Abstract
The common practice of cultivating rice through transplanting requires considerably higher amount of water, which poses several challenges, particularly in the present era of global climate change and decreasing availability of water. The direct-sown rice (DSR) presents a promising/resource-saving alternative for cultivation of rice in the current scenario of changing climatic conditions. While some local cultivars are well-adapted for DSR, there has been limited success in breeding for DSR varieties. This is mainly because of the lack of comprehensive knowledge about the adaptability of rice to changing climatic conditions. Comprehensive analysis of methylome and transcriptome profiles of rice [Nagina-22 (N-22) and IR-64] grown by transplanting as well as direct-sowing deciphered epigenetic regulation of genes involved in adaptive plasticity. Expression of a large number of genes correlated with DNA (de)methylation in N-22 under DSR conditions implies that (de)methylation of DNA base is associated with the adaptation of N-22 to DSR conditions. Hypomethylation of genes in leaves of N-22 showing their up-regulated expression under DSR conditions suggests epigenetic modification to be a key factor in adaptive plasticity of N-22. Post-translational modification of proteins, particularly histone modifications, and changes in chromatin architecture contribute to adaptability of N-22 under adverse climatic conditions. The finding might help in developing more resilient/efficient DSR cultivars suitable for changing global climate.
Keywords
Oryza sativa, Direct-sowing, Adaptability, DNA methylation, Gene × environment interaction
Introduction
Rice (Oryza sativa L.) is a staple food crop for a significant portion of the global population, particularly in the Asian regions. Being a semi-aquatic plant, rice thrives in flooded or saturated soil conditions. Cultivation of rice is widely practiced through transplanting, which requires plenty of water and poses ecological, economic, and social challenges. Flooded rice fields are considered to be the source of methane emission and associated with water losses due to evaporation and percolation [1] resulting in low water-productivity [2] and lesser ecological efficiency/integrity [3]. In addition, transplanted puddled rice (TPR) is responsible for imbalanced dosage of natural resources as well as shortage of labor. To address these issues, one of the potential issues includes development and adoption of direct-sown rice (DSR) cultivars [4,5].
DSR requires less water and labor (12–35%), causes significant reduction in methane gas emissions (10–90%), improves soil physical properties, promotes aeration, and reduces soil compaction. DSR involves less drudgery and production cost with comparable yields to traditional transplanted rice, which make it an attractive/sustainable option for the farmers. However, DSR is susceptible to water-scarcity mainly because the field lacks protection of tilled soil surface (lesser compaction), which enhances water infiltration and reduces retention of water in soil [6]. Higher infestation of nematodes and weeds in DSR fields are some of the main challenges faced by the farmers [7,8]. A variety initially bred for TPR, may not be suitable for direct-sown conditions, as it might result in suboptimal yield (50-60% yield losses). The rice varieties bred for DSR should ideally possess traits to make them well-adapted to fluctuating conditions including early/germination vigor, weed competitiveness, drought tolerance, nematode resistance, etc. [9]. The productivity of DSR can be made comparable to that of TPR by implementing effective weed and nematode management [10]. Replacing TPR with improved DSR would align with the goals of sustainable and resource-efficient agriculture to feed burgeoning global population [11].
Nagina-22 (N-22), a drought and heat-tolerant landrace of aus rice from Assam, India, is one of the rice cultivars suitable for DSR [12]. N-22 is highly tolerant to drought and heat stress, serving as a donor in breeding programmes for drought tolerance [13]. Being a short duration (90–95 days) cultivar, N-22 escapes the negative impacts of extended drought stress, which often occurs at reproductive stage of crop growth. The choice of N-22 for the present study has been a strategic approach based on the knowledge from earlier reports on its performance under abiotic stresses, its usage in the development of stress-tolerant rice varieties [14,15] as well as for studies on DSR traits [12,16]. In contrast, IR-64 (a high yielding rice variety) has been bred for irrigated and transplanted conditions, which is sensitive to drought stress, particularly at reproductive stage [17], and fails to perform well on direct-sowing [16].
While transcriptomic analyses have been successful in identifying candidate genes involved in plant growth and development under stressful conditions [18,19] as well as DSR conditions [12,16], only limited information on epigenomics of rice under DSR conditions has been available. Cytosine (de)methylation in DNA has been a basic epigenetic mechanism [20,21] that plays pivotal roles in regulation of gene expression in response to environmental stresses [22-25]. DNA methylation level is sensitive to environmental cues, and it gets patterned dynamically in response to stress in various crop plants including rice [25-31]. Repetitive exposure to abiotic stress might result in stable epigenetic markers [28] which might form a basis for stress memory allowing plants to be more prepared for subsequent stress events [32,33]. Root system architecture (RSA) plays crucial roles in plant's adaptability to fluctuating environmental conditions/abiotic stresses [16,34,35]. Better understanding of the molecular basis of (epi)genetic plasticity might help breeders in developing varieties better equipped to thrive/perform under changing climatic conditions having enhanced tolerance to abiotic stresses [36].
Materials and Methods
Plant materials and growth conditions
Nagina-22 (a landrace well-performing under direct-sown as well as transplanted conditions) and IR-64 (a high-yielding rice variety suited for transplanted/irrigated conditions) were used for epigenomic and transcriptomic analyses after continuously growing by direct-sowing and transplanting [by sowing seeds directly at a depth of 2 cm in 12" plastic pots containing soil with moisture content of approximately 12% (direct-sowing, having only half of the moisture content in the nursery soil) or by nursery raising, uprooting the seedlings, followed by transplanting in the puddled soil in pots with frequent irrigation] for several generations at the experimental farm of Indian Agricultural Research Institute, New Delhi, India. TPR pots were irrigated on alternate days, particularly in the absence of rainfall using tap water. DSR pots were grown under rain-fed conditions; however, in the absence of rainfall life-saving irrigation was given to the plants/pots. Plant tissue (root and leaf) samples were collected (in Kharif 2021) at the reproductive stage of growth in nine biological replications from the plants grown under TPR and DSR conditions. The tissue samples were immediately frozen in liquid nitrogen and stored in ultradeep freezer (at -80°C) for further molecular analyses.
Assessment of root architecture
To assess architectural changes in root of the rice cultivars grown in Kharif 2021 by direct-sowing and transplanting, roots were cut at the root–shoot junction and separated from the rest of the plant. Roots were properly washed with water to remove any soil/debris adhering to the roots and placed in a tray filled with water (1 cm depth). The roots in the tray were scanned using a desktop scanner with a resolution of 600 dpi (Epson 100XL flatbed scanner). Scanned images were analyzed for different morphological parameters using WinRHIZO Pro software (v2009, Regent Instruments, Montreal, Canada) for quantitative analyses of root traits like length, number, diameter, surface area, etc. to have insights on the adaptability of rice cultivars under different methods of planting.
Assessment of agronomic performance of rice cultivars
The agronomic performance of the rice cultivars was assessed in Kharif 2022 (after seven consecutive years of growing by the same method of planting (direct-sowing or transplanting) to understand the effects of method of planting on yielding potential of the crop. For this, the number of tillers for each plant was recorded at the age of 50 days for N-22 and 60 days for IR-64 (at the same stage of vegetative growth). The number of panicles per plant, test-weight of seeds, and grain yield per pot were analyzed in three replications in Kharif 2021 as well as 2022.
DNA/RNA isolation, library preparation, WGBS/transcriptome sequencing
Genomic DNA isolated from leaf and root tissues of contrasting rice cultivars in three replications, pooled and used for the preparation of whole-genome bisulfite sequencing (WGBS) libraries and sequenced using paired-end 150 bp chemistry at the Illumina Hiseq2500 platform with 25× coverage. The WGBS raw data submitted at NCBI SRA database under Bio-Project ID: SUB11780954. Total RNAs extracted from leaf and root tissues were used to prepare transcriptome libraries and sequenced using PE 150 bp chemistry at Illumina HiSeq 2500 platform. The raw sequence data submitted at NCBI SRA dataset under BioProject ID: SUB9557360 were utilized for transcriptome analysis. Hisat2 and Stringtie package were used to map clean reads on the rice reference genome and expression values were calculated as FKPM (Fragments Per Kilobase of transcript per Million mapped reads).
