations, and DNA ploidy [10,11].
Progression spans from spontaneous regression to aggressive disease with metastatic dissemination that can be fatal. NBs are staged, and their risk levels are assessed using the International Neuroblastoma Risk Group Staging System (INRGSS) as summarized in Table 1, which defines risk based on image-defined risk factors (IDRF) as validated by the Children’s Oncology Group [10,11]. Most NBs with serious morbidity and mortality are high-risk (HR-NB). This “high-risk” designation is primarily assigned to patients who either have tumors with MYCN amplification or patients older than 18 months with metastatic disease per the INRGSS [10,11]. NBs account for approximately 15% of all pediatric cancer mortality in 2025 as the most common pediatric extracranial solid tumor [12–14]. Survival is dependent on the age at diagnosis, with a 5-year OS at 91% in infants that drops to an event-free survival of 48% in toddlers older than 18 months [4]. The National Comprehensive Cancer Network (NCCN) states that long-term survival for high-risk neuroblastomas remains at approximately 50%, and relapse rates are high [15,16]. Prognosis is strongly affected by tumor biology; the most notable MYCN amplification, which is strongly associated with negative outcomes. Metaiodobenzylguanidine (mIBG) is a norepinephrine analog that is highly sensitive and specific for detecting NB. It is taken up by over 90% of NB cells, which makes it a radiotracer for the disease. The Curie score standardizes the interpretation of mIBG scintigraphy and is used in NB management for prognostic stratification, especially after induction. It allows tracking of NBs over time to assess response and residual disease or relapse. The prognostic relevance of this scoring system was demonstrated in the Children's Oncology Group A3973 study, where a post-induction Curie score greater than 2 was independently associated with significantly inferior event-free survival in patients with stage 4 disease. Additional prognostic and therapeutic determinants to be discussed in this review include segmental chromosomal aberrations, ALK alterations, and GD2 expression [17,18].
|
Stage |
Age |
Key Tumor Features |
Risk Group |
Notes |
|
L1 |
Any |
Localized tumor, no IDRFs, favorable biology |
Low |
May spontaneously regress. Usually amenable to surgical resection only; minimal risk of metastasis |
|
L2 |
Any |
Locoregional tumor, ≥1 IDRF, may have unfavorable biology |
Intermediate / High (dependent on biology/age) |
Often requires chemotherapy before surgery; higher surgical risk |
|
M |
<18 months ≥18 months |
Distant metastasis, favorable biology (non-amplified MYCN) Distant metastasis, unfavorable biology (MYCN amplification, segmental chromosomal alterations) |
Intermediate
High |
Infants with limited metastasis may respond well to chemo Requires intensive multimodal therapy (chemo, surgery, radiation, immunotherapy) |
|
MS |
<18 months |
Metastasis limited to liver, skin, minimal bone marrow involvement; favorable biology |
Low / Intermediate |
Often resolves with minimal therapy or observation; excellent prognosis |
In 2010, the identification of GD2’s high expression on nearly all NB cells provided the first major breakthrough in NB immunotherapy [22]. The ANBL0032 trial demonstrated that anti-GD2 monoclonal antibody dinutuximab, in combination with cytokines and isotretinoin, improved the 2-year event-free survival from 46% to 66% when compared to isotretinoin alone [19,20]. Since the success of anti-GD2 monoclonal antibodies, the field has seen an explosion of novel immunotherapeutic approaches over the past several years. CAR-T targeting GD2 has shown to be particularly promising, with third-generation GD2-CAR T cells incorporating dual costimulatory domains and inducible safety switches that demonstrate a 63–66% overall response rate and 68% 5-year overall survival (OS) in relapsed/refractory patients that had been pretreated [21–23]. Other antigens are also being targeted by CAR T-cell platforms to address NB heterogeneity [26]. Rational combination strategies have become a central focus of contemporary NB immunotherapy development. Clinical trials are currently underway for combinations of anti-GD2 antibodies with checkpoint inhibitors such as nivolumab (anti-PD-1), cytokines (IL-15, IL-21), epigenetic modulators (EZH2 inhibitors), and metabolic modulators [24–27]. Additionally, biomarker-guided treatments that check ALK mutation status, immune infiltration profiles, cytokine signatures, and GD2 expression levels for individual tumors are being utilized more frequently to guide therapy [24–26]. Platforms like armored CAR T-cells, bispecific antibodies, antibody-drug conjugates, among others, are some of the more novel therapeutic options in development for NB [25,27].
Even with the developing therapies, there are still several roadblocks for HR-NB treatments that prevent long-term survivability. HR-NB’s immunosuppression and molecular complexity continue to create challenges in its therapeutic approach, with 5-year event-free survival still only reaching 50% of affected pediatric individuals [4,5,24]. HR-NB’s infiltration of myeloid cells, downregulation of MHC class I expression, secretion of immunosuppressive mediators such as galectin-1, and low mutational burden create the tumor’s immunosuppressive microenvironment [28–31]. This behavior limits therapeutic efficacy. For instance, CAR-T is restricted by antigen loss following CAR-T, T-cell exhaustion, decreased CAR-T persistence, and immunosuppressive myeloid populations continue to limit long-standing responses to treatment [20,21,28]. HR-NBs additionally have extensive redundancy and multiple interconnected pathways that require a rational combination of therapies to overcome resistance [28]. The toxicity of many of these therapies, such as neuropathic pain, also limit treatment due to tolerability [27,32]. Socioeconomic limitations to necessary agents and resources for research and clinical trials require coordinated international effort and early regulatory engagement [3,14,25,27].
This review aims to synthesize the current evidence on the most recent developments in immunotherapeutic strategies for NB with an emphasis on GD2-targeted therapies, CAR T-cell platforms, combination approaches, and biomarker-driven personalization. By achieving this, we hope to facilitate current understanding of NB, so that further advancements may be made to improve the outcomes for the affected pediatric population.
Molecular/Genetic Features
Given the high level of differentiation required by these multipotent embryonic cells, the cellular process of differentiation is very complex and consists of a multitude of genes [3,6,7]. The alteration of a gene implicated in these differentiating pathways can then lead to the formation of neuroblasts [33]. With the wide variety of genes involved neural crest development, no genetic marker has been singled out to drive the formation of NB [34]. Instead, multiple molecular biomarkers, pathways and therapeutic targets such as ALK, MYCN, GPC2, mTOR, GD2, and B7-H3 have been implicated in NB and its treatment [2,35–42]. Figure 1 illustrates the tumor biology and receptor targets relevant for this review. These biomarkers have a range of expression frequencies in NBs that make them choice targets for therapy. ALK is altered in around 14–21.5% of HR-NBs, with higher percentages of ALK alteration on relapsed patients. MYCN is amplified in about 36–38% of HR-NBs [35,43,44], and GPC2 is expressed in 50–90% of all NBs [38,45]. B7-H3 is expressed in 74–96% of NBs with limited expression in normal tissues [46–48]. GD2 is expressed in about 96–100% of all NBs and is considered a hallmark target for immunotherapy [37,49,50]. There is currently limited published data on the precise percentages of overlap between the mentioned biomarkers (ALK, MYCN, GD2, GPC2, B7-H3) in NB. However, there is high overlap and regulation between their roles for the development of NB [38,42,44,49,51,52]. These biomarkers, the mechanisms by which they are involved in HR-NB development, and some of their relationships with each other are discussed further to develop an understanding of the mechanisms behind the developing therapies for NB.
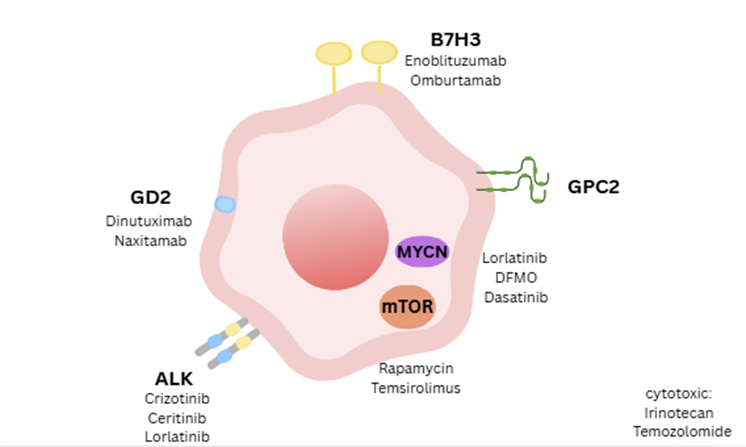
ALK is a gene encoding a tyrosine kinase receptor and is typically expressed during nervous system development. It is a current developing target for immunotherapy as the most mutated single gene in primary NB [30,51,53]. ALK-activating mutations have recently been found in up to 21.5% of HR-NB tumors, a percentage higher than the normally attributed 9–10% [51–53]. ALK is overactivated outside its usual expression in the context of specific development, leading to downstream signals being amplified to cause abnormal proliferation and blockage of cell differentiation [34,53]. Heritable mutations in ALK following this pathway have been found to cause the majority of hereditary NB cases [36]. In a genomic analysis completed by the Children’s Oncology Group in September, clonal and subclonal ALK mutations, as well as ALK amplification, were found to independently predict inferior outcomes for patient, with a 5-year survival that drops from the 66.3% of ALK wild-type tumors to a 37.7% in ALK-mutated tumors, and even lower to 25% for ALK-amplified tumors [51].
ALK’s role in NB development comes from its function as an oncogene. Point mutations (usually R1275Q and F1174L) or genomic amplification cause ligand-independent dimerization to constitutively activate ALK [52,54]. Activated ALK then autophosphorylates specific tyrosine residues in its intracellular domain to create docking sites for adaptor proteins with SH2 domains [55–57]. This then initiates multiple parallel signaling cascades including ERK-ETV5-RET, PI3K/AKT/mTOR, STAT3, and JAK/STAT pathways, among others, to create a critical oncogenic axis [54] and promote differentiation and cell-cycle arrest [29,34,53]. ALK mutations additionally upregulates POSTN (periostin) and WNT signaling in a feedforward loop that increases focal adhesion and extracellular matrix (ECM) genes and promotes tumor growth and maintenance [58].
It is important to note that a large majority of the pathways and signals can be activated and promoted through multiple mechanisms and can involve several biomarkers. For example, B7-H3 (CD276) is an immune checkpoint protein that is highly expressed in NBs with limited expression in normal tissue [46,48]. It activates STAT3 through the JAK2/STAT3 pathway, which integrates with ALK-driven STAT3 activation, and contributes to chemoresistance in NB [46,48,59]. Another example is MYCN, which directly targets GPC2 transcription, which then also activates the WNT B-catenin pathway [38,45]. ALK mutations cooperate with MYCN in a bidirectional positive feedback loop through several interconnected pathways to mutually promote their expression [29,54]. Their coexpression works to upregulate S-phase kinase-associated protein 2 (SKP2), an E3 ubiquitin ligase that targets the CDK inhibitor p27 for degradation, maintaining neuroblast proliferation [55,60,61]. This then decreases ECM integrity and enhances cell invasion to cause complete penetrance of NB in mouse models [29,54,62]. Outcomes were more inferior with simultaneous ALK and MYCN amplification in HR-NB, with specific ALK F1174L mutation showing to have a synergistic effect with worse outcomes when present with concurrent amplification of MYCN [51,52,63].
As briefly mentioned previously, MYCN status is considered one of the more important features of NB tumors due to its association with poor prognosis, especially in HR-NB [64]. MYCN amplification is found in around 25% of all NB cases [65]. MYCN is an oncogene that encodes for the transcription factor N-Myc, a basic helix-loop-helix leucine zipper transcription factor that forms heterodimers with myc-associated factor X (MAX) to regulate gene expression [66,67]. The MYCN-MAX heterodimer binds to DNA at enhancer boxes sequences that are both canonical and non-canonical. This regulates transcription and activation of an extensive list of genes, proteins, and pathways, including GPC2 and mTOR pathway components, that influence cellular processes through numerous mechanisms including cell cycle progression, metabolic reprogramming, differentiation, angiogenesis, tumor invasion, apoptosis, and metastasis in the context of NB development [68–70]. More specifically N-Myc is involved with spliceosome machinery and increasing RNA and protein production [71]. MYCN amplification also causes cellular undifferentiation and increased mitotic and karyorrhectic activities [72,73]. It is worth mentioning that MYCN is stabilized by mTOR signaling, creating a bidirectional regulatory loop [74,75]. This creates therapeutic vulnerability in MYCN-amplified NBs as mTOR kinase activity must be completely blocked to effectively destabilize MYCN [74–77]. Overall, those with an unfavorable histology designation and an amplified MYCN status showed the poorest clinical outcomes [72].
GD2 ganglioside is a glycosphingolipid found on a broad spectrum of human cancers, both pediatric and adult [78] but limited to peripheral pain fibers (C-fibers), melanocytes, and some central nervous system structures in normal tissue [32,79]. GD2 biosynthesis begins with ceramides then becomes glucosylceramide (via GCS). It then consecutively becomes lactosylceramide, then GM3 (via GM3 synthase/ST3GAL5), then GD3 (via GD3 synthase/ST8SIA1), and finally becomes GD2 (via GD2 synthase/B4GALNT1 [80,81]. Its expression on normal tissue surfaces is associated with the more severe side effects of anti-GD2 immunotherapy, such as neuropathic pain [32]. Gangliosides are sphingolipids that physiologically function in cell recognition and the regulation of signaling proteins including epidermal growth factor (EGF) receptor and vascular endothelial growth factor (VEGF) receptor [37,82]. The exact function of GD2 itself in normal tissue is not yet completely understood, but it is known to function in cell signaling [78]. In a wide spectrum of cancers, including NB, GD2 is believed to escalate progression by increasing tumor cell proliferation, motility, migration, and invasion [37,83]. In NBs, the expression patterns of GD2 can vary and higher tumor derived GD2 levels have been linked to accelerated progression and lower survival rates in patients [84].
Nevertheless, 98–100% of NB tumors have been found to express GD2 at some level [37,50,85]. Since GD2 is highly sensitive to NB tissue and is only expressed in a small amount of normal tissue, it is an ideal target antigen for immunotherapy [37,50,85]. The biosynthesis of GD2 biosynthesis includes sialylation by GM3 synthase (ST3GAL5) and GD3 synthase (ST8SIA1), which is why GD2 expression can be modulated by sialic acid availability and histone deacetylase activity [86]. GD2 acts as a signaling modulator within lipid rafts in the plasma membrane of NB cells [37,80]. In the lipid rafts, it interacts with tyrosine kinases and signal transducers to enhance phosphorylation and induces SRC-dependent activation of signaling cascades that go on to promote tumor progression and maintenance [85,87–89]. This pathway is mechanistically different from growth-factor-induced signaling [37,83]. GD2 additionally contributes to immune evasion by functioning as an immune checkpoint that induces T-cell dysfunction [80].
Histologic Classification
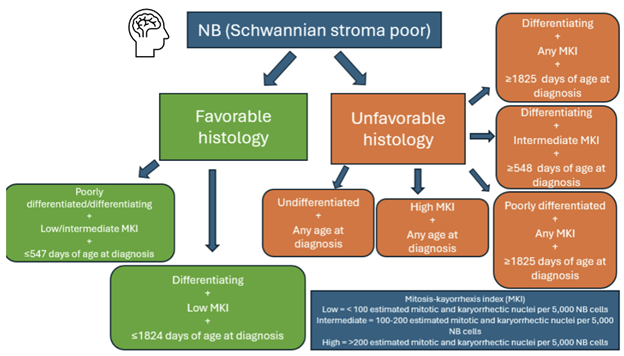
Under the microscope, neuroblastomas present as uniform, small round blue tumor cells [90]. Another popular diagnostic feature includes “Homer-Wright rosettes”. This name represents the neural fibrils and bundles of cells seen in neuroblastomas described by James Wright in his landmark paper in 1910 [91]. In the present day, neuroblastomas have a range of histopathologic diversity that can impact the prognosis and course of treatment of the disease. The International Neuroblastoma Pathology Classification (INPC) system is recommended by National Comprehensive Cancer Network® (NCCN®) guidelines to predict outcomes and guide treatment intensity. NCCN advises that histologic classification based on INPC should be made before initiation of therapy [92]. The classification can be seen broken down in Figure 2. Histologically, the diagnosis of neuroblastoma is completed by distinguishing it from peripheral neuroblastic tumors including ganglioneuroblastoma (intermixed), ganglioneuroblastoma (nodular), and ganglioneuroma [93]. NBs are identified as Schwannian stroma poor, then further classified as favorable or unfavorable depending on the grade of neuroblastic differentiation, mitosis-kayorrhexis index (MKI), and age at diagnosis [94]. Neuroblastic differentiation of NB cells includes undifferentiated, poorly differentiated, or differentiating while the MKI is the estimated mitotic and karyorrhectic nuclei per 5,000 neuroblastoma cells. Below 100 is considered low, intermediate between 100–200, and over 200 is high [95]. NB tumors fall under the Favorable Histology category if they fall into one of two groups. The first group includes poorly differentiated or differentiating subtypes with low or intermediate MKI and less than or equal to 547 days of age at diagnosis [95]. The first group includes poorly differentiated or differentiating subtypes with low or intermediate MKI and less than or equal to 547 days of age at diagnosis [94]. The second group includes tumors with the differentiating subtype with low MKI and less than or equal to 1,824 days of age at diagnosis [95]. The Unfavorable Histology tumors can fall within 5 groups; 1) undifferentiated subtype at any age, 2) high MKI at any age, 3) poorly differentiated subtype with any MKI with more than or equal to 548 days of age at time of diagnosis, 4) differentiating subtype with intermediate MKI with mora than or equal to 548 days of age at diagnosis, and 5) differentiating subtype with any MKI with more than or equal to 1,825 days of age at diagnosis [94]. All of these factors must be considered and determined of NB tumor before treatment can begin, thus contributing to the complexity of NB management and course of treatment.
Current and Ongoing Therapies
The extensive crosstalk and redundancy in signaling networks, as discussed above, explains why single-agent therapies often fail in NB [96]. The current consensus treatment regimen for high-risk neuroblastoma can be found in Table 2. It consists of intensive multimodal therapy divided into three sequential phases, achieving an estimated 5-year event-free survival rate of 51% [15]. Induction therapy involves 5–6 cycles of multiagent chemotherapy (preferably ANBL12P1 [97] or ANBL1531 [98] regimens per the Children’s Oncology Group), followed by surgical resection of the primary tumor with a goal of gross total resection, yielding approximately 80% partial response or better [15,97]. Consolidation therapy includes tandem autologous stem cell transplantation with thiotepa/cyclophosphamide followed by dose-reduced carboplatin/etoposide/melphalan (demonstrating superior 3-year event-free survival of 61.6% versus 48.4% for single transplant), along with consolidative radiotherapy at 21.6 Gy to the primary site and residual metastatic disease [99–101]. Post-consolidation therapy consists of immunotherapy, often an anti-GD2 monoclonal antibody (dinutuximab) combined with sargramostim, and a retinol, generally isotretinoin, for six 28-day cycles, which significantly improved 2-year event-free survival to 66% compared to 46% with isotretinoin alone [19,102], with interleukin-2 no longer recommended based on recent data showing no benefit and increased toxicity [20,49,103,104]. For eligible patients achieving partial response or better following immunotherapy, continuation therapy with eflornithine (also known as DFMO) for up to 2 years is an additional treatment option (category 2B recommendation) that has recently become FDA approved [105] and is supported by non-randomized data demonstrating improved event-free survival (hazard ratio 0.48) and overall survival (hazard ratio 0.32) compared to historical controls [106–108].
|
Phase |
Treatment / Modalities |
Typical Agents |
|
Induction |
Intensive multi-agent chemotherapy (5-6 rounds)
|
Platinum agents (cisplatin, carboplatin), alkylators (cyclophosphamide, ifosfamide), topoisomerase inhibitors (etoposide, topotecan), anthracyclines (doxorubicin) Or locoregional disease |
|
Consolidation |
High-dose myeloablative chemotherapy + autologous stem cell transplant (ASCT/HSCT)
|
Bu-Mel (busulfan + melphalan) or CEM (carboplatin, etoposide, melphalan) + stem-cell rescue post-myeloablative chemo
|
|
Maintenance / Post-consolidation |
Anti-GD2 immunotherapy
|
Dinutuximab, dinutuximab beta, naxitamab ± sargramostim (GM-CSF) Isotretinoin / 13-cis-retinoic acid |
The majority of novel therapeutics seek to either improve outcomes, decrease toxicity, or address treatment resistance [109]. Based on current literature, targeted therapies can be split into two categories based on their mechanism of action. One category includes agents centered around tumor destruction through the enhancement of the immune system. These agents recruit and activate the immune system to destroy tumor cells. This category would include anti-GD2 antibodies, immune checkpoint inhibitors, CAR-T, cytokine therapies, and other cellular immunotherapies [15,24]. The other category includes agents centered around inhibition of oncogenic signaling activity. These agents include ALK and mTOR pathway inhibitors, multi-kinase inhibitors, MYCN-targeted therapies, cell cycle inhibitors, epigenetic modulators, and metabolic modulators, among others [15,31,40]. Chemoimmunotherapy regimens currently utilize the differing mechanisms to target HR-NB from both sides, and the different approaches are part of the consideration of ongoing approaches towards combination therapies [26]. As previously discussed, recent advances include anti-GD2 immunotherapy, targeted therapy for ALK-driven tumors, and GD2-targeted CAR-T, among other immunotherapies [25]. These and other novel therapies will now be discussed in further detail.
Immunotherapy with anti-GD2 monoclonal antibodies
Currently, anti-GD2 monoclonal antibodies (dinutuximab, dinutuximab beta) are the standard of care for HR-NB maintenance and relapse treatment [104,110,111]. Anti-GD2 antibodies function by decreasing phosphorylation to downregulate multiple signaling pathways including the PI3K/AKT/mTOR pathway [88,112]. Dinutuximab and dinutuximab-beta are the main anti-GD2 antibodies utilized clinically at this time, but naxitamab has also become more popular and has gained FDA approval in 2023 [113] for relapsed or refractory high-risk neuroblastoma in bone or bone marrow, when combined with GM-CSF [114,115]. Clinical trials based around naxitamab have increased, and show higher response rates compared to the general treatment regime for HR-NB. A phase 2 trial demonstrated a 50% overall response rate in heavily pretreated patients with residual disease, including 38% complete responses and 58% bone compartment responses, with manageable toxicities mainly consisting of grade 3 hypotension and pain [114,116]. Naxitamab-based chemoimmunotherapy regimens have achieved response rates of 64–85% in patients with refractory or progressive neuroblastoma [117]. In another phase I/II trial with a small cohort, complete response was noted in 75%, with five-year progression-free and overall survival rates being 38% and 64% respectively [118].
The anti-GD2 antibodies’ antitumor mechanism of action works mainly through the activation of the antibody-dependent cell-mediated cytotoxicity (ADCC) pathway. This is often enhanced using granulocyte-macrophage colony-stimulating factor (GM-CSF) and is a part of the current consensus treatment [119]. GM-CSF, known as the drug sargramostim, enhances the sensitivity of the response by activating and expanding myeloid effector populations and is a common adjuvant in HR-NB treatment to anti-GD2 antibodies [120,121]. However, due to the downsides of GM-CSF, such as its high levels of toxicity, cost, and its ability to blunt some anti-tumor immunity, researchers have consistently searched for alternative cytokines to utilize [122]. While G-CSF had previously been offered as a suitable alternative to GM-CSF [123], clinical data has sufficiently found that there is a clinically significant difference between using the two cytokines, and a worse outcome when no cytokine is used at all [120].
There have additionally been studies centered around immunocytokines and engineered cytokine fusions linking anti-GD2 antibodies to IL-15 or IL-21, among others, that demonstrate superior preclinical activity compared to the IL-2-based constructs previously mentioned, with complete tumor regressions in immunocompetent mouse models using enhanced CD8+ T cell responses and favorable modulation of the tumor microenvironment [124–130]. One study studied trifunctional antibody-cytokine fusion protein formats by combining two interleukins, either IL-15 and IL-7 or IL-17 and IL-21, and saw improved potency in inducing JAK-STAT pathway activation, providing a promising candidate for future drug development [130]. Another ongoing area of investigation focuses on other fusion antibodies, such as the combination of anti-GD2 antibodies with SIRPα domains to block CD47-mediated signal to overcome innate immune checkpoint resistance, demonstrating enhanced phagocytosis and NK cell-mediated killing in preclinical models [24,131–133].
CAR-T therapies
Recent phase 1/2 clinical trials of GD2-targeted chimeric antigen receptor T cells (GD2-CART01) have shown encouraging results in pediatric patients with refractory and high-risk metastatic NBs. GD2-direced CAR T cells are patient derived T-lymphocytes that are chemically engineered to express a CAR recognizing the neuroblastoma-associated antigen GD2, enabling for the targeted destruction of GD2-positive tumor cells [21]. As GD2 is abundantly expression on NB with limited expression on normal tissues, it serves as an ideal immunotherapeutic target [21,35]. Initial findings from the trial found an overall response rate of 63% among the 27 patients studied, including 9 complete remissions, as well as detectable CAR T-cell among patients for up to 30 months. From the patients who received the recommended doses, the 5-year OS and event free survival were found to be 68% and 53%, respectively [22,23].
Based on the high response rates and sustained CAR T-cell persistence observed in the initial cohort, investigators broadened the study with 19 additional participants and continued follow-up, enabling a more extensive long-term evaluation. GD2-CART01 continued significant clinical activity, with an overall response of ~66% and complete remission in up to 40% of patients [22]. Notably, GD2-CART01 persistence was detectable in ~64% of patients for at least 12 months, suggesting sustained immunological surveillance. The study concluded that in patients with refractory or relapsing NB after conventional treatments, immunotherapy targeting GD2-CART01 can induce clinically significant and durable remissions while maintaining a favorable safety profile [21,22].
Current CAR-T related preclinical focus is on armored CAR T-cells with targets outside of GD2, bicistronic CARs, and synthetic extracellular vesicles (SyntEVs), GD2IL18CART, which has added IL18 to GD2 CAR T-cells, has demonstrated higher levels of IFN-γ and TNF-α release in comparison to GD2CART01, and it is now being prepared for clinical investigation [134]. CXCR2-armored GPC2 CAR-T is also under investigation. The CXCR2 cytokine armor has been found to simultaneously enhance trafficking and reduce myeloid-derived suppressor cells (MDSCs), providing the T cells with improved infiltration and targeting of the tumors [109]. GPC2-targeted CAR T cells with NFAT-inducible IL-15/IL-21 have also been found to prolong survival without inducing hypercytokinemia-related mortality in mouse models [135,136].
Other immunotherapies
A portion of HR-NB’s therapeutic resistance has recently been attributed to tumor-derived extracellular vesicles that remodel the tumor microenvironment by simultaneously inhibiting natural killer cell (NK) maturation and suppressing macrophages [137]. This diminishes ADCC and decreases the efficacy of medications focused on tumor destruction and immune system upregulation. In November 2025, nontumor-derived GPC2+ SyntEVs were developed as enhancers for CAR-T with GD2 or albumin-binding domains [138]. Serial infusions of these after GPC2 CAR-T boosted peripheral CAR T-cell persistence in patient-derived xenografts, and the GD2-targeting SyntEVs showed decreased levels of antigen downregulation [138]. Bicistronic CARs that target both GPC2 and B7-H3 have also been under development and aim to address GD2 resistant therapies overcome [139]. Currently, these CARs have shown prolonged resistance to exhaustion compared to single-antigen CARs and hold further implications for combination therapies [139].
Other possible therapies that aim to address GD2-resistant HR-NB by modulating GD2 expression, synthesis, and signaling pathways are being investigated. EZH2 inhibitors, which are epigenetic modulators, are about to undergo clinical testing in combination with anti -GD2 antibodies [140]. NB cells become resistant to anti-GD2 therapy by reducing expression of GD3 synthase (ST8SIA1) and downregulating GD2 expression by transitioning from adrenergic to mesenchymal NB cells [140]. Inhibiting EZH2 rewires these mesenchymal NB cells and restores GD3 synthase and GD2 surface expression [141]. This resensitizes cells to anti-GD2 antibody therapy [140]. Another study has found that combining histone deacetylase (HDAC) inhibitors (i.e. vorinostat) with sialic acid supplementation upregulates GD2 and could be utilized to boost anti-GD2 immunotherapy in NB tumors with less GD2 expression [86]. The shift towards prioritization of GPC2 and B7-H3 also highlights alternative treatment to GD2-centered treatments and has with both targets demonstrating great preclinical validation. The ongoing clinical development of these platforms will be highly important in determining the potential of these alternative targets in comparison to GD2-centered treatments [25,26,47,48,136].
Pathway-directed therapies for HR-NB
ALK inhibitors have become a good alternative for the subset of HR-NB patients with ALK positive disease. Combined with the ANBL0532 trial, discussed in the genetics section previously, an argument is building for the early integration of ALK inhibitors in patients with ALK mutation, although safety and efficacy data for ALK inhibitors is still not sufficient enough for adoption outside of its current utilization in clinical trials during induction therapy [15,26,141]. The current ALK inhibitors include crizotinib, ceretinib, ensartinib, alectinib, and loratinib. Lorlatinib is a relatively newer drug as a third-generation ALK/ROS1 inhibitor currently in clinical trials. It has been designed for high penetration of the blood brain barrier and is effective against resistance mutations compared to previous generations [142].
Lorlatinib’s mechanism of action works mainly by downregulating the G2/M cell cycle kinases and repressing MYCN expression [142]. Phase 1 testing of lorlatinib with relapsed/refractory ALK-driven NB was completed in the NANT2015-02 trial and established recommended phase 2 dosing of 115 mg/m² in children and 150 mg in adults, with the majority of its toxicity profiles being metabolic effects (hypertriglyceridemia 90%, hypercholesterolemia 79%, weight gain 87%) [143]. Notably, single-agent response rates reached 30% in pediatric patients and 67% in adults, with 48% of responders achieving complete MIBG responses [143]. Further research found that when combined with topotecan/cyclophosphamide, the response rate for lorlatinib increased to 63% in children, supporting synergistic activity [144]. The drug is now in phase 3 testing by the Children's Oncology Group for integration into frontline therapy.
Despite initial sensitivity to ALK inhibition, ALK-mutant tumors can develop resistance through kinase mutations and oncogenic cooperation with MYCN. This resistance mechanism enables cooperative signaling between ALK and MYCN, further amplifying resistance to maintain oncogenic mechanisms despite ALK inhibition [61]. These adaptive mechanisms sustain proliferative and survival pathways despite ALK inhibition, highlighting that ALK-targeted therapy fails as a single therapy measure and can only benefit a subset of patients.
Combinations with ALK inhibitors are currently considered a possible alternative for tumors with resistance against ALK inhibitors due to their synergistic effects [145]. The NANT consortium phase 1/2 trial found that combining lorlatinib with topotecan and cyclophosphamide, two chemotherapy drugs, achieved a 50% response rate in pretreated patients [145]. Studies combining lorlatinib with the MDM2 inhibitor idasanutlin have demonstrated improved effects, inducing complete tumor regression and significantly delayed regrowth in ALK-amplified models [145]. Other newer ALK inhibitors, such as ESK 440, are also under development as possible alternatives for HR-NBs resistant to treatments currently available. ESK440 is a dual ALK/FAK inhibitor that has shown either equal or enhanced efficacy compared to loratinib alone and retained activity against loratinib-resistant cell lines in preclinical models [29]. A dual ALK/ATR inhibition combination has also developed, utilizing the high levels of replication stress induced by ALK signaling and removing the defense ATR signaling would normally provide to reduce tumors [146,147]. The preclinical trial demonstrated that 14-day ALK/ATR inhibition achieved complete tumor regression and promoted differentiation to neuronal/Schwann cell lineages [146].
Another combination addressing the increasing resistance in HR-NB therapy is based on disrupting the PI3K/Akt/mTOR pathway, a common focus for NB pathogenesis [12,148]. The clinical trial ITCC-053 focuses on the combination of crizotinib with temsirolimus, an mTOR inhibitor, for ALK/MET-aberrated relapsed/refractory NBs. It has currently completed phase 1b and has established a recommended phase 2 dose as crizotinib 150 mg/m² twice daily with temsirolimus 40 mg/m²/week. However, the grade 3 toxicities, as well as the need for dose reductions due to CYP3A4 interactions, have raised concerns for the tolerability and utility of the combination [149–151]. Preclinical data investigating mTOR inhibition in combination with chemotherapy in NB and in other pediatric tumors implies that mTOR inhibition may overcome relative resistance to ALK inhibitors, but trials are ongoing, and clinical validation is still required for HR-NB at this time [149,151–153].
Treatments for MYCN-amplified relapsed/refractory neuroblastoma
Given the central role that MYCN amplification has in driving aggressive tumor behavior, targeted immunotherapies have increasingly advanced to improve outcomes in relapsed or refractory NB. MYCN-amplified tumors are increasingly dependent on the mTOR signaling complex for cell proliferation, a serine-threonine protein kinase composed of mTORC1 and mTORC2 subunits. mTORC1 has been largely studied for its ability to promote anabolic processes through the signaling conduction of nutrient availability and cellular energy status, while mTORC2 focuses on the insulin/IGF-1 pathway to increase cell proliferation and regulate survival [154,155]. Rapamycin, under the generic name of sirolimus, is an mTOR inhibitor that has shown extensive inhibition in the proliferation of NB cells through the initiation of G1 cell-cycle arrest [156].
NB cells are particularly subjected to the pro-oncogenic effects of autophagy, a mechanism that can allow NBs to sustain survival under metabolic stress, including the recycling of damaged organelles to support energy homeostasis to avoid cell death despite a nutrient or oxygen-limited environment [157]. mTOR is a negative regulator of autophagy, where activated mTOR promotes anabolic metabolism and inhibits autophagy initiation [158]. Rapamycin mediated inhibition of mTORC1 functions to reduce anabolic signaling and shift to catabolic survival pathways, ultimately reducing proliferative capacity. When metabolic stress becomes excessive, autophagy can no longer sustain survival and rendering NB cells become largely vulnerable to growth inhibition by the mTORC1 blockade. While prolonged rapamycin treatment has shown to induce insulin resistance, impair glucose homeostasis, and even block T-cell activation, such effects reflect the spectrum of mTOR’s influence in cellular physiology and highlights why this pathway remains a compelling candidate for approaching MYCN-amplified NB [159].
Further advancements in treating MYCN amplified tumors have been made with the combination of dasatinib plus rapamycin alongside traditional therapies of irinotecan plus temozolomide (RIST). The clinical trial evaluated 124 patients to randomly receive either RIST or standalone therapy of irinotecan plus temozolomide. Specifically, among MYCN amplified-tumors, the median progression-free survival was 6 months in the RIST group while only 2 months in the control group. The trial also demonstrated acceptable and comparable toxicity profiles between the two groups, primarily consisting of hematological adverse events such as thrombocytopenia and anemia [96].
Conclusion: Challenges and Further Investigation
Over the past two decades, HR-NB immunotherapy has seen substantial developments, improving life expectancy from 10–20% in the 1990s to over 50% currently. However, HR-NB still lacks long-standing curative treatment. Current research is centered around addressing the ongoing therapeutic challenges investigating high variability in NB and HR-NB expression and the complex interconnected relationships of the factors and pathways in these tumors [109]. Ongoing challenges in current HR-NB therapy include dose-limiting toxicities such as neuropathic pain, the varying levels of biomarker expression and immunosuppression within NBs that cause resistance to its treatment, and the emergence of late relapses and central nervous system metastases [27,117,160]. Collectively, these resistance mechanisms highlight the limitations of the standard therapy for NBs and emphasize the need to overcome conventional treatment modalities. Multi-targeting approaches are currently considered the most promising in addressing the resistance mechanisms that NB has developed and are highlighted as high priority for investigation in the Third Neuroblastoma Drug Development Strategy Forum [26]. Studies such as the one on CXCR2-armored GPC2 CAR T-cells have shown the importance of studying both therapeutic efficacy as well as the resistance mechanisms of NB [127]. Epigenetic reprogramming of tumor microenvironment through chromatin-modifying agents such as EZH2 inhibition, SyntEVs, and other approaches to enhance or alter anti-GD2 antibody efficacy, have shown potential for clinical translation as well [25]. Moving forward, improvements in HR-NB outcomes depend on further addressing the key gaps in the current knowledge and clinical approaches. The major areas that require investigation and prioritization can be sorted as either further understanding of the disease itself or further development of novel treatments. Due to the heterogeneity of NB, precision-based therapies utilizing the biomarkers discussed (GD2, GPC2, B7-H3, MYCN, ALK, etc.) is a major focus for HR-NB treatment. It would simultaneously reduce major toxicities and allow for a more focused dismantling of HR-NBs. To have more precise therapies, an understanding of the whole picture of NB is required. The molecular and genetic complexity of HR-NB, with the numerous signaling pathways, oncogenic cooperation, chemokines such as CXCR2, the feedback loops and axis, and the synergistic relationships between all these factors, among others, are all areas that still need further investigation at this time. Future progress will depend on improving overall survival and highlights the necessity of continued international collaboration to also address progress that has alternatively been slowed due to cost and access to resources. From the approval of dinutuximab to the identification of novel targets and checkpoints, the consistent progress of the past decades provides the confidence that continued investigation of HR-NB, and its therapeutic interventions will lead towards a world where the treatment of HR-NB will be curative and long-standing, instead of being palliative and short-lived.
Author Disclosure Statement
The authors have no disclosures or conflicts.
References
2. Kameneva P, Artemov AV, Kastriti ME, Faure L, Olsen TK, Otte J, et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat Genet. 2021 May;53(5):694–706.
3. Ponzoni M, Bachetti T, Corrias MV, Brignole C, Pastorino F, Calarco E, et al. Recent advances in the developmental origin of neuroblastoma: an overview. J Exp Clin Cancer Res CR. 2022 Mar 11;41(1):92.
4. Karalexi MA, Servitzoglou M, Papadakis V, Kachanov D,Česen Mazič M, Baka M, et al. Survival patterns of childhood neuroblastoma: an analysis of clinical data from Southern-Eastern European countries. Eur J Cancer Prev Off J Eur Cancer Prev Organ ECP. 2023 May 1;32(3):254–63.
5. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007 Jun 23;369(9579):2106–20.
6. Parham DM. Neuroectodermal and neuroendocrine tumors principally seen in children. Am J Clin Pathol. 2001 Jun;115 Suppl:S113-28.
7. Gonzalez Malagon SG, Liu KJ. Linking neural crest development to neuroblastoma pathology. Development. 2022 Aug 1;149(15):dev200331.
8. He GQ, He SJ, Jing XY, Dai YL, Guo X, Gao J, et al. Dissecting neuroblastoma heterogeneity through single-cell multi-omics: insights into development, immunity, and therapeutic resistance. Oncogene. 2026 Jan;45(2):123–39.
9. Ni Cheallaigh L, Liu JF, Ball-Gamble A, Walker D, Ritzmann TA, Shanmugavadivel D. Spotting childhood abdominal tumours: a systematic review and meta-analysis of the clinical presentation. Arch Dis Child. 2026 Jan 19;111(2):158–66.
10. Irwin MS, Naranjo A, Zhang FF, Cohn SL, London WB, Gastier-Foster JM, et al. Revised Neuroblastoma Risk Classification System: A Report From the Children’s Oncology Group. J Clin Oncol Off J Am Soc Clin Oncol. 2021 Oct 10;39(29):3229–41.
11. Cohn SL, Pearson ADJ, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol Off J Am Soc Clin Oncol. 2009 Jan 10;27(2):289–97.
12. Sharma R, Yadav J, Bhat SA, Musayev A, Myrzagulova S, Sharma D, et al. Emerging Trends in Neuroblastoma Diagnosis, Therapeutics, and Research. Mol Neurobiol. 2025 May;62(5):6423–66.
13. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin. 2025;75(1):10–45.
14. Maris JM. Recent Advances in Neuroblastoma. N Engl J Med. 2010 Jun 10;362(23):2202–11.
15. Bagatell R, Park JR, Acharya S, Aldrink J, Allison J, Alva E, et al. Neuroblastoma, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024 Aug 1;22(6):413–33.
16. Olsen HE, Campbell K, Bagatell R, DuBois SG. Trends in conditional survival and predictors of late death in neuroblastoma. Pediatr Blood Cancer. 2020 Oct;67(10):e28329.
17. Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans MEC, et al. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci. 2012 Nov 20;109(47):19190–5.
18. Thakur A, Dijkstra A. Hallmarks of Neuroblastoma—Pathophysiology, Diagnosis, and Therapeutic Interventions. Cancer Nexus. 2025;1(2):e70007.
19. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 Antibody with GM-CSF, Interleukin-2, and Isotretinoin for Neuroblastoma. N Engl J Med. 2010 Sep 30;363(14):1324–34.
20. Yu AL, Gilman AL, Ozkaynak MF, Naranjo A, Diccianni MB, Gan J, et al. Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin Cancer Res Off J Am Assoc Cancer Res. 2021 Apr 15;27(8):2179–89.
21. Bufalo FD, Angelis BD, Caruana I, Baldo GD, Ioris MAD, Serra A, et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N Engl J Med. 2023 Apr 5;388(14):1284–95.
22. Locatelli F, Pagliara D, De Ioris MA, Becilli M, Del Baldo G, Serra A, et al. GD2-targeting CAR T cells in high-risk neuroblastoma: a phase 1/2 trial. Nat Med. 2025 Nov;31(11):3689–99.
23. Li CH, Sharma S, Heczey AA, Woods ML, Steffin DHM, Louis CU, et al. Long-term outcomes of GD2-directed CAR-T cell therapy in patients with neuroblastoma. Nat Med. 2025 Apr;31(4):1125–9.
24. Anderson J, Majzner RG, Sondel PM. Immunotherapy of Neuroblastoma: Facts and Hopes. Clin Cancer Res. 2022 Aug 2;28(15):3196–206.
25. Shokouhfar M, Darzi A, Ameli F, Nami MT, Khorasani SK, Eini P, et al. Immunotherapeutic advances in pediatric neuroblastoma: Overcoming resistance through biomarker-guided combinations. Biomed Pharmacother. 2026 Jan 31;196:119020.
26. DuBois SG, Moreno L, Anderson J, Asgharzadeh S, Bagatell R, Beck-Popovic M, et al. Accelerating Drug Development for Neuroblastoma: Consensus Statement From the Third Neuroblastoma Drug Development Strategy Forum. Pediatr Blood Cancer. 2025 Sep;72(9):e31831.
27. DuBois SG, Moreno L, Bagatell R, Cheung NK, Gray JC, Locatelli F, et al. Paediatric Strategy Forum for medicinal product development of agents targeting GD2 ganglioside in children and adolescents with cancer. Eur J Cancer Oxf Engl 1990. 2025 Dec 9;231:116093.
28. Kennedy PT, Zannoupa D, Son MH, Dahal LN, Woolley JF. Neuroblastoma: an ongoing cold front for cancer immunotherapy. J Immunother Cancer. 2023 Nov 22;11(11):e007798.
29. Chugh S, Tien JC, Hon J, Kenum C, Mannan R, Cheng Y, et al. Therapeutic benefit of the dual ALK/FAK inhibitor ESK440 in ALK-driven neuroblastoma. Neoplasia N Y N. 2025 Feb;60:100964.
30. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013 Mar;45(3):279–84.
31. Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, et al. Neuroblastoma Arginase Activity Creates an Immunosuppressive Microenvironment That Impairs Autologous and Engineered Immunity. Cancer Res. 2015 Aug 1;75(15):3043–53.
32. Mastrangelo S, Rivetti S, Triarico S, Romano A, Attinà G, Maurizi P, et al. Mechanisms, Characteristics, and Treatment of Neuropathic Pain and Peripheral Neuropathy Associated with Dinutuximab in Neuroblastoma Patients. Int J Mol Sci. 2021 Nov 23;22(23):12648.
33. Tomolonis JA, Agarwal S, Shohet JM. Neuroblastoma pathogenesis: deregulation of embryonic neural crest development. Cell Tissue Res. 2018 May;372(2):245–62.
34. Wulf AM, Moreno MM, Paka C, Rampasekova A, Liu KJ. Defining Pathological Activities of ALK in Neuroblastoma, a Neural Crest-Derived Cancer. Int J Mol Sci. 2021 Oct 29;22(21):11718.
35. Lee JW, Son MH, Cho HW, Ma YE, Yoo KH, Sung KW, et al. Clinical significance of MYCN amplification in patients with high-risk neuroblastoma. Pediatr Blood Cancer. 2018 Oct;65(10):e27257.
36. Mossë YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as the Major Familial Neuroblastoma Predisposition Gene. Nature. 2008 Oct 16;455(7215):930–5.
37. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front Oncol. 2020 Jul 7;10:1000.
38. Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, et al. Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell. 2017 Sep 11;32(3):295–309.e12.
39. Maparu K, Chatterjee D, Kaur R, Kalia N, Kuwar OK, Attri M, et al. Molecular crosstalk between GPCR and receptor tyrosine-protein kinase in neuroblastoma: molecular mechanism and therapeutic implications. Med Oncol. 2025 Mar 23;42(5):131.
40. Li N, Spetz MR, Li D, Ho M. Advances in immunotherapeutic targets for childhood cancers: A focus on glypican-2 and B7-H3. Pharmacol Ther. 2021 Jul;223:107892.
41. Danilenko M, Nath S, Baines J, Gordon F, Merugu S, Allinson LM, et al. Detection of Targetable Genetic Abnormalities in Neuroblastoma Circulating Tumour DNA. Int J Mol Sci. 2025 Sep 27;26(19):9466.
42. Irwin MS, Goldsmith KC. Current and Emerging Biomarkers: Impact on Risk Stratification for Neuroblastoma. J Natl Compr Cancer Netw JNCCN. 2024 Aug;22(6):e247051.
43. Campbell K, Gastier-Foster JM, Mann M, Naranjo AH, Van Ryn C, Bagatell R, et al. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Cancer. 2017 Nov 1;123(21):4224–35.
44. Morgenstern DA, Kao PC, Moreno L, Staunton J, Bagatell R, Berlanga P, et al. Factors associated with survival following relapse of high-risk neuroblastoma: A study from the International Neuroblastoma Risk Group (INRG) Data Commons. J Clin Oncol. 2025 Jun;43(16_suppl):10052.
45. Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci U S A. 2017 Aug 8;114(32):E6623–31.
46. Pulido R, Nunes-Xavier CE. Hopes on immunotherapy targeting B7-H3 in neuroblastoma. Transl Oncol. 2023 Jan;27:101580.
47. Dondero A, Morini M, Cangelosi D, Mazzocco K, Serra M, Spaggiari GM, et al. Multiparametric flow cytometry highlights B7-H3 as a novel diagnostic/therapeutic target in GD2neg/low neuroblastoma variants. J Immunother Cancer. 2021 Apr;9(4):e002293.
48. Rasic P, Jeremic M, Jeremic R, Dusanovic Pjevic M, Rasic M, Djuricic SM, et al. Targeting B7-H3-A Novel Strategy for the Design of Anticancer Agents for Extracranial Pediatric Solid Tumors Treatment. Molecules. 2023 Apr 11;28(8):3356.
49. Ladenstein R, Pötschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018 Dec;19(12):1617–29.
50. Terzic T, Cordeau M, Herblot S, Teira P, Cournoyer S, Beaunoyer M, et al. Expression of Disialoganglioside (GD2) in Neuroblastic Tumors: A Prognostic Value for Patients Treated With Anti-GD2 Immunotherapy. Pediatr Dev Pathol. 2018;21(4):355–62.
51. Berko ER, Naranjo A, Daniels AA, McNulty SN, Krytska K, Druley T, et al. Frequency and Clinical Significance of Clonal and Subclonal Driver Mutations in High-Risk Neuroblastoma at Diagnosis: A Children’s Oncology Group Study. J Clin Oncol. 2025 May 10;43(14):1673–84.
52. Bellini A, Pötschger U, Bernard V, Lapouble E, Baulande S, Ambros PF, et al. Frequency and Prognostic Impact of ALK Amplifications and Mutations in the European Neuroblastoma Study Group (SIOPEN) High-Risk Neuroblastoma Trial (HR-NBL1). J Clin Oncol. 2021 Oct 20;39(30):3377–90.
53. Rosswog C, Fassunke J, Ernst A, Schömig-Markiefka B, Merkelbach-Bruse S, Bartenhagen C, et al. Genomic ALK alterations in primary and relapsed neuroblastoma. Br J Cancer. 2023 Apr;128(8):1559–71.
54. Lopez-Delisle L, Pierre-Eugène C, Louis-Brennetot C, Surdez D, Raynal V, Baulande S, et al. Activated ALK signals through the ERK-ETV5-RET pathway to drive neuroblastoma oncogenesis. Oncogene. 2018 Mar;37(11):1417–29.
55. Sattu K, Hochgräfe F, Wu J, Umapathy G, Schönherr C, Ruuth K, et al. Phosphoproteomic analysis of anaplastic lymphoma kinase (ALK) downstream signaling pathways identifies signal transducer and activator of transcription 3 as a functional target of activated ALK in neuroblastoma cells. FEBS J. 2013 Nov;280(21):5269–82.
56. Lee C, Jia Y, Li N, Sun X, Ng K, Ambing E, et al. Crystal structure of the ALK (anaplastic lymphoma kinase) catalytic domain. Biochem J. 2010 Sep 15;430:425–37.
57. Aboualizadeh F, Yao Z, Guan J, Drecun L, Pathmanathan S, Snider J, et al. Mapping the Phospho-dependent ALK Interactome to Identify Novel Components in ALK Signaling. J Mol Biol. 2021 Nov 19;433(23):167283.
58. Huang M, Fang W, Farrel A, Li L, Chronopoulos A, Nasholm N, et al. ALK upregulates POSTN and WNT signaling to drive neuroblastoma. Cell Rep. 2024 Mar 26;43(3):113927.
59. Liu H, Tekle C, Chen YW, Kristian A, Zhao Y, Zhou M, et al. B7-H3 silencing increases paclitaxel sensitivity by abrogating Jak2/Stat3 phosphorylation. Mol Cancer Ther. 2011 Jun;10(6):960–71.
60. Kramer M, Ribeiro D, Arsenian-Henriksson M, Deller T, Rohrer H. Proliferation and Survival of Embryonic Sympathetic Neuroblasts by MYCN and Activated ALK Signaling. J Neurosci Off J Soc Neurosci. 2016 Oct 5;36(40):10425–39.
61. Tucker ER, Poon E, Chesler L. Targeting MYCN and ALK in resistant and relapsing neuroblastoma. Cancer Drug Resist. 2019 Sep 19;2(3):803–12.
62. Shi T, Mukae K, Shindo M, Onuki R, Nakazawa A, Takenobu H, et al. Coexpression of MYCN and ALK Induces Neuroblastoma-Like Tumors From Human iPS Cell-Derived Cranial Neural Crest Cells. Genes Cells. 2026 Jan;31(1):e70083.
63. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012 Jul 10;22(1):117–30.
64. Bartolucci D, Montemurro L, Raieli S, Lampis S, Pession A, Hrelia P, et al. MYCN Impact on High-Risk Neuroblastoma: From Diagnosis and Prognosis to Targeted Treatment. Cancers. 2022 Sep 12;14(18):4421.
65. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013 Oct;3(10):a014415.
66. Bhardwaj N, Das G, Srinivasan R. Neuroblastoma-derived v-myc avian myelocytomatosis viral related oncogene or MYCN gene. J Clin Pathol. 2023 Aug;76(8):518–23.
67. Chen Y, Yang H, Xiao L, Yan N, Zhang M. Molecular regulation and therapeutic targeting of MYCN in neuroblastoma: a comprehensive review. Front Cell Dev Biol. 2025;13:1683331.
68. Hu X, Zhou Y, Hill C, Chen K, Cheng C, Liu X, et al. Identification of MYCN non-amplified neuroblastoma subgroups points towards molecular signatures for precision prognosis and therapy stratification. Br J Cancer. 2024 May;130(11):1841–54.
69. Alptekin A, Ye B, Yu Y, Poole CJ, van Riggelen J, Zha Y, et al. Glycine decarboxylase is a transcriptional target of MYCN required for neuroblastoma cell proliferation and tumorigenicity. Oncogene. 2019 Dec;38(50):7504–20.
70. Liu Y, Liu D, Wan W. MYCN-induced E2F5 promotes neuroblastoma cell proliferation through regulating cell cycle progression. Biochem Biophys Res Commun. 2019 Mar 26;511(1):35–40.
71. Hsu TYT, Simon LM, Neill N, Marcotte R, Sayad A, Bland CS, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015 Sep 17;525(7569):384–8.
72. Goto S, Umehara S, Gerbing RB, Stram DO, Brodeur GM, Seeger RC, et al. Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the Children’s Cancer Group. Cancer. 2001 Nov 15;92(10):2699–708.
73. Schramm A, Köster J, Marschall T, Martin M, Shimada H, Stram DO, et al. Identification of subsets of neuroblastomas by combined histopathologic and N-myc analysis. J Natl Cancer Inst. 1995 Oct 4;87(19):1470–6.
74. Kling MJ, Griggs CN, McIntyre EM, Alexander G, Ray S, Challagundla KB, et al. Synergistic efficacy of inhibiting MYCN and mTOR signaling against neuroblastoma. BMC Cancer. 2021 Sep 26;21(1):1061.
75. Schwermer M, Fielitz K, et al. Next-generation RNA sequencing reveals differential expression of MYCN target genes and suggests the mTOR pathway as a promising therapy target in MYCN-amplified neuroblastoma. Int J Cancer. 2013 Feb 1;132(3):E106-15.
76. Johnsen JI, Segerström L, Orrego A, Elfman L, Henriksson M, Kågedal B, et al. Inhibitors of mammalian target of rapamycin downregulate MYCN protein expression and inhibit neuroblastoma growth in vitro and in vivo. Oncogene. 2008 May 1;27(20):2910–22.
77. Vaughan L, Clarke PA, Barker K, Chanthery Y, Gustafson CW, Tucker E, et al. Inhibition of mTOR-kinase destabilizes MYCN and is a potential therapy for MYCN-dependent tumors. Oncotarget. 2016 Sep 6;7(36):57525–44.
78. Dobrenkov K, Cheung NKV. GD2-targeted immunotherapy and radioimmunotherapy. Semin Oncol. 2014 Oct;41(5):589–612.
79. Mora J, Climent A, Roldán M, Flores MC, Varo A, Perez-Jaume S, et al. Desensitizing the autonomic nervous system to mitigate anti-GD2 monoclonal antibody side effects. Front Oncol. 2024 May 15;14:1380917.
80. Machy P, Mortier E, Birklé S. Biology of GD2 ganglioside: implications for cancer immunotherapy. Front Pharmacol. 2023;14:1249929.
81. Cao S, Hu X, Ren S, Wang Y, Shao Y, Wu K, et al. The biological role and immunotherapy of gangliosides and GD3 synthase in cancers. Front Cell Dev Biol. 2023;11:1076862.
82. Krengel U, Bousquet PA. Molecular Recognition of Gangliosides and Their Potential for Cancer Immunotherapies. Front Immunol. 2014 Jul 21;5:325.
83. Cavdarli S, Groux-Degroote S, Delannoy P. Gangliosides: The Double-Edge Sword of Neuro-Ectodermal Derived Tumors. Biomolecules. 2019 Jul 27;9(8):311.
84. Valentino L, Moss T, Olson E, Wang HJ, Elashoff R, Ladisch S. Shed tumor gangliosides and progression of human neuroblastoma. Blood. 1990 Apr 1;75(7):1564–7.
85. Wu ZL, Schwartz E, Seeger R, Ladisch S. Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res. 1986 Jan;46(1):440–3.
86. van den Bijgaart RJE, Kroesen M, Wassink M, Brok IC, Kers-Rebel ED, Boon L, et al. Combined sialic acid and histone deacetylase (HDAC) inhibitor treatment up-regulates the neuroblastoma antigen GD2. J Biol Chem. 2019 Mar 22;294(12):4437–49.
87. Groux-Degroote S, Rodríguez-Walker M, Dewald JH, Daniotti JL, Delannoy P. Gangliosides in Cancer Cell Signaling. Prog Mol Biol Transl Sci. 2018;156:197–227.
88. Furukawa K, Ohmi Y, Hamamura K, Ohkawa Y, Hashimoto N, Tajima O, et al. GD2 is a Crucial Ganglioside in the Signal Modulation and Application as a Target of Cancer Therapeutics. Cancer Sci. 2025 Apr;116(4):862–70.
89. Tong W, Maira M, Gagnon M, Saragovi HU. Ligands Binding to Cell Surface Ganglioside GD2 Cause Src-Dependent Activation of N-Methyl-D-Aspartate Receptor Signaling and Changes in Cellular Morphology. PloS One. 2015;10(8):e0134255.
90. Neuroblastoma [Internet]. [cited 2025 Dec 9]. Available from: https://www.pathologyoutlines.com/topic/adrenalneuroblastoma.html.
91. Rothenberg AB, Berdon WE, D’Angio GJ, Yamashiro DJ, Cowles RA. Neuroblastoma—remembering the three physicians who described it a century ago: James Homer Wright, William Pepper, and Robert Hutchison. Pediatr Radiol. 2009 Feb 1;39(2):155–60.
92. Bagatell R, Park JR, Acharya S, Aldrink J, Allison J, Alva E, et al. NCCN Guidelines Index Table of Contents Discussion. 2025.
93. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer. 1999 Jul 15;86(2):349–63.
94. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999 Jul 15;86(2):364–72.
95. Joshi VV, Rao PV, Cantor AB, Altshuler G, Shuster JJ, Castleberry RP. Modified histologic grading of neuroblastomas by replacement of mitotic rate with mitosis karyorrhexis index. A clinicopathologic study of 223 cases from the Pediatric Oncology Group. Cancer. 1996 Apr 15;77(8):1582–8.
96. Corbacioglu S, Lode H, Ellinger S, Zeman F, Suttorp M, Escherich G, et al. Irinotecan and temozolomide in combination with dasatinib and rapamycin versus irinotecan and temozolomide for patients with relapsed or refractory neuroblastoma (RIST-rNB-2011): a multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2024 Jul;25(7):922–32.
97. Granger MM, Naranjo A, Bagatell R, DuBois SG, McCune JS, Tenney SC, et al. Myeloablative Busulfan/Melphalan Consolidation following Induction Chemotherapy for Patients with Newly Diagnosed High-Risk Neuroblastoma: Children’s Oncology Group Trial ANBL12P1. Transplant Cell Ther. 2021 Jun;27(6):490.e1-e8.
98. Children’s Oncology Group. A Phase 3 Study of 131I-Metaiodobenzylguanidine (131I-MIBG) or ALK Inhibitor Therapy Added to Intensive Therapy for Children With Newly Diagnosed High-Risk Neuroblastoma (NBL). clinicaltrials.gov; 2025 Oct [cited 2025 Dec 10]. Report No.: NCT03126916. Available from: https://clinicaltrials.gov/study/NCT03126916.
99. Park JR, Kreissman SG, London WB, Naranjo A, Cohn SL, Hogarty MD, et al. Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA. 2019 Aug 27;322(8):746–55.
100. Seif AE, Naranjo A, Baker DL, Bunin NJ, Kletzel M, Kretschmar CS, et al. A pilot study of tandem high-dose chemotherapy with stem cell rescue as consolidation for high-risk neuroblastoma: Children’s Oncology Group study ANBL00P1. Bone Marrow Transplant. 2013 Jul;48(7):947–52.
101. George RE, Li S, Medeiros-Nancarrow C, Neuberg D, Marcus K, Shamberger RC, et al. High-Risk Neuroblastoma Treated With Tandem Autologous Peripheral-Blood Stem Cell–Supported Transplantation: Long-Term Survival Update. J Clin Oncol. 2006 Jun 20;24(18):2891–6.
102. Kreissman SG, Villablanca JG, Diller L, London WB, Maris JM, Park JR, et al. Response and toxicity to a dose-intensive multi-agent chemotherapy induction regimen for high risk neuroblastoma (HR-NB): A Children’s Oncology Group (COG A3973) study. J Clin Oncol. 2007 Jun 20;25(18_suppl):9505.
103. Ladenstein R, Pötschger U, Valteau-Couanet D, Luksch R, Castel V, Ash S, et al. Investigation of the Role of Dinutuximab Beta-Based Immunotherapy in the SIOPEN High-Risk Neuroblastoma 1 Trial (HR-NBL1). Cancers. 2020 Jan 28;12(2):309.
104. Desai AV, Gilman AL, Ozkaynak MF, Naranjo A, London WB, Tenney SC, et al. Outcomes Following GD2-Directed Postconsolidation Therapy for Neuroblastoma After Cessation of Random Assignment on ANBL0032: A Report From the Children’s Oncology Group. J Clin Oncol Off J Am Soc Clin Oncol. 2022 Dec 10;40(35):4107–18.
105. FDA. FDA approves eflornithine for adult and pediatric patients with high-risk neuroblastoma. Maryland: FDA; 2023 Dec 14 [cited 2025 Dec 10]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-eflornithine-adult-and-pediatric-patients-high-risk-neuroblastoma.
106. Oesterheld J, Ferguson W, Kraveka JM, Bergendahl G, Clinch T, Lorenzi E, et al. Eflornithine as Postimmunotherapy Maintenance in High-Risk Neuroblastoma: Externally Controlled, Propensity Score-Matched Survival Outcome Comparisons. J Clin Oncol. 2024 Jan 1;42(1):90–102.
107. Sholler GLS, Ferguson W, Bergendahl G, Bond JP, Neville K, Eslin D, et al. Maintenance DFMO Increases Survival in High Risk Neuroblastoma. Sci Rep. 2018 Sep 27;8(1):14445.
108. Sholler G. A Phase II Preventative Trial of DFMO (Eflornithine HCl) as a Single Agent in Patients With High Risk Neuroblastoma in Remission [Internet]. clinicaltrials.gov; 2024 Aug [cited 2025 Dec 10]. Report No.: NCT02395666. Available from: https://clinicaltrials.gov/study/NCT02395666.
109. Giudice AM, Roth SL, Matlaga S, Cresswell-Clay E, Mishra P, Schürch PM, et al. Reprogramming the neuroblastoma tumor immune microenvironment to enhance GPC2 CAR T cells. Mol Ther J Am Soc Gene Ther. 2025 Sep 3;33(9):4552–69.
110. Wieczorek A, Śladowska K, Lode HN. Efficacy and Safety of Anti-GD2 Immunotherapy with Dinutuximab Beta in the Treatment of Relapsed/Refractory High-Risk Neuroblastoma. Target Oncol. 2025 Jul 1;20(4):551–68.
111. Furman WL, Federico SM, McCarville MB, Shulkin BL, Davidoff AM, Krasin MJ, et al. A Phase II Trial of Hu14.18K322A in Combination with Induction Chemotherapy in Children with Newly Diagnosed High-Risk Neuroblastoma. Clin Cancer Res. 2019 Nov 1;25(21):6320–8.
112. Durbas M, Horwacik I, Boratyn E, Kamycka E, Rokita H. GD2 ganglioside specific antibody treatment downregulates PI3K/Akt/mTOR signaling network in human neuroblastoma cell lines. Int J Oncol. 2015 Sep;47(3):1143–59.
113. FDA. FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow. Maryland: FDA; 2024 Aug 9 [cited 2025 Dec 11]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-naxitamab-high-risk-neuroblastoma-bone-or-bone-marrow.
114. ASCO. Naxitamab-based chemoimmunotherapy for resistant high-risk neuroblastoma: Results of “HITS” phase II study. New York: ASCO; [cited 2025 Dec 11]. Available from: https://www.asco.org/abstracts-presentations/206943.
115. Lu S, Wang J, Chen Y, Liang H, Zhu J, Yang T, et al. The combination of naxitamab with or without sintilimab (anti-PD-1) for the treatment of refractory/progressive neuroblastoma: A prospective, non-randomized, multicenter trial. J Clin Oncol. 2025 Jun [cited 2025 Dec 11];43:e22014.
116. Mora J, Chan GCF, Morgenstern DA, Amoroso L, Nysom K, Faber J, et al. The anti-GD2 monoclonal antibody naxitamab plus GM-CSF for relapsed or refractory high-risk neuroblastoma: a phase 2 clinical trial. Nat Commun. 2025 Feb 14;16(1):1636.
117. Olgun N, Arayici ME, Kızmazoglu D, Cecen RE. Assessment of Chemo-Immunotherapy Regimens in Patients with Refractory or Relapsed Neuroblastoma: A Systematic Review with Meta-Analysis of Critical Oncological Outcomes. J Clin Med. 2025 Jan 31;14(3):934.
118. Kushner BH, Modak S, Mauguen A, Basu EM, Roberts SS, Cheung NKV. Naxitamab plus stepped-up dosing of granulocyte-macrophage colony-stimulating factor for primary refractory high-risk neuroblastoma: results of a phase I/II trial. J Hematol Oncol. 2025 Nov 26 ;18(1):114.
119. Barry WE, Jackson JR, Asuelime GE, Wu HW, Sun J, Wan Z, et al. Activated Natural Killer Cells in Combination with Anti-GD2 Antibody Dinutuximab Improve Survival of Mice after Surgical Resection of Primary Neuroblastoma. Clin Cancer Res. 2019 Jan 1;25(1):325–33.
120. Mora J, Modak S, Kinsey J, Ragsdale CE, Lazarus HM. GM-CSF, G-CSF or no cytokine therapy with anti-GD2 immunotherapy for high-risk neuroblastoma. Int J Cancer. 2024 Apr 15;154(8):1340–64.
121. Fukuda M, Horibe K, Furukawa K. Enhancement of in vitro and in vivo anti-tumor activity of anti-GD2 monoclonal antibody 220-51 against human neuroblastoma by granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor. Int J Mol Med. 1998 Oct;2(4):471–5.
122. Park MY, Lim BG, Kim SY, Sohn HJ, Kim S, Kim TG. GM-CSF Promotes the Expansion and Differentiation of Cord Blood Myeloid-Derived Suppressor Cells, Which Attenuate Xenogeneic Graft-vs.-Host Disease. Front Immunol. 2019 Feb 26;10:183.
123. Martinez Sanz P, van Rees DJ, van Zogchel LMJ, Klein B, Bouti P, Olsman H, et al. G-CSF as a suitable alternative to GM-CSF to boost dinutuximab-mediated neutrophil cytotoxicity in neuroblastoma treatment. J Immunother Cancer. 2021 May;9(5):e002259.
124. Nguyen R, Zhang X, Sun M, Abbas S, Seibert C, Kelly MC, et al. Anti-GD2 Antibodies Conjugated to IL15 and IL21 Mediate Potent Antitumor Cytotoxicity against Neuroblastoma. Clin Cancer Res. 2022 Sep 1;28(17):3785–96.
125. Nguyen R, Moustaki A, Norrie JL, Brown S, Akers WJ, Shirinifard A, et al. Interleukin-15 Enhances Anti-GD2 Antibody-Mediated Cytotoxicity in an Orthotopic PDX Model of Neuroblastoma. Clin Cancer Res. 2019 Dec 15;25(24):7554–64.
126. Nguyen R, Houston J, Chan WK, Finkelstein D, Dyer MA. The role of interleukin-2, all-trans retinoic acid, and natural killer cells: surveillance mechanisms in anti-GD2 antibody therapy in neuroblastoma. Cancer Immunol Immunother CII. 2018 Apr;67(4):615–26.
127. Sechler JM, Barlic J, Grivel JC, Murphy PM. IL-15 alters expression and function of the chemokine receptor CX3CR1 in human NK cells. Cell Immunol. 2004 Aug;230(2):99–108.
128. Rhode PR, Egan JO, Xu W, Hong H, Webb GM, Chen X, et al. Comparison of the super agonist complex, ALT-803, to IL-15 as cancer immunotherapeutics in animal models. Cancer Immunol Res. 2016 Jan;4(1):49–60.
129. Boersma B, Poinot H, Pommier A. Stimulating the Antitumor Immune Response Using Immunocytokines: A Preclinical and Clinical Overview. Pharmaceutics. 2024 Aug;16(8):974.
130. Möller AM, Vettermann S, Baumann F, Pütter M, Müller D. Trifunctional antibody-cytokine fusion protein formats for tumor-targeted combination of IL-15 with IL-7 or IL-21. Front Immunol. 2025;16:1498697.
131. Schuurmans F, Wittner A, van den Bijgaart RJE, Tahk S, Boros MGM, Looman MWG, et al. Development of aGD2-SIRPα fusion antibodies targeting neuroblastoma and the innate immune checkpoint receptor CD47. Mol Cancer Ther. 2025 Oct 7.
132. Zhang Y, He B, Zou P, Wu M, Wei M, Xu C, et al. Targeted release of a bispecific fusion protein SIRPα/Siglec-10 by oncolytic adenovirus reinvigorates tumor-associated macrophages to improve therapeutic outcomes in solid tumors. J Immunother Cancer. 2025 Apr 1;13(4):e010767.
133. Bouwstra R, van Meerten T, Bremer E. CD47-SIRPα blocking-based immunotherapy: Current and prospective therapeutic strategies. Clin Transl Med. 2022 Aug;12(8):e943.
134. Fischer-Riepe L, Kailayangiri S, Zimmermann K, Pfeifer R, Aigner M, Altvater B, et al. Preclinical Development of CAR T Cells with Antigen-Inducible IL18 Enforcement to Treat GD2-Positive Solid Cancers. Clin Cancer Res. 2024 Aug 15;30(16):3564–77.
135. Okada R, Reyes-González JM, Rodriguez C, Kondo T, Oh J, Sun M, et al. GPC2-Targeted CAR T Cells Engineered with NFAT-Inducible Membrane-Tethered IL15/IL21 Exhibit Enhanced Activity against Neuroblastoma. Cancer Immunol Res. 2025 Sep 2;13(9):1363–73.
136. Giudice AM, Matlaga S, Roth SL, Gladney W, Groff D, Hofmann TJ, et al. D3-GPC2-Directed CAR T Cells Are Safe and Efficacious in Preclinical Models of Neuroblastoma and Small Cell Lung Cancer. Clin Cancer Res. 2025 Dec 15;31(24):5276–93.
137. Liu X, Wills CA, Chen L, Zhang J, Zhao Y, Zhou M, et al. Small extracellular vesicles induce resistance to anti-GD2 immunotherapy unveiling tipifarnib as an adjunct to neuroblastoma immunotherapy. J Immunother Cancer . 2022 Apr 28 [cited 2025 Dec 12];10(4):e004399.
138. Giudice AM, Matlaga S, Roth SL, Pascual-Pasto G, Schürch PM, Rouin G, et al. Target antigen-displaying extracellular vesicles boost CAR T cell efficacy in cell and mouse models of neuroblastoma. Sci Transl Med. 2025 Nov 19;17(825):eads4214.
139. Tian M, Cheuk AT, Wei JS, Abdelmaksoud A, Chou HC, Milewski D, et al. An optimized bicistronic chimeric antigen receptor against GPC2 or CD276 overcomes heterogeneous expression in neuroblastoma. J Clin Invest. 2022 Aug 15;132(16):e155621.
140. Mabe NW, Huang M, Dalton GN, Alexe G, Schaefer DA, Geraghty AC, et al. Transition to a mesenchymal state in neuroblastoma confers resistance to anti-GD2 antibody via reduced expression of ST8SIA1. Nat Cancer. 2022 Aug;3(8):976–93.
141. Moreno L, Barone G, DuBois SG, Molenaar J, Fischer M, Schulte J, et al. Accelerating drug development for neuroblastoma: Summary of the Second Neuroblastoma Drug Development Strategy forum from Innovative Therapies for Children with Cancer and International Society of Paediatric Oncology Europe Neuroblastoma. Eur J Cancer Oxf Engl 1990. 2020 Sep;136:52–68.
142. Matkar S, East MP, Stuhlmiller TJ, Witek GM, Farrel A, Pastor S, et al. Kinome Reprogramming of G2/M Kinases and Repression of MYCN Contribute to Superior Efficacy of Lorlatinib in ALK-Driven Neuroblastoma. Mol Cancer Ther. 2025 Sep 2;24(9):1389–401.
143. Goldsmith KC, Park JR, Kayser K, Malvar J, Chi YY, Groshen SG, et al. Lorlatinib with or without chemotherapy in ALK-driven refractory/relapsed neuroblastoma: phase 1 trial results. Nat Med. 2023 May;29(5):1092–102.
144. ASCO. Phase I trial of lorlatinib in combination with topotecan/cyclophosphamide in children with ALK-driven refractory or relapsed neuroblastoma: A new approaches to neuroblastoma therapy consortium study. Atlanta: ASCO; 2022 [cited 2025 Dec 12]. Available from: https://www.asco.org/abstracts-presentations/206953.
145. Tucker ER, Jiménez I, Chen L, Bellini A, Gorrini C, Calton E, et al. Combination Therapies Targeting ALK-aberrant Neuroblastoma in Preclinical Models. Clin Cancer Res. 2023 Apr 3;29(7):1317–31.
146. Borenäs M, Umapathy G, Lind DE, Lai WY, Guan J, Johansson J, et al. ALK signaling primes the DNA damage response sensitizing ALK-driven neuroblastoma to therapeutic ATR inhibition. Proc Natl Acad Sci U S A. 2024 Jan 2;121(1):e2315242121.
147. Szydzik J, Lind DE, Arefin B, Kurhe Y, Umapathy G, Siaw JT, et al. ATR inhibition enables complete tumour regression in ALK-driven NB mouse models. Nat Commun. 2021 Nov 24;12(1):6813.
148. Benchia D, Bîcă OD, Sârbu I, Savu B, Farcaș D, Miron I, et al. Targeting Pathways in Neuroblastoma: Advances in Treatment Strategies and Clinical Outcomes. Int J Mol Sci. 2025 May 15;26(10):4722.
149. ASCO. Schellekens K. A phase 1b study of crizotinib in combination with temsirolimus in pediatric ALK- or MET-aberrated relapsed or refractory neuroblastoma (ITCC-053): Results of the phase 1 part. Netherlands: ASCO; 2023 [cited 2025 Dec 12]. Available from: https://www.asco.org/abstracts-presentations/217985.
150. Foster JH, Voss SD, Hall DC, Minard CG, Balis FM, Wilner K, et al. Activity of Crizotinib in Patients with ALK-Aberrant Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group Study (ADVL0912). Clin Cancer Res. 2021 Jul 1;27(13):3543–8.
151. Tucker ER, Danielson LS, Innocenti P, Chesler L. Tackling Crizotinib Resistance: The Pathway from Drug Discovery to the Pediatric Clinic. Cancer Res. 2015 Jul 15;75(14):2770–4.
152. Boni J, Leister C, Burns J, Cincotta M, Hug B, Moore L. Pharmacokinetic profile of temsirolimus with concomitant administration of cytochrome p450-inducing medications. J Clin Pharmacol. 2007 Nov;47(11):1430–9.
153. Gupta AA, Xue W, Harrison DJ, Hawkins DS, Dasgupta R, Wolden S, et al. Addition of temsirolimus to chemotherapy in children, adolescents, and young adults with intermediate-risk rhabdomyosarcoma (ARST1431): a randomised, open-label, phase 3 trial from the Children’s Oncology Group. Lancet Oncol. 2024 Jul;25(7):912–21.
154. Kennedy BK, Lamming DW. The mechanistic Target of Rapamycin: The grand conducTOR of metabolism and aging. Cell Metab. 2016 Jun 14;23(6):990–1003.
155. Panwar V, Singh A, Bhatt M, Tonk RK, Azizov S, Raza AS, et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. 2023 Oct 2;8(1):375.
156. Sehgal SN. Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc. 2003 May 1;35(3, Supplement):S7–14.
157. Huang S, Gu S. Targeting autophagy in neuroblastoma. World J Pediatr Surg. 2020 Aug 27;3(3):e000121.
158. Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. J Clin Invest. 2015 Jan 2;125(1):25–32.
159. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017 Mar 9;168(6):960–76.
160. Chan GCF, Chan CM. Anti-GD2 Directed Immunotherapy for High-Risk and Metastatic Neuroblastoma. Biomolecules. 2022 Feb 24;12(3):358.