Abstract
According to its primary structure, the PITG_06016 gene encodes for one of the 7 glucokinases present in Phytophthora infestans PiGlcK-1, the causal agent of late blight disease. Currently, there are no structural studies of any of its enzymes, hence the determination of the three-dimensional (3D) structure of PiGlcK-1 becomes a significant contribution to the deduction of its functions, its interaction with ligands, and possible regulatory mechanisms. In this work we present the first structural model obtained by in silico tools for PiGlcK-1. For the prediction of this model, different algorithms were used to find the best annealing, refinement, and qualitative evaluation of them. A structural comparison of the predicted model with other structures of crystallized glucokinases enzymes allowed us to identify both the regions of interaction with their classical substrates (glucose and ATP) and those involved in the binding of other substrates such as fructose and ADP. In addition, we propose a possible recognition region of PPi, an activator of kinase activity that includes the GXGE motif, conserved in enzymes of the ribokinase (RK) family which distinguishes this PiGlcK-1 from a classical glucokinase. Accordingly, these findings suggest PPi-binding motif as potential targets for the development of inhibitors of PiGlcK-1 activity.
Keywords
Phythophtora infestans, Glucokinase, Modeling structural, Molecular docking, PPi-binding motif
Abbreviations
ATP: Adenosine Triphosphate; ADP: Adenosine Diphosphate; PPi: Pyrophosphate; NCBI: National Center for Biotechnology Information; ΔG: Gibbs free energy; NADPH: Nicotinamide Adenine Dinucleotide Phosphate reduced
Introduction
Phytophthora infestans is the causal agent of late blight disease, which affects potato and tomato crops worldwide, that brings about significant economic losses in the production of these crops [1]. The PITG_06016 gene codes for one of the 7 glucokinases present in this phytopathogen [2]. This glucokinase, known as PiGlcK-1, is very important due to its high degree of expression, precisely at the infectious stages of its life cycle [3]. PiGlcK-1 was also the first P. infestans glucokinase to be biochemically characterized; based on its sequence, it would belong to the glucokinase A group of the hexokinase family. Certainly, the characterization of PiGlcK-1 has revealed the versatility of this enzyme in the phosphorylation of both glucose and fructose, as well as in the utilization of ATP, ADP, and PPi as phosphoryl donors [4].
These findings raise the need for further structural analyses on PiGlcK-1. In this sense, a better understanding of the three-dimensional (3D) structure of this protein, as well as the spatial description of the possible binding sites to various ligands would contribute to the knowledge of potential targets for the design of inhibitors of the enzymatic activity of PiGlcK-1.
However, experimental determination of protein structures remains a costly and time-consuming challenge. In fact, to date no P. infestans enzyme has been structurally characterized. Conversely, bioinformatics tools offer an alternative that allows prediction of 3D protein structures by molecular dynamics and homology modeling making it faster, cheaper, and highly reliable. The reliability of these tools is based on the use of databases of solved protein structures that can serve as homology templates in the simulation and structural prediction of proteins [5].
In this work we present the first model obtained by in silico tools of the 3D structure of PiGlcK-1, and the binding sites to its classical substrates glucose and ATP, to fructose and ADP, and a possible binding site to PPi that is proposed as a promising target for the design of an inhibitor of this enzyme.
Materials and Methods
Sequence availability
The amino acid sequence of the protein PiGlcK-1 (accession number XP_002998228), glucokinase of Escherichia coli (EcGlcK)(accession number AAA_ 64506.1), glucokinase of Saccharomyces cereviceae S288C (ScGlcK)(accession number NP_009890.1), glucokinase of Homo sapiens (HsGlcK)(accession number AAB_97681.1) and ADP-specific glucokinase of Pyrococcus furiosus (PfGlcK) (WP_011011427.1) were obtained from the NCBI database. NCBI BLAST (Basic Local Alignment Search Tool) [6] was used to compare the primary structure of PiGlcK-1 with glucokinases from other organisms. PSIPRED [7] was used to know its secondary structure.
Structural analysis of PiGlcK-1 protein
The 3D structure of the protein PiGlcK-1 was obtained by homology modeling using the Phyre2 [8], LOMETS [9], and I-TASSER [10] algorithms. Each simulation obtained was refined with Galaxy Refine [11] to the maximum allowed, in some cases up to three times. The validation of structural stability was performed by qualitative evaluation by ModFOLD6 [12] and Ramachandran plot analysis obtained with SwissModel [13]. The topology of the PiGclK-1 model was obtained with Pro-origami [14]. The visualization, comparison and editing of the models were performed in Chimera 1.15.1 [15].
Docking
Molecular docking was carried out in SwissDock [16] using the refined PiGlcK-1 model and substrates obtained from PubChem [17]: glucose (107526), fructose (2723872), ATP/Mg+2 (5957), ADP/Mg+2 (6022), and PPi/Mg+2 (644102). In each case, the best energetic fits, such as model interaction energy of the complex bound to PiGlcK-1 and fullfitness values, were evaluated. Model visualization and editing were performed with Chimera 1.15.1 [15].
Results
Comparative analysis of the primary and secondary structure of PiGlcK-1
A comparative analysis of the primary structure of P. infestans glucokinase-1 with different glucokinases available in the database, not phylogenetically related to PiGlcK-1, yielded different levels of identity. Interestingly, glucokinase from E. coli (EcGlcK) presented the highest identity with PiGlcK-1, showing 98% alignment and a fairly significant E value (number chance hits expected in a database of a particular size) of 5E-47. On the other hand, the ScGlcK and HsGlcK showed very low identity values 48% and 7% respectively, with respect to PiGlcK-1 and little significant E values. Finally, PfGlcK did not yield any relevant results.
Coil, helix, and strands constitute the secondary structure of PiGlcK-1. The secondary structure could be used to validate the tertiary structure and was compared with other non-phylogenetically related glucokinases (Supplementary Figure 1).
Tertiary structure modeling
The models obtained from the structure of PiGlcK-1 in three different algorithms (Phyre2, LOMETS and I-TASSER) were highly satisfactory, obtaining overall scores greater than 0.4 and a p-value cut-off of less than 0.001 which are considered very good (Table 1).
| Model | Favored region (%) | Confidence and p-value | Global model quality score |
|---|---|---|---|
| Phyre2 | |||
| Before refinement | 91.50 | 4.42 E-7 | 0.6220 |
| After refinementa | 97.95 | 1.93 E-7 | 0.6412 |
| LOMETS | |||
| Before refinement | 93.45 | 5.14 E-8 | 0.6718 |
| After refinement b | 96.30 | 5.22 E-8 | 0.614 |
| I-TASSER | |||
| Before refinement | 81.60 | 3.56 E-8 | 0.6803 |
| After refinement c | 96.01 | 2.83 E-8 | 0.6854 |
| a2 time, b3 time, c3 time | |||
I each case, the models were refined until the best structure was obtained, according to the Ramachandran graph. The best model, based on its higher stereochemical quality, was that given by Phyre2, favoring 97.95% of the amino acids in the Ramachandran regions. This value renders the 3D model of the PiGlcK-1 protein for this work potentially reliable and of good quality (Figure 1).
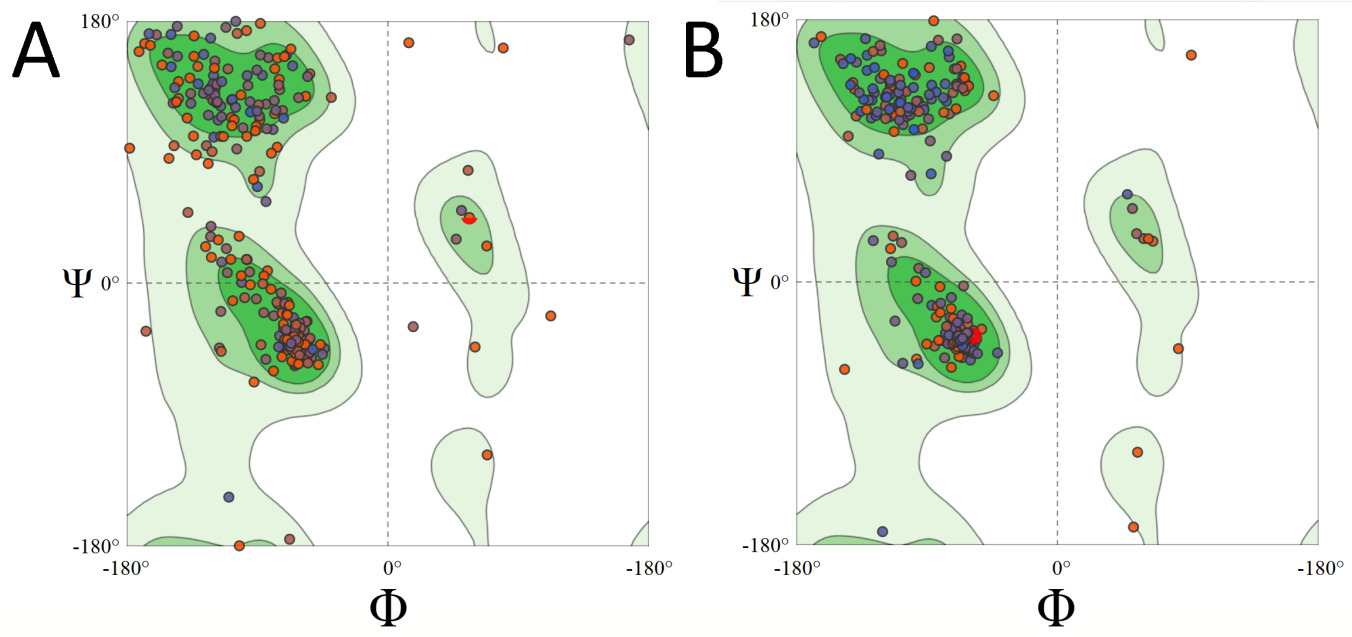
Figure 1: Ramachandran plot of PiGlcK-1 model for Phyre2 before (A) and after (B) refinement.
The predicted model indicated that the monomeric structure was composed of two domains, a small domain that includes residues 7 to 128 and a large domain formed by residues 135 to 343. Both domains were consecutively labeled from N-terminal to C-terminal and linked by a hinge (Figure 2).
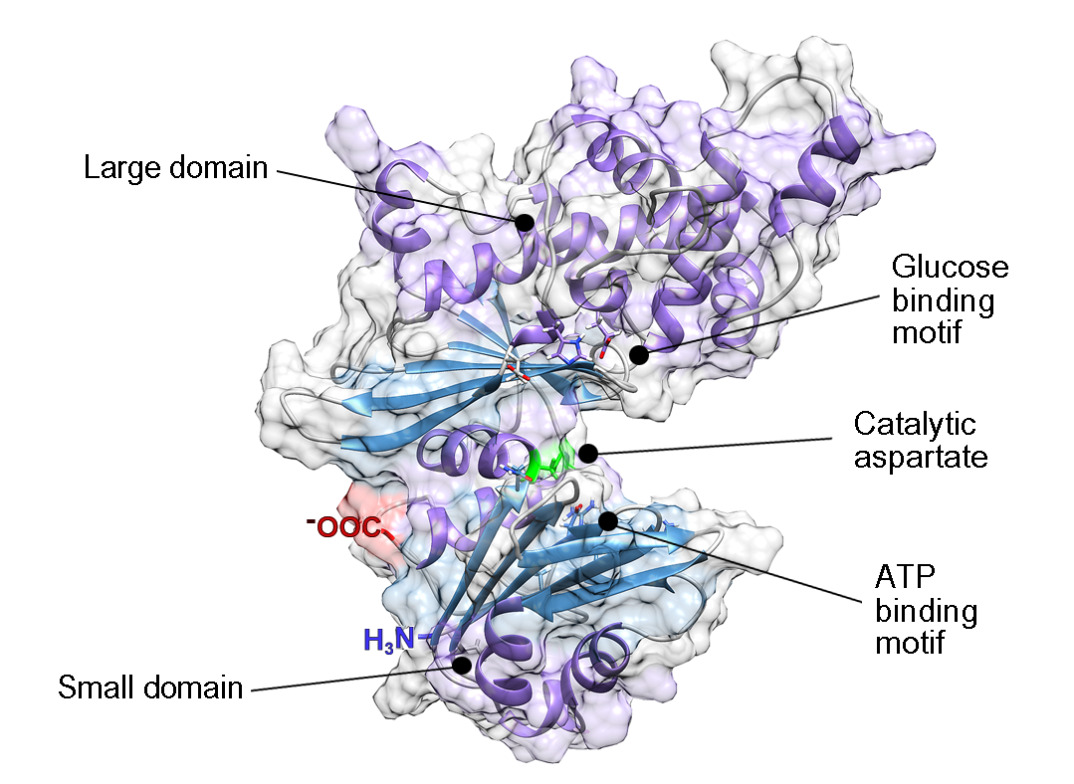
Figure 2: PiGlcK-1 model 3D predicted by Phyre2. PiGlcK-1 monomer showing the glucose binding motif in the large domain, the ATP binding motif in the small domain and the catalytic aspartate binding motif in the hinge region.
These results were compared with those obtained by crystallography for EcGlcK (PDB: 1SZ2), ScGlcK (PDB: 6P4X), HsGlcK (PDB: 1V4S) and PfGlcK (PDB: 1UA4) (Supplementary Figure 2).
The topology obtained by Pro-Origami (Supplementary Figure 3) showed that the small domain consists of 4 α-helices and 7 β-sheet distributed in two mixed central sheets; one of them of 5 stranded β (β1, β2, β3, β4 and β7) with β2 anti-parallel to the rest and another one formed by 2 β-sheet (β5 and β6) anti-parallel to each other. These sheets are flanked by 4 α-helices (α1, α2, α3 and α4).
On the other hand, the large domain consists of 10 α-helices and 6 β-sheet organized in a single mixed central sheet containing all 6 chains β (β8, β9, β10, β11, β12 and β13) with β8 and β10 anti-parallel to the rest. This sheet is flanked by 10 α-helices (α5, α6, α7, α8, α9, α10, α11, α12, α13 and α14) (Supplementary Figure 3).
Molecular docking studies
In order to know the active site of the PiGlcK-1 a docking was performed with its classic substrates glucose and ATPMg2+. This analysis showed that the glucose binding site is in the large domain while the ATP binding site is in the small domain; both molecules interact with an extensive network of hydrogen bonds within the active site (Figure 3). These results coincide with those reported for EcGlcK [18], GlcK from Trypanosoma cruzi (TcGlcK) [19] and the 3D model of the complex GlcK-Mg2+-ATP-glucose (GMAG) of human [20].
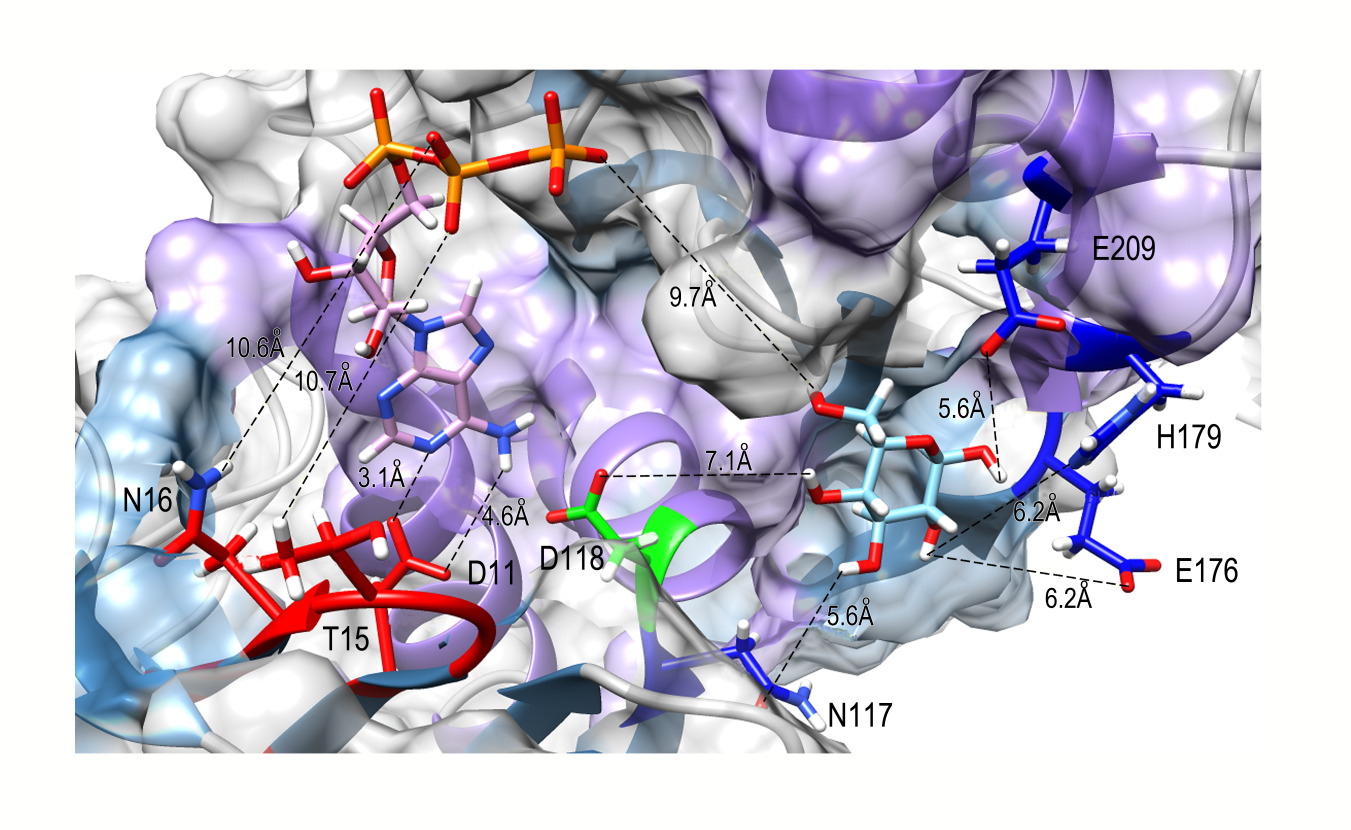
Figure 3: The binding site region between PiGlcK-1 and glucose and ATP. Docking result using Swiss Dock. Amino acids in PiGlcK-1 involved in the interaction with their ligands are highlighted in red (ATP) and blue (glucose). Catalytic aspartate is highlighted in green.
In this sense, the hydrogen bonds with the best values of ΔG and fullfitness were obtained from the residues Glu 209; Glu 176, His 179; Asn 117, and Asp 118 with atoms O1; O2; 03 and O4 of glucose with a ΔG of -5.27 kcal/mol and a fullfitness value of -1746.84 (Figure 3). In addition, for the β-phosphoryl group of ATP, the best values were obtained with the amino acids Thr 15 and Asn 16, and the γ-phosphoryl interacting with the O6 atom of glucose with an energy of -11.69 kcal/mol and a fullfitness value of -2187.34 (Figure 3). Notably, all the interacting residues described above are highly conserved in group A GlcKs from diverse organisms [4].
Fructose-binding site
We have previously demonstrated experimentally that PiGlcK-1 is able to phosphorylate other sugars such as fructose [4]. To identify a possible binding site for this hexose, we compared the fructose binding site with that of glucose, obtaining that this sugar interacts with the same amino acid residues described for glucose in this work. Fructose binding, as for glucose, occurred in the large domain and hinge region of PiGlcK-1 with an energy of -5.742 kcal/mol and a fullfitness value of -1728.80 (Figure 4A).
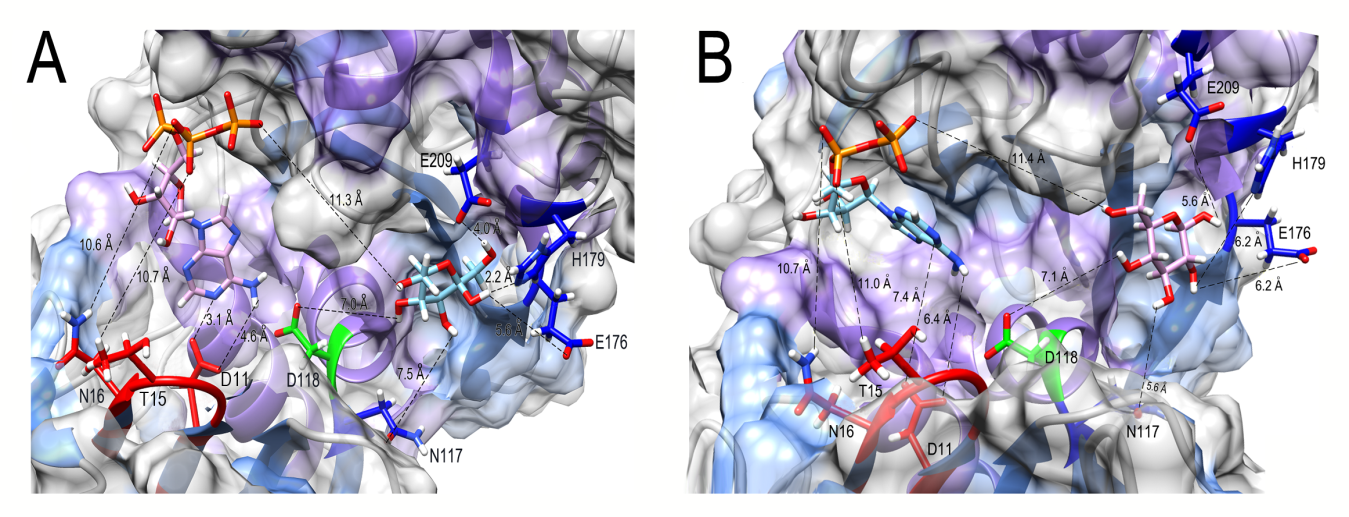
Figure 4: Docking result using Swiss Dock for fructose (A) and ADP (B) to PiGlcK-1. (A) Amino acids in PiGlcK-1 involved in the interaction with their ligands are highlighted in red (ATP) and blue (fructose). (B) Amino acids in PiGlcK-1 involved in the interaction with their ligands are highlighted in red (ADP) and blue (glucose).
Identification of ADP and PPi-binding site
One of the particularities of PiGlcK-1 is to use ADP or PPi as a phosphoryl donor for the formation of hexose- 6-phosphate. Additionally, PPi proved to be an efficient activator of PiGlcK-1 when the phosphoryl donors are ATP or ADP [4]. Initially, we determined the binding site of ADP by performing a docking of this ligand to PiGlcK-1.
Accordingly, it was found that PiGlcK-1 interacts with the same residues present in the small domain described for ATP with an energy of -10.59 kcal/mol and a fullfitness of -2041.78 (Figure 4B).
For its part, the best docking obtained for PiGlcK-1-PPi was located in a region other than the ATP-binding and ADP-binding region, this being the larger domain with an energy of -5.425 kcal/mol and a fullfitness of -2150.14 (Figure 5A). Interestingly, the amino acids involved in this interaction, Lys 282, Thr 180 and Arg 210 (Figure 5B), have already been reported for the PPi-dependent kinase from Thermotoga maritima [21]. Markedly, PiGlcK-1 also has the conserved GXGD(E) motif consisting of Gly 156, Leu 157, Gly 158 and Glu 159. This motif has been described in the ADP-dependent kinases of the Ribokinase family (RK) [22,23] in a region very close to the previously described PPi-binding motif, thus suggesting that the residues involved in phosphoryl binding in RKs could also be participating in interaction with PPi in PiGlcK-1. However, although this motif is shown to bind ADP, it is unlikely in our case since the ADP would be in a region other than the hinge region.
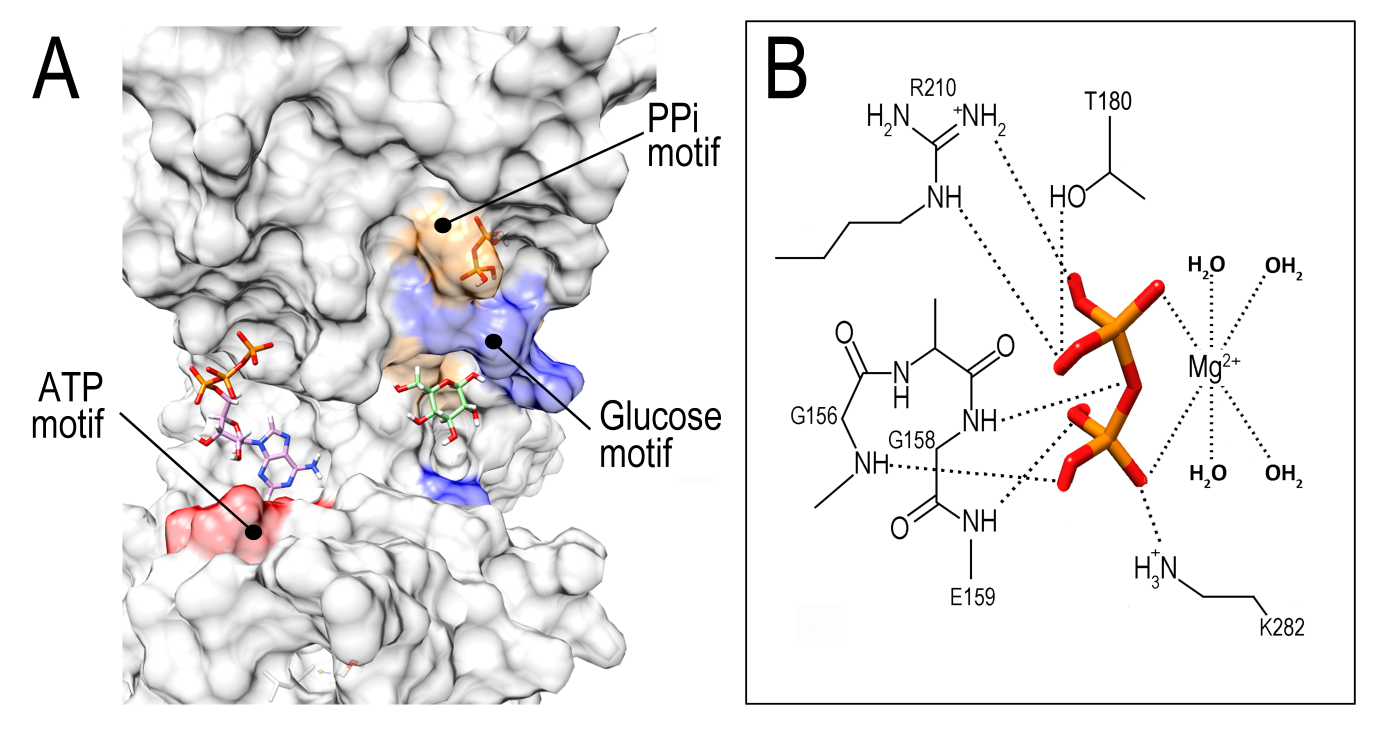
Figure 5: Docking result using Swiss Dock for PPi to PiGlcK-1. (A) Overview of the PPi binding site (orange) distinct from that of ATP (red) and glucose (blue). (B) Close-up of the PPi binding region showing the GXGE motif and specific amino acids involved in PPi recognition.
Discussion
Using molecular modeling and simulation methods, we constructed the 3D structural model for PiGlcK-1. This model was validated and structurally compared with glucokinases from other organisms, verifying that PiGlcK-1 folds similarly to GlcK from E. coli, S. cerevisiae and H. sapiens (Supplementary Figure 2), whose crystallographic structures have already been described.
With our model of PiGlcK-1, we were able to verify that the monomeric structure of the enzyme is highly conserved as it is composed of a large domain and a small domain connected to each other by a hinge region. These domains are made of β-sheet, flanked by α-helices, and a cavity between both lobes where the catalytic site is located, similar to many hexose kinases from several phylogenetically unrelated organisms [18,20,24].
Molecular docking assays on PiGlcK-1 verified that ATP and glucose do indeed interact via hydrogen bonds with the residues that form the active site of the protein, as it has been described for several hexokinases and glucokinases [24]. The glucose binding site comprises well-conserved residues in the hinge region (Asp 118, Asn 117) and the large domain (Glu 176, His 179, Glu 209). In contrast, ATP binds mainly to residues Asp 11, Thr 15, and Asn 16 of the small domain with the γ-phosphoryl group pointing toward the hinge region, as it has been reported for hexokinase and glucokinase enzymes from various organisms [24].
In each case, the distance between the enzyme and its substrates were calculated on a rigid model, so the value of these exceeds 2 Å. However, it is worth considering the intrinsic flexibility of different GlcKs [18,24] which has reached in P. furiosus a displacement of 12 Å when comparing the protein in the presence and absence of glucose [25]. Thus, the interactions between the ligands and the amino acid residues in PiGlcK-1 proposed here are highly consistent with the crystallographic structures mentioned above.
Furthermore, it has been observed that fructose from the plant, like glucose, is important for the metabolism of P. infestans during the establishment of infection, where its concentration in potato and tomato leaves is higher than that of glucose [26-28]. Due to the absence of a specialized fructokinase (EC 2.7.1.4) in P. infestans [28] this sugar must be obligatorily phosphorylated by PiGlcK-1.
Therefore, after verifying in the docking of PiGlcK-1 with glucose and ATP that these were located in the active site, interacting with amino acid residues conserved for other kinases, the docking was carried out for fructose as well as for the phosphoryl donors ADP and PPi.
As expected, both fructose and ADP are able to form hydrogen bonds with the same amino acid residues described for their glucose and ATP pairs, respectively, validating that these can be utilized under certain physiological conditions during the life cycle of P. infestans. In this case, when glucose or ATP concentrations are low, P. infestans could obtain energy and/or NADPH through glycolysis or the pentose phosphate pathway. These routes could function using fructose and ADP as substrates, thus ensuring that the enzyme remains active during the establishment of infection regardless of the availability of glucose or ATP.
On the contrary, the docking for PPi, located in the large domain, close to the hinge region but in a site different from the ATP/ADP binding site (Figure 5A), would explain its role as an activator of PiGlcK-1 activity in the presence of ATP or ADP [4]. This activating role of PPi might be linked to a possible stabilization of a catalytically more active conformation of the enzyme, although more assays are needed to corroborate this hypothesis.
The importance of the role played by PPi in P. infestans has been evidenced by several facts such as : i) P. infestans can produce PPi through multiple biosynthesis and hydrolysis reactions [29]; ii) significant expression of pyruvate phosphate dikinase (PPDK), 6-phosphofructose- 1-kinase, and 6-phosphofructose-2-kinase all dependent on PPi [3,28,30]; iii) it has been suggested that the majority of the glycolytic flux in the hyphal sporulation stage of Phytophthora cinnamomi occurs via PPDK [31]; iv) PPDK may play a more relevant role than pyruvate kinase (PYK) in glycolysis of P. infestans due to a higher expression of PPDK relative to PYK [32]; v) pyrophosphate stimulates calcium uptake in diverse organisms including P. infestans, which is required for its growth and development [33].
Although the true value of PPi in the metabolism of P. infestans remains to be clarified, it is important to consider the binding motif of this ligand when designing potential inhibitors of PiGlcK-1 activity as it constitutes a promising target for attack in the search for a solution to the threat posed by late blight.
Conclusion
In this work we have presented the first in silico structural model of glucokinase-1 from the phytopathogen P. infestans (PiGlcK-1). The resulting structure was shown to be of high quality, which allows it to be used in docking analyses for other ligands. We confirmed that the folding of this protein is similar to that of other glucokinases from different organisms and we were able to identify the binding sites for its substrates glucose and ATP, as well as for the ligands fructose, ADP, and PPi reported for the first time for a classical glucokinase of the GlcK-A group.
Altogether, these findings lay the groundwork for the design of future inhibitors of PiGlcK-1 enzymatic activity that would aid in the control of late blight disease. It is important to underscore the need to establish a new classification within a more diverse group of kinases, one that considers not only the primary sequence of PiGlcK-1, but also its 3D structure.
Acknowledgments
We are very grateful to experiment.com and especially to Charlie Kinsella, Brian Repez, Amy Collete, and Pureum Kim for their valuable financial contribution to this project.
Conflicts of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this paper.
References
2. Haas BJ, Kamoun S, Zody MC, Jiang RHY, Handsaker RE, Cano LM, et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461(7262):393-98.
3. Judelson HS. Metabolic diversity and novelties in the Oomycetes. Annu Rev Microbiol. 2017;71(1):21-39.
4. Villalobos-Piña L, Balza H, Acosta H, Andrade- Alviarez D, Rojas A, Avilan L, et al. Characterization of glucokinase-1 from Phytophthora infestans, a versatile enzyme. Fungal Genom Biol. 2020;10 (100001):1-9.
5. Petrey D, Honig B. Protein structure prediction: in roads to biology. Mol Cell. 2005; 20(6):811-19.
6. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Research. 2008;36:W5-W9.
7. Buchan DWA, Jones DT. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019;47:W402-W407.
8. Kelley L, Mezulis S, Yates C, Wass MN, Sternberg M. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6).
9. Wu S, Zhang Y. LOMETS: a local meta-threadingserver for protein structure prediction. Nucleic Acids Res. 2007;35(10):3375-82.
10. Jianyi Y, Renxiang Y, Ambrish R, Dong X, Jonathan P, Yang Z. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):1-5.
11. Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013;41.
12. Maghrabi AHA, McGuffin LJ. ModFOLD6: an accurate web server for the global and local quality estimation of 3D models of proteins. Nucleic Acids Res. 2017;45:W416-W421.
13. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296-W303.
14. Stivala A, Wybrow M, Wirth A, Whisstock JC, Stuckey PJ. Automatic generation of protein structure cartoons with Pro-origami. Bioinformatics. 2011;27(23):3315-16.
15. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605-12.
16. Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270-W277.
17. Kim S, Chem J, Cheng T, Gindulyte A, He S, Li Q. PubChem, in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021; 49(D1):D1388-D1395.
18. Lunin VV, Li Y, Schrag JD, Iannuzzi P, Cygler M, Matte A. Crystal structures of Escherichia coli ATP-dependent glucokinase and its complex with glucose. J Bacteriol. 2004;186(20):6915-27.
19. Cordeiro AT, Cáceres AJ, Vertommen D, Concepción JL, Michels PAM, Versées W. The crystal structure of Trypanosoma cruzi glucokinase reveals features determining oligomerization and anomer specificity of hexose-phosphorylating enzymes. J Mol Biol. 2007;372(5):1215-1226.
20. Zhang J, Li C, ShiT, Chen K, Shen X, Jiang H. Lys169 of human Glucokinase is a determinant for glucose phosphorylation: implication for the atomic mechanism of glucokinase catalysis. PLoS One. 2009;4(7):1-12.
21. Nagata R, Fujihashi M, Sato P, Atomi H, Miki K. Identification of a pyrophosphate-dependent kinase and its donor selectivity determinants. Nat Commun. 2018;9:1765:1-8.
22. Park J, Gupta RS. Adenosine kinase and ribokinasethe RK family of proteins. Cell Mol Life Sci. 2008;65:2875-96.
23. Cabrera R, Babul J, Guixé V. Ribokinase family evolution and the role of conserved residues at the active site of the PfkB subfamily representative, Pfk2 from Escherichia coli. Arch Biochem Biophys. 2010;502:23-30.
24. Van Schaftingen E. Hexokinase/Glucokinase. Encyclopedia of Biochemistry 3rd Edition. 2020.
25. Ito S, Fushinobu S, Jeong J, Yoshioka I, Koga S, Shoun H, et al. Crystal structures of an ADP-dependent glucokinase from Pyrococcus furiosus. Implications for a sugar-induced conformational change in ADP-dependent kinase. J Mol Biol. 2003;(331):871-83.
26. Engstrom K, Stromberg A. Changes in sugar content during induction of systemic acquired resistance to late blight caused by Phytophthora infestans (Mont) de Bary in potato. J Phytopathol. 1996;144:33-36.
27. Daniele E, Dommes J, Hausman, JF. Carbohydrates and resistance to Phytophthora infestans in potato plants.Acta Physiol Plant. 2003;25:171-78.
28. Judelson HS, Tani S, Narayan RD. Metabolic adaptation of Phytophthora infestans during growth on leaves, tubers and artificial media. Mol Plant Pathol. 2009;10(6):843-55.
29. Garc-a-Bayona L, Garavito MF, Lozano GL, Vasquez JJ, Myers K, Fry WE, et al. De novo pyrimidine biosynthesis in the oomycete plant pathogen Phytophthora infestans. Gene. 2014;537(2):312-21.
30. Ah-Fong AM, Kim KS, Judelson HS. RNA-seq of life stages of the oomycete Phytophthora infestans reveals dynamic changes in metabolic, signal transduction, and pathogenesis genes and a major role for calcium signaling in development. BMC Genomics. 2017;18(198):1-21.
31. Marshall J, Anthony R, Ashton A. Isolation and characterization of four genes encoding pyruvate, phosphate dikinase in the oomycete plant pathogen Phytophthora cinnamomi. Curr Genet. 2001;(1):73-81.
32. Abrahamian M, Kagda M, Ah-Fong AM, Judelson HS. Rethinking the evolution of eukaryotic metabolism: novel cellular partitioning of enzymes in stramenopiles links serine biosynthesis to glycolysis in mitochondria. BMC Evol Biol. 2017;17(241):1-16.
33. Okorokov LA, Sysuev VA, Kulaev IS. Pyrophosphatestimulated uptake of calcium into the germlings of Phytophthora infestans. Eur J Biochem. 1978;83:507- 11.