WGBS read processing, sequence alignment and identification of 5-mc
The adaptor sequences and low-quality reads were trimmed/removed from the raw reads using FastQC toolkit. The high-quality filtered reads for each library were mapped to the rice reference genome using Bismark software on default parameters [37]. Bisulfite conversion efficiency and error-rate was checked by alignment of the reads on rice chloroplast genome. Methylation status at every C in the genome was determined at a P-value cut-off of 0.005. The content of 5-methylcytosine (5-mC) in all the reads at the same cytosine site (methylation level, percentage of reads/cytosines showing methylation) was also determined.
Identification of differentially methylated regions
For screening of differentially methylated regions (DMRs) under direct-sown conditions over transplanting in the rice cultivars, the frequency of mCs was determined in a bin size of 100 bp using CGmap tools. The bins showing ³ 2-fold change in methylation with q-value of <0.05 were considered as DMRs. For annotation of DMRs in the genic territories, GeneDMRs of R package was used [38,39].
Gene ontology analysis
GO enrichment analysis for different categories (biological process, molecular function, and cellular component) of the differentially methylated genes was performed using AgriGO v2 software. Identification of overrepresented GO terms was performed by comparing a query list of genes and the corresponding GO terms with a background list of the query genes. The background genes representing all annotated genes in the rice genome and GO annotations were obtained from the Rice Genome Annotation Project database.
Correlation analysis for DNA methylation and gene expression
A correlation between gene expression and methylation was performed by comparing (de)methylation of the DMR-associated gene with differential expression of the gene. Plotting methylation density on differentially expressed gene (based on Fragments Per Kilobase of transcript per Million values) was performed for correlation analysis. The correlation between differentially expressed gene (p ≤ 0.05, fold change ≥ 2) and the differentially methylated gene (≥ 1.5-fold change, q-value ≤ 0.05) on direct-sowing over transplanting within the rice cultivar was analyzed at three different (CG, CHG, and CHH) sequence contexts by estimating methylation level differences at different parts (promoter, gene-body and intergenic region) of the gene.
RT-qPCR validation of differentially expressed genes
Expression level of selected differentially methylated and differentially expressed genes in leaf as well as root of the contrasting rice cultivars was validated using RT-qPCR analysis. Total RNAs extracted from three biological replicates were used for the expression analysis, as mentioned elsewhere [30,31]. Sequence details of the primers used for the RT-qPCR analysis are provided in (Supplementary Table S1).
Results
Root architectural changes and agronomic performance of rice
Root growth was recorded to be significantly affected on growing by direct-sowing (Figure 1). A significant reduction (~41% decrease in number and length) in roots of rice at reproductive stage was recorded when IR-64 was grown by direct-sowing compared to that recorded on transplanting. However, a minor change in root architecture was observed in the case of N-22 on its growing by direct-sowing or transplanting with the difference being <8.5%. A significant decrease (>48%) in root length was recorded in the case of IR-64 grown by direct-sowing, while the reduction was only ~ 9% in the case of N-22 (Figure 2a). More than 39% reduction in the number of roots was observed for IR-64 on direct-sowing, whereas it was only ~10% in the case of the N-22 (Figure 2b). Similarly, on direct-sowing a considerable decrease in both root surface area (Figure 2c) and root volume (Figure 2d) was observed for the rice cultivars [IR-64 (>50% and >46%, respectively) and N-22 (~20% and ~9%, respectively)].
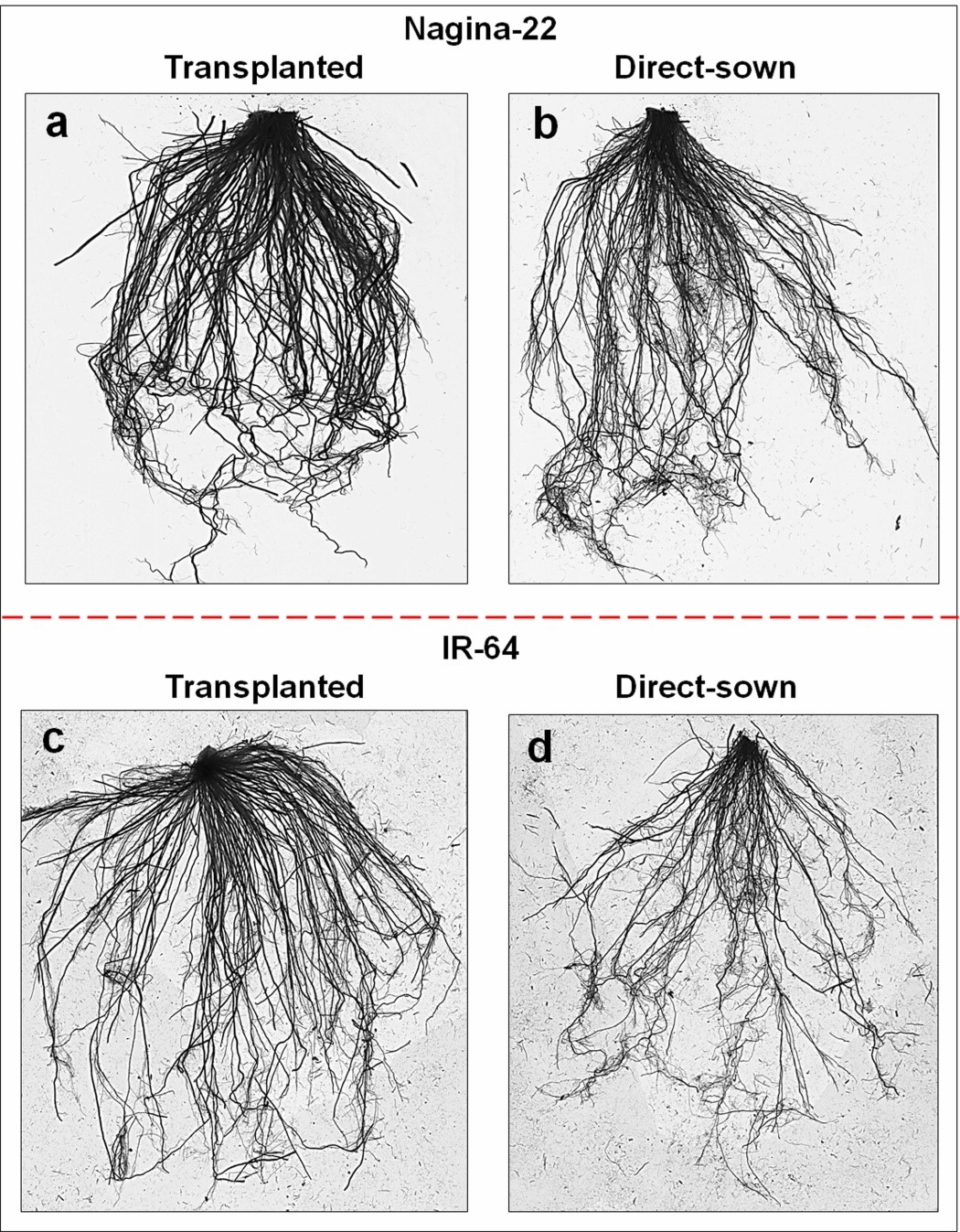
Figure 1. Root architectural changes in rice cultivars showing the effect of different methods of planting (direct-sowing and transplanting) at reproductive stage. (a) Nagina-22 (N-22) grown by transplanting, (b) N-22 grown by direct-sowing, (c) IR-64 grown by transplanting, (d) IR-64 grown by direct-sowing.
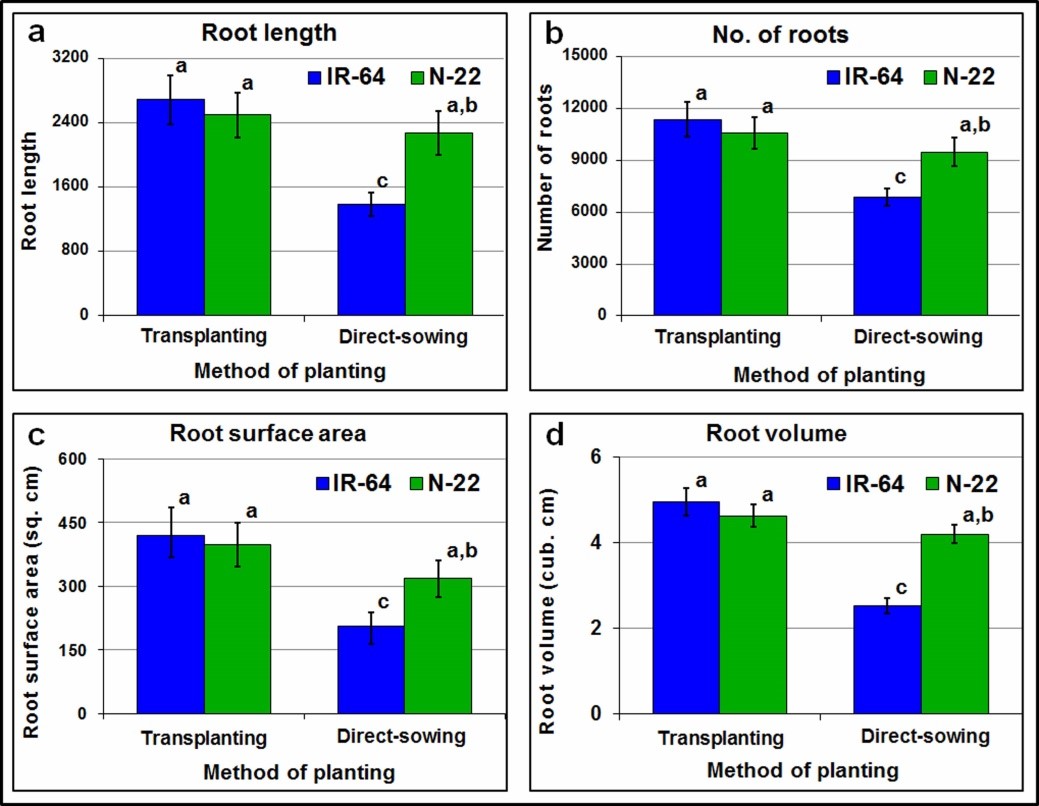
Figure 2. Growth and development of roots in rice (IR-64 and N-22) cultivars grown by different methods of planting (direct-sowing and transplanting). (a) Comparative analysis of root length for the rice cultivars; (b) number of roots for the contrasting rice cultivars; (c) surface area of roots; and (d) root volume for the rice cultivars. The mean value (n= 3) followed by different lower-case letters are significantly different (p <0.05), error bar represents the standard deviation (± SD).
A significant (41%) reduction in the number of tillers was observed in the case of IR-64 on direct-sowing compared to that on transplanting, while it was only 11% in the case of rice cultivar N-22. This indicates that the planting method has a notable impact on growth and development of plants as well as tillering in these rice cultivars. A significant effect of the method of planting was also observed on the number of panicles (Figure 3a) as well as on the overall agronomic performance of these rice cultivars (Figure 3b). While the reduction in the number of tillers bearing panicle was considerably higher (>47%), >18 reduction in test weight of the seeds was observed in case of IR-64 on direct-sowing. Ultimately, these resulted in a significant reduction in the grain yield, which was >60% in the case of IR-64 while only ~24% reduction in the case of N-22 (Figure 3b).
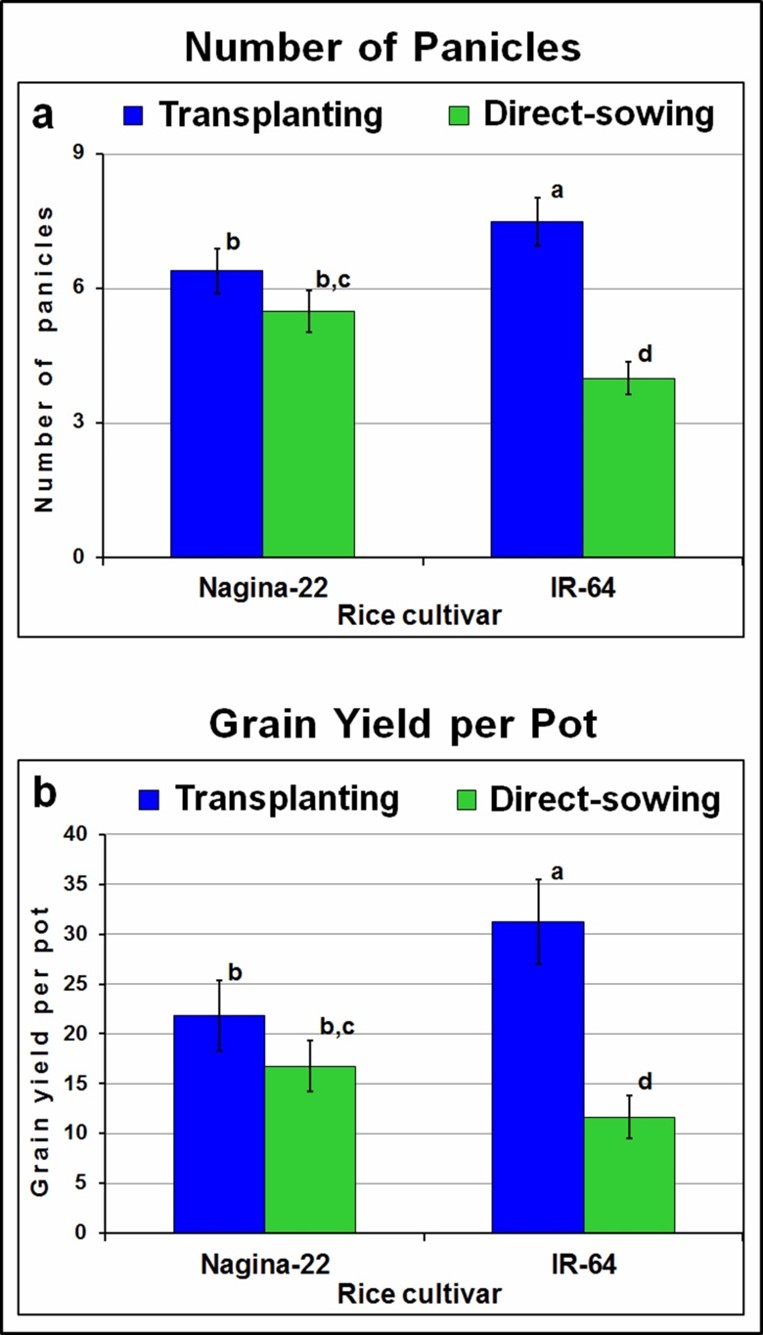
Figure 3. Method of planting affecting agronomic performance of rice cultivars (IR-64 and N-22) grown by direct-sowing and transplanting. (a) Number of panicles per plant; (b) grain yield per pot. Mean (n= 3) followed by different lower-case letters are significantly different (p ≤ 0.05), error bar represents standard deviation (± SD).
Bisulfite conversion and mapping efficiency
Sufficiently higher (>99.5%) bisulfite-conversion efficiency was observed on bisulfite treatment of the DNA for WGBS analysis. On average 38 million clean reads for each sample and the average mapping efficiency rate of 63% was obtained (Supplementary Table S2). A noticeable (~1.5%) increase in 5-mC content was observed in roots on growing the rice cultivars by direct-sowing over transplanting. However, a significant increase in 5-mC content in leaf was observed in the case of N-22 only. The mapping efficiency was observed to be ~10% lesser in case of the root tissue samples in both the rice cultivars. The content of 5-mC was found to vary between 15-17% in the rice genome depending on the cultivar, tissues, and methods of planting.
Changes in methylation landscape on changing the method of planting
Differentially methylated cytosines (DmCs) in different sequence contexts were identified on direct-sowing (treatment) over transplanting (control) of the rice cultivars at each of the cytosine locations. An increase (~3.0%) in methylation in CHH context, while a decrease (~5.5-6.5%) in CG and CHG contexts in the leaf of IR-64 was observed on direct-sowing. On the other hand, in the leaf of N-22 only a minor increase in methylation in the CG context but decrease (~0.5%) in methylation was observed in CHG and CHH contexts on direct sowing. In roots of the rice cultivars, a significant decrease (1.0-5.0%) in methylation was observed in the CG context, while contrasting pattern was observed in the CHG and CHH contexts with hypomethylation in CHG context but hypermethylation in CHH context in N-22 under DSR conditions.
Differential methylation in genomic regions
Genomic regions were classified into four parts, according to the rice genome annotation, including proximal promoter (<1 Kb), distal promoter (>1 Kb), gene-body, and intergenic regions for analyzing DNA methylation patterns. The maximum changes in DNA methylation were observed in the promoter regions of N-22 on direct-sowing compared to that on transplanting. An increase in DNA methylation in the proximal (<1 Kb) promoter in leaf of N-22 (~2.0% in CG, ~1.0% in CHG, and ~3.0% in CHH context) compared to that in the leaf of IR-64 on growing by direct-seeding provided insights into the epigenetic responses of the rice cultivars to the method of planting (Figure 4a). Whereas a decrease in DNA methylation in the distal promoter (~2.0% in CG, ~1.0% in CHG, and ~1.5% in CHH context) in leaf of N-22 was observed. While only a minor change (~0.5% in different context) in DNA methylation in the gene-body in leaf of N-22 on direct-sowing compared to that leaf of IR-64 was observed, significant changes in methylation in the intergenic region of N-22 (particularly in root, Figure 4a) compared to that in IR-64 demonstrated the involvement of DNA methylation. Comparative analysis of DmCs in different parts of the genes on direct-sowing (treatment) and transplanting (control) in the contrasting rice cultivars at the reproductive stage of growth indicated about 75% of the changes in the promoter region, while 18-21% in the intergenic regions and 4-7% in the gene-body (Figure 4).
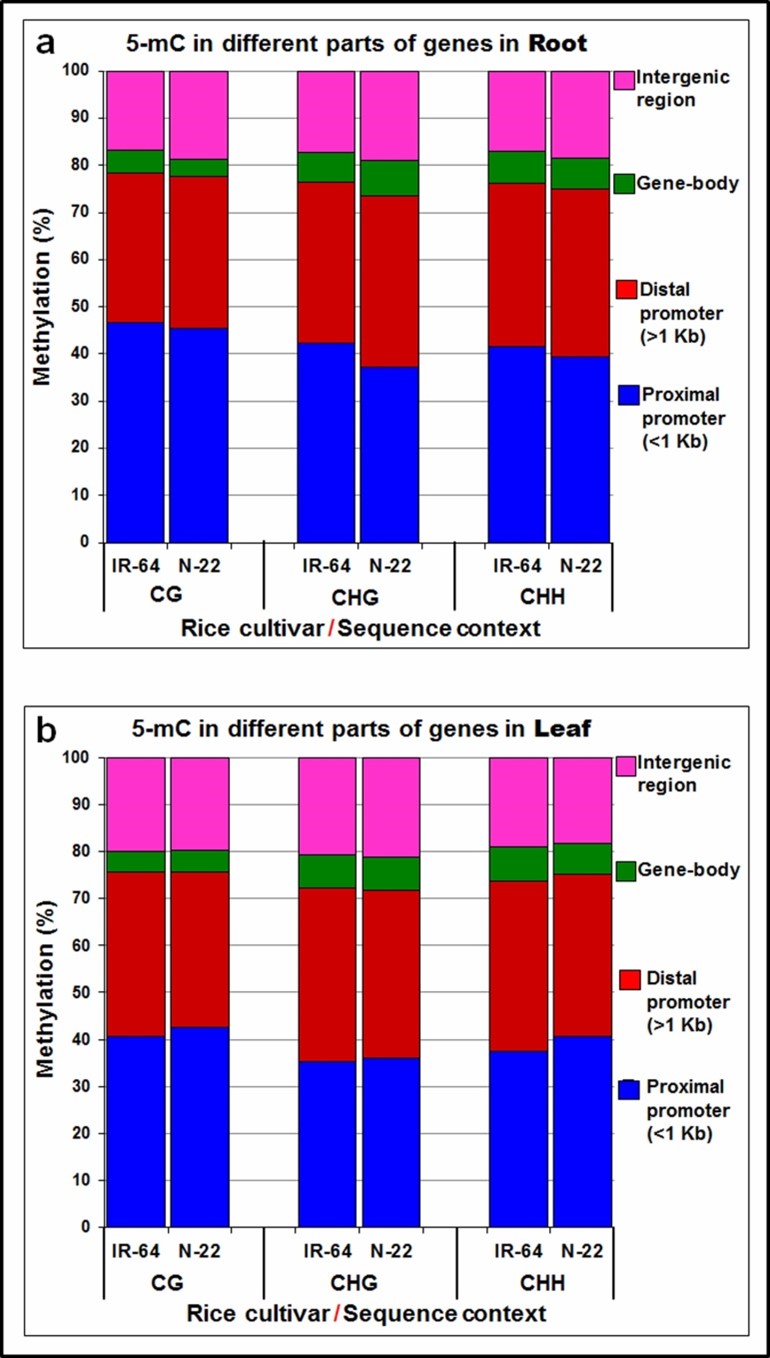
Figure 4. Distribution of 5-methylcytosine (5-mC) in different parts of the gene/sequence context in rice cultivars on direct-sowing (treatment) over transplanting (control) in (a) root and (b) leaf of contrasting rice (IR-64 and Nagina-22) cultivar at reproductive stage of growth.
Comparative analysis revealed more hypomethylated DMRs (13,704) in the leaf of N-22 (Figure 5a), while a higher number of hypermethylated DMRs in leaf (15,739) as well as root (21,506) of IR-64 on direct-sowing. The DMRs were distributed in different sequence contexts with the maximum in CG context. The higher number of DMRs in all three sequence contexts in leaf and root of IR-64 presents more extensive/cultivar-specific epigenetic response to direct-sowing compared to that in N-22 (Figure 5).
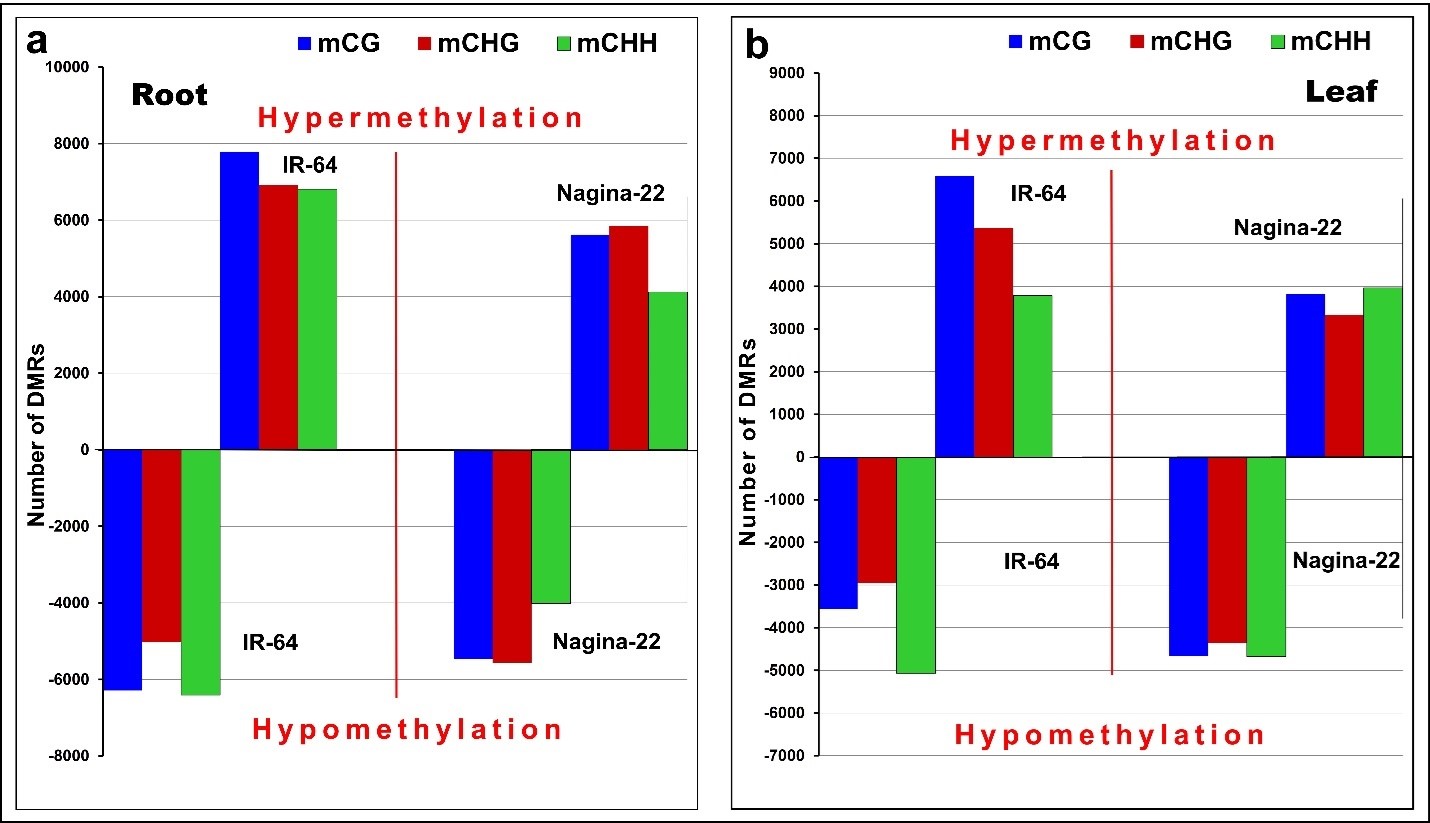
Figure 5. Distribution of differentially methylated regions (DMRs) in different sequence contexts in rice cultivars on direct-sowing over transplanting. (a) Hyper- and hypo-methylation in root; (b) hyper- and hypo-methylation in leaf of the rice (IR-64 and Nagina-22) cultivars. Bins showing difference in methylation at q-value <0.05 and >1.5-fold change in methylation between the methods of planting were considered as DMRs.
About 24,000 genes were differentially methylated in the leaf of N-22 (with ~11,000 genes hypermethylated and ~13,000 genes hypomethylated), while ~27,000 genes were differentially methylated in the leaf of IR-64 under DSR conditions (with ~14,000 hypermethylated and ~13,000 hypomethylated genes). The major difference was observed in CHH context in terms of the number of hypermethylated genes between N-22 and IR-64 in leaves. In roots of N-22, a total of ~27,500 genes were differentially methylated (with ~14,000 genes hypermethylated and ~13,500 genes hypomethylated), while ~34,500 (~19,000 hypermethylated and ~16,000 hypomethylated) genes were differentially methylated in roots of IR-64 under direct-sown conditions.
Gene ontology analysis of differentially methylated gene
GO analysis of differentially methylated (in CG context) genes and its possible effects on gene expression when grown by direct-sowing indicated role of DNA methylation in modulating biological/molecular/cellular processes. Large number of differentially expressed genes (DEGs) for the GO (biological process) terms like ‘regulation of transcription’ and ‘protein/amino acid phosphorylation’ (post-translational modification of protein) were hypomethylated in CG context in roots of IR-64, while these genes were hypermethylated in roots of N-22 under DSR conditions. The GO terms and the associated DEGs for the biological processes like ‘RNA-dependent DNA replication’ and ‘chromatin assembly/disassembly’ were hypomethylated in CHH context in roots of IR-64, while these genes were hypermethylated in the leaf of N-22 on direct-sowing. GO analysis of the differentially methylated genes (DMGs) in the leaf of IR-64 showed hypomethylation in CG context of a large number of genes associated with biological process. On the contrary, the genes were hypermethylated in CG context in leaf of N-22 under DSR conditions. Hypomethylation of the DEGs associated with ‘regulation of transcription’ was observed in CHG context in leaf of IR-64, while hypermethylation of DEGs was observed in CHG context in leaf of N-22 on direct-sowing. Thus, DNA (de)methylation-mediated modulation in expression of genes associated with adaptability of N-22 under fluctuating climatic conditions was observed.
Effect of methylation of promoter on gene expression
Hypomethylation of promoter in CG context in roots was associated with up-regulated expression of ~260 genes in N-22. On the other hand, ~770 hypermethylated promoters showed down-regulated expression of the genes in IR-64. Some of the genes hypermethylated/down-regulated in CHG context in promoter region in leaf of N-22, but hypomethylated in leaf of IR-64, are presented in Figure 6. Hypermethylation of promoter of ~240 genes were associated with down-regulated expression exclusively in roots of N-22. Similarly, in the CHG context hypomethylation of promoters was associated with up-regulated expression of ~270 genes in roots of N-22, compared to ~660 genes hypermethylated at promoter/down-regulated in roots of IR-64. Hypermethylation of promoter in CHG context was associated with down-regulated expression of ~320 genes exclusively in roots of N-22, while hypomethylation of promoter was associated with transcriptional expression of ~220 genes in roots of N-22, compared to ~720 down-regulated genes in roots of IR-64. Hypermethylation of genes caused down-regulated expression, while hypomethylated genes were up-regulated in roots of the contrasting rice genotypes on direct-sowing (Figure 7). Interestingly, the genes hypermethylated/down-regulated in roots of N-22 were hypomethylated/up-regulated in roots of IR-64 and vice versa (Figure 8a).
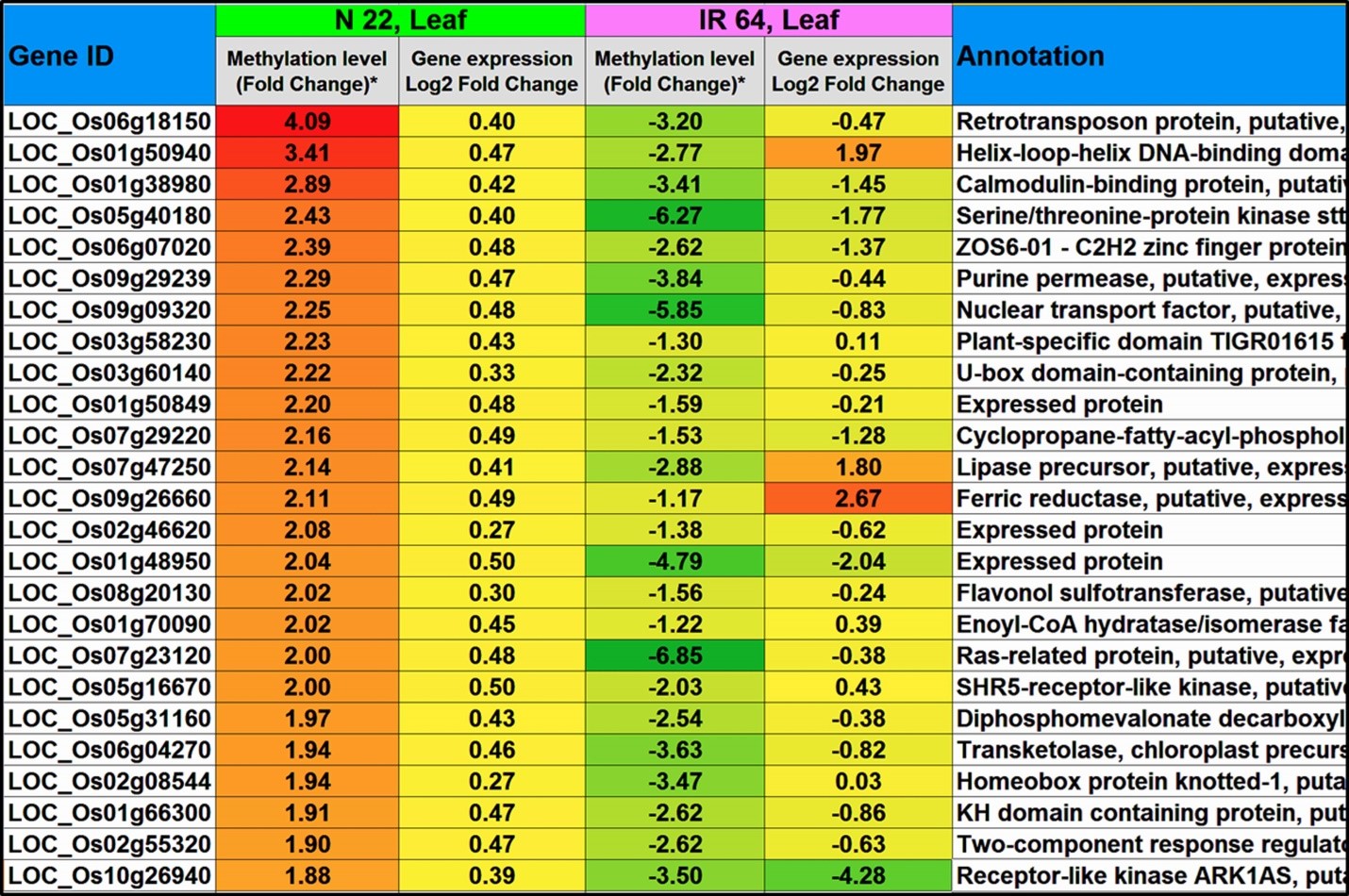
Figure 6. Top 25 genes hypermethylated at promoter in CHG context and down-regulated in leaf of N-22, but hypomethylated and up-regulated in leaf of IR-64 under direct-sown condition over transplanting.
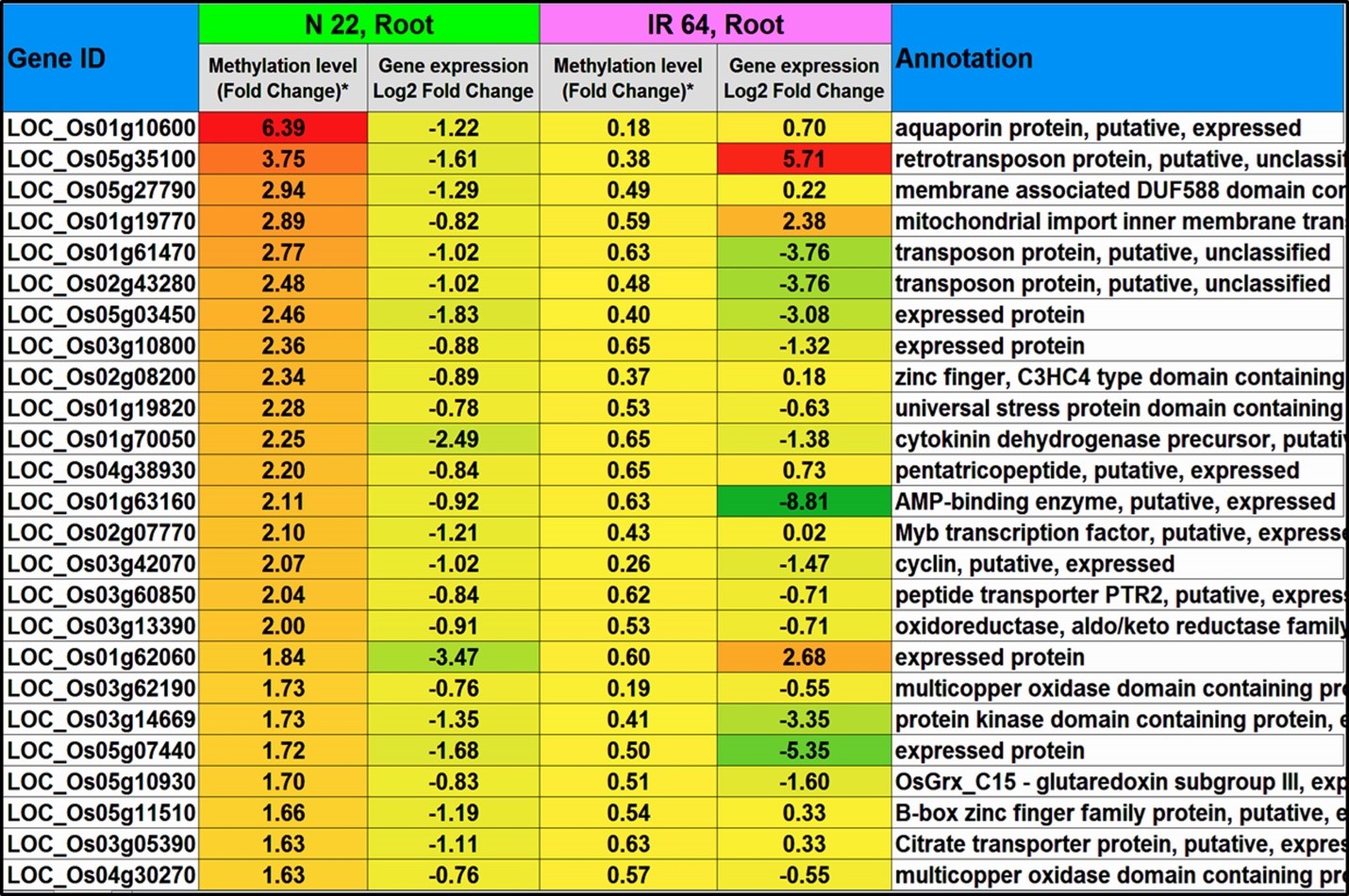
Figure 7. Top 25 genes hypermethylated at promoter in CHH context and down-regulated in roots of N-22, but hypomethylated in roots of IR-64 under direct-sown condition over transplanting.
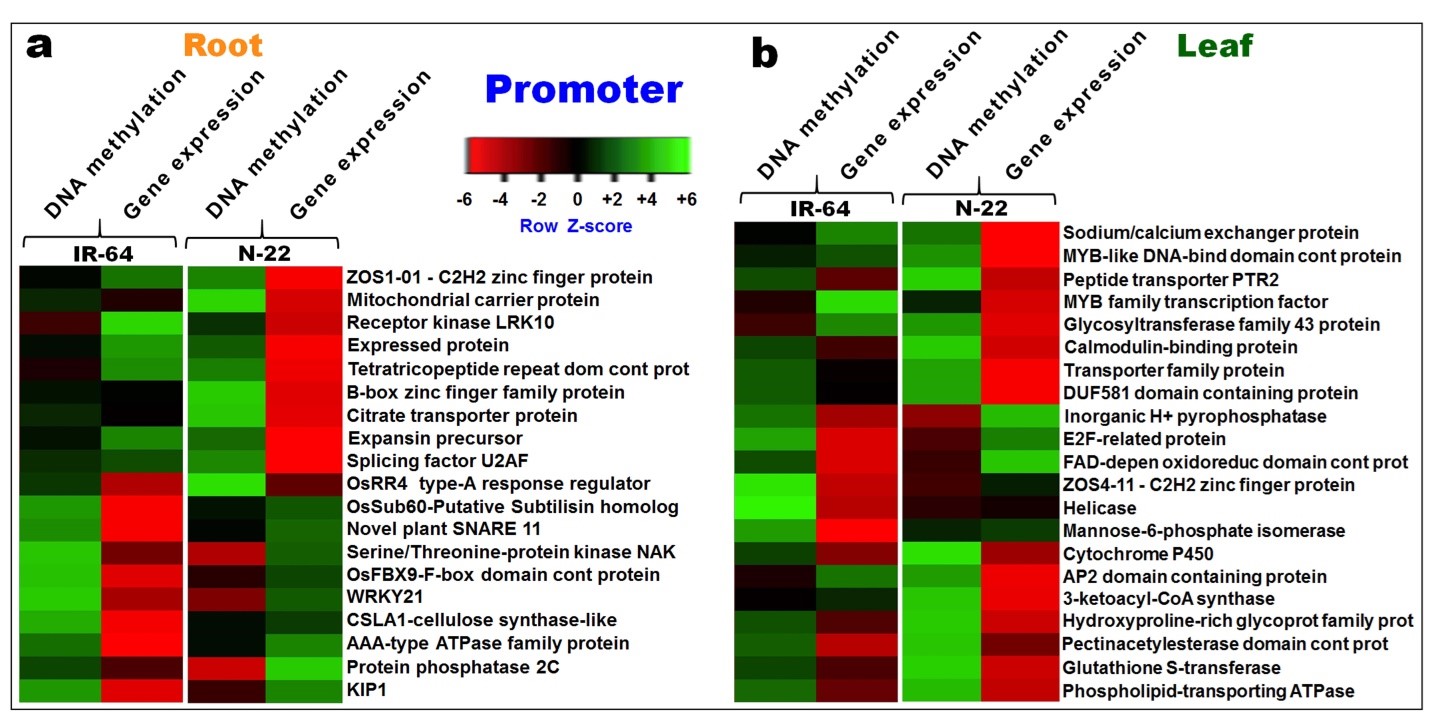
Figure 8. Heat map depicting differential methylation of DNA in CG context at promoter and its effect on gene expression. (a) Root and (b) leaf of contrasting rice (IR-64 and N-22) cultivars on direct-sowing over transplanting.
In leaf, hypomethylation of promoters in CG context was observed to be associated with up-regulated expression of ~710 genes in N-22, while hypermethylation of ~550 promoters resulted in down-regulated expression of the genes in the leaf of IR-64. Among these hypomethylated promoters/genes, ~500 genes were up-regulated exclusively in the leaf of N-22. Among the hypermethylated promoters/genes in the CG context, ~530 genes were down-regulated exclusively in the leaf of N-22. Among the hypomethylated promoters/genes in CHG context, ~450 genes were up-regulated exclusively in the leaf of N-22, while hypermethylation of promoters in the CHG context was associated with down-regulated expression of ~590 genes in the leaf of N-22. About ~310 hypomethylated promoters/genes were associated with up-regulated expression in leaf of IR-64.
Similarly, hypomethylation of promoters/genes in the CHH context was associated with up-regulated expression of ~390 genes in the leaf of N-22, while ~250 hypermethylated promoters/genes showed down-regulated expression in leaf of IR-64. Hypermethylation of promoters in CHH context was observed to be associated with down-regulated expression of ~450 genes in the leaf of N-22, whereas ~410 hypermethylated promoters were associated with transcriptional repression of the genes in the leaf of IR-64. Thus, methylation of promoter caused down-regulated expression of the genes. Hypermethylation of genes caused down-regulated expression, while hypomethylation caused up-regulated expression of the genes in the leaf of N-22 on growing by direct-sowing. Although contrasting patterns of gene expression was observed between the rice cultivars, more efficient regulation of gene expression through DNA methylation was observed in the case of N-22 compared to that in IR-64 (Figure 8b).
Effect of gene-body methylation on expression of gene
Some of the genes showed differential methylation at the gene-body and differential expression in roots of the rice cultivars under direct-sown conditions. The genes showing possible regulation through DNA methylation at the gene-body in CG context included those for phosphatidylinositol transfer, transporter, transcription factor, receptor-like kinase, cyclin, auxin efflux carrier, and ATP binding protein (Figure 9a). Several of the genes were hypomethylated in CG context at the gene-body in the leaf of N-22 but only about 50% of them showed up-regulated expression (Figure 9b). The genes coding for DNA-directed RNA polymerase RPABC1 subunit, RNA recognition motif-containing protein, F-box domain-containing protein. Leucine-rich repeat family protein, cell division cycle protein 23, and pentatricopeptide showed up-regulated/ differential methylation/expression in the leaf of N-22 under DSR conditions (Figure 9b).
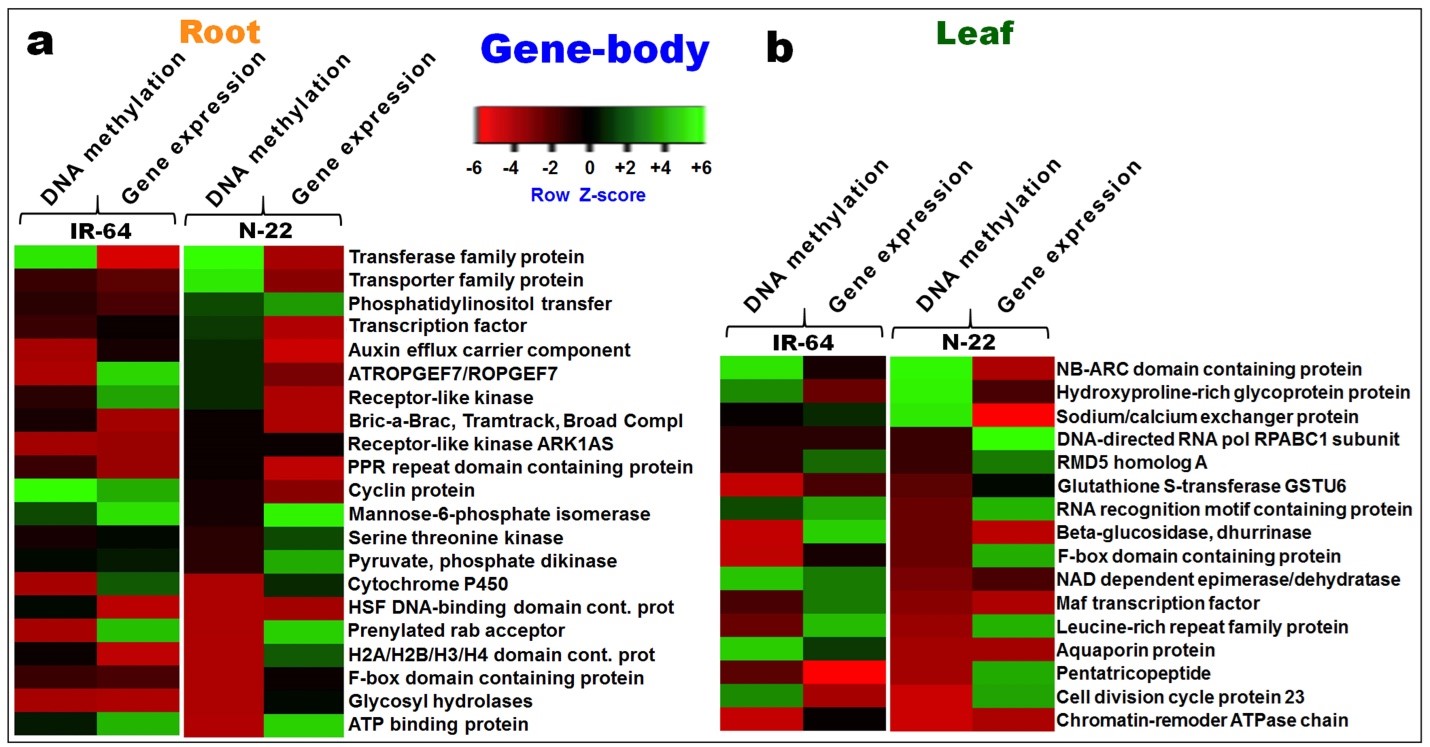
Figure 9. Heat map depicting differential methylation of DNA in CG context at gene-body and its effect on gene expression. (a) Root and (b) leaf of contrasting rice (IR-64 and N-22) cultivars on direct-sowing over transplanting.
Validation of gene expression by RT-qPCR
A set of seven DEGs selected for leaf and root were validated by RT-qPCR for their expression, which showed the expression pattern similar to that observed on transcriptome analysis. This validated trustworthiness of the transcriptome data and the effects of DNA (de)methylation on gene expression (Figure 10).
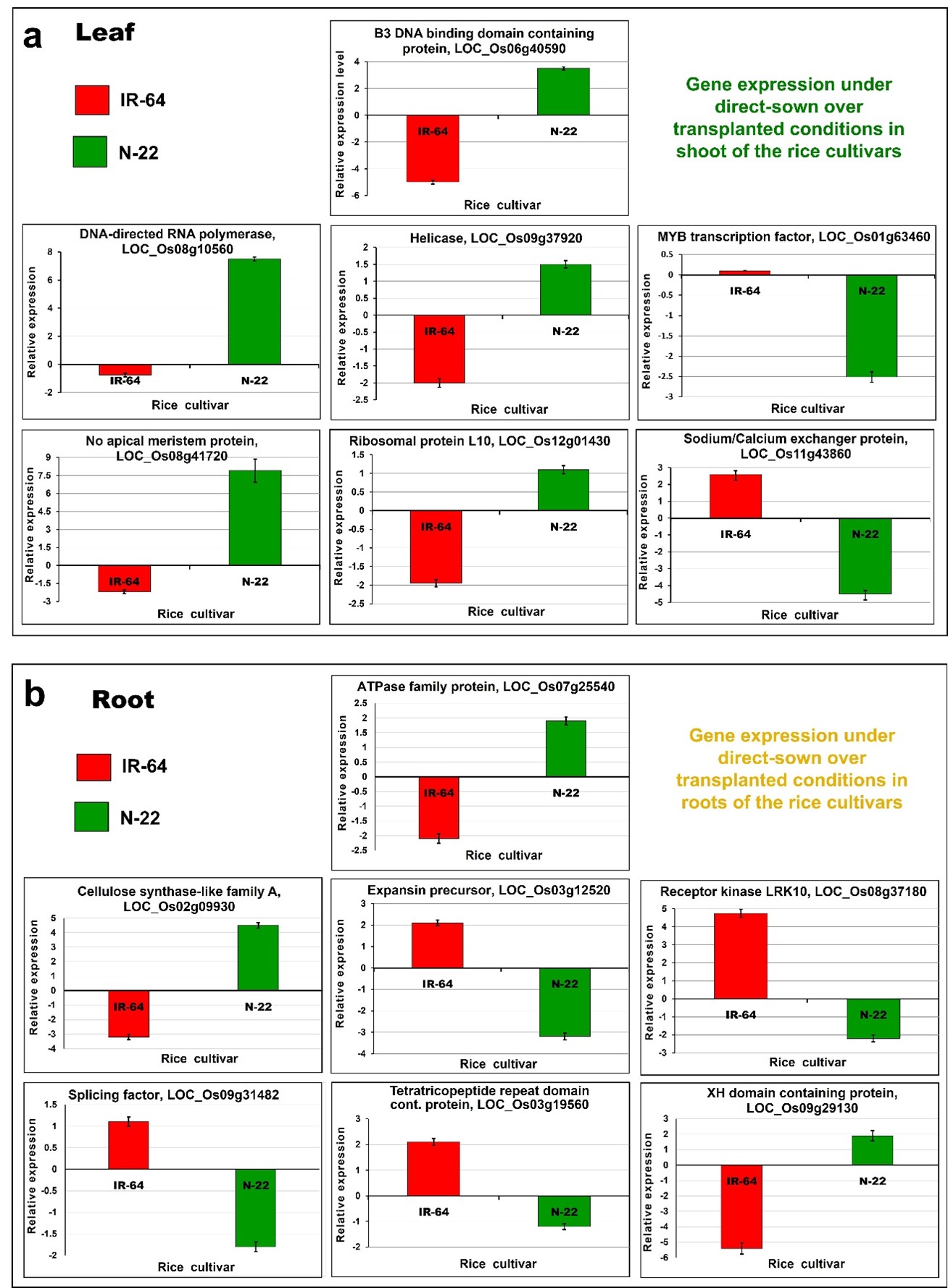
Figure 10. Validation of differential expression of selected genes due to DNA methylation using RT-qPCR analysis. (a) Leaf and (b) root of the contrasting rice [IR-64 and Nagina-22 (N-22)] cultivars grown by direct-sowing and transplanting. Expression indicates the mean value (n=3), and error bar represents standard deviation (± SD).
Discussion
As epigenetic modifications are reported to play important roles in regulating the expression of genes involved in abiotic/biotic stress tolerance [24,40], the study aimed at deciphering the role of DNA (de)methylation in modulating adaptive plasticity of rice, through gene × environment interactions under changing climatic conditions, particularly when grown by direct-sowing. N-22 showcases better adaptive responses in roots with only minor alterations in the parameters of root system architecture (Figure 1). N-22 exhibited better adaptive responses with a significant reduction in RSA parameters, which was comparatively higher in case of IR-64 on direct-sowing (Figure 2).
These findings on changes in RSA are in agreement with an earlier report by Shrestha et al. [17]. N-22 maintains its root parameters without showing significant reduction, which indicates appreciable adaptive plasticity of N-22 (Figures 2). The effects of such adaptive responses of rice cultivars could be observed in terms of their yielding potential. More importantly, our comparative analysis on grain yield of the rice cultivars under DSR conditions indicated >60% lower yield of IR-64 (Figure 3).
While no considerable change in 5-mC content (at whole genome level) was recorded in leaf of the rice cultivars on direct-sowing, an increase in 5-mC content in roots of the rice cultivars indicates global hypermethylation of genome under DSR conditions. A decrease in methylation in CG and CHG contexts (but increase in CHH context) was observed in leaf of IR-64, while the absence of a significant change in methylation in N-22 suggests stable responses under changing conditions on direct-sowing. This confirms the involvement of epigenetic modifications in modulating adaptive plasticity of rice, particularly under stressful conditions, which corroborate with the earlier findings on differential expression of genes in rice grown by different methods of planting [12]. The observed stability in methylation in roots of N-22 and dynamic changes in roots of IR-64 highlights the complexity of epigenetic responses in different rice varieties under specific environmental conditions.
Changes in 5-mC content in different parts of gene highlights major change (increase in methylation in CG and CHG contexts) observed in promoter region, particularly in leaf of N-22 on direct-sowing (Figure 4a). A decrease in methylation in CHG and CHH contexts specifically in the proximal promoter region of genes in roots of N-22 on direct-sowing, suggests that the methylation alterations are responsive to fluctuating climatic conditions on direct-sowing (Figure 4b). These findings on (de)methylation-mediated epigenetic regulation of gene expression are consistent with earlier reports [41-45]. Differential patterns of hyper- and hypomethylated DMRs in roots of IR-64 and N-22, as well as more hypomethylated DMRs in leaves of N-22 on direct-sowing highlight the complexity of gene × environment interactions, tissue-specific responses, and role of epigenetic modifications in adaptation of rice to changing environmental conditions (Figure 5).
Hypermethylation at promoter, specifically in CG and CHG contexts, was observed in the genes associated with the GO terms 'protein amino acid phosphorylation' and 'regulation of transcription' in both leaf and root tissues of N-22 when it is grown by direct-sowing. The genes associated with the GO terms 'RNA-dependent DNA replication' and 'chromatin assembly/disassembly' highlight tissue-specific epigenetic regulation in leaf of N-22 on direct-sowing. In roots of N-22, the genes associated with the GO terms did not exhibit any change in methylation level. However, a contrasting result was observed in roots of IR-64 where the genes for the mentioned GO terms were hypomethylated in CHH on direct-sowing. Tissue-specific modulation of DNA methylation and its impact on gene expression are being documented [46]. Tissue-specific modulation of DNA methylation in response to developmental stages and environmental conditions is a well-established and significant aspect of plant biology as reported earlier [25,47,48].
On direct-sowing, more genes tend to get hypermethylated (leading to down-regulation) in leaves of IR-64, while more genes tend to get hypomethylated in leaves of N-22 (resulting in up-regulation of the gene) (Figure 6). In roots of N-22, a smaller number of genes were regulated through DNA methylation compared to that in leaves. Moreover, among the epigenetically regulated genes in roots, notable hypermethylation in CHH context resulted in down-regulation of gene (Figure 7). Maximum differential methylation in promoter region indicates that (de)methylation of promoter plays pivotal role in epigenetic regulation of gene highlighting the importance of epigenetic mechanisms in N-22 on its ability to changing environmental conditions. The higher number of hypermethylated DMRs in CHH context in the leaf of N-22 under DSR conditions, compared to IR-64 (Figure 5), indicates the involvement of the RNA-directed DNA methylation pathway (RdDM) which is reported to be associated with de novo DNA methylation in silencing or switching-on the genes in response to stressful conditions [25,27,29]. This suggests that epigenetic regulation, particularly through DNA methylation, in leaves contributes significantly to adaptability of N-22 on direct-sowing [16].
The changes in DNA methylation were predominantly observed in CG context in the genic region. Sodium/calcium exchanger (LOC_Os02g21009) appeared to be hypermethylated at the coding region. The sodium/calcium exchanger gene was hypermethylated and down-regulated in leaves of N-22 whereas it was less methylated and expressed in leaves of IR-64 on direct-sowing (Figure 9). Differential methylation of sodium/calcium exchanger gene at gene-body in N-22 and IR-64 suggest potential correlation between DNA methylation and gene expression in response to environmental cues. In a similar manner, about 15 other important genes (playing role in stress response pathways) exhibiting differential methylation at gene-body (Figure 9), suggest that epigenetic regulation is an important regulatory mechanism under stressful conditions. Different effects of CG methylation at gene-body on the expression of gene in roots of contrasting rice cultivars suggest a complex interplay between epigenetic modifications and gene regulation (Figure 9). Methylation at different genomic regions can lead to distinct outcomes in terms of gene expression [49], which might play a role in adaptability of organism under changing environmental conditions [50]. The positive as well as negative correlation observed between DNA methylation (especially gene-body methylation) and gene expression in the present study align with the earlier reports [28,38,39,51-55]. The observed correspondence between methylation of promoter and gene expression level (up-/down-regulated) in N-22 adds depth to our understanding on epigenetic regulation of gene expression under stress. Lack of such correlation between promoter methylation and gene expression in IR-64 suggests that such functional relationship might be highly genotype-specific. Also, the differential expression of genes related to growth regulation, nutrient reservoir activities, translational machinery, carbohydrate metabolic processes, proteolytic activities, and chromatin assembly/epigenetic modifications highlights multilayered nature of gene regulation [12]. Differential methylation/expression of genes associated with histone modifications, noncoding RNAs, and chromatin architecture have been reported to play significant roles in shaping the expression patterns of genes [56-58]. The adaptability of plants under changing climatic conditions is a complex process shaped by dynamic interplay between genetic factors, environmental signals, and regulatory epigenetic modifications.
Conclusions
Dynamics of gene × environment interaction, appearing as epigenetic switching on/off of genes, is a potential mechanism influencing the fitness of rice under fluctuating environmental conditions. This study represents a pioneering report providing new insights into epigenetic regulation of gene expression and its impact on adaptive plasticity of rice under changing climatic conditions under DSR conditions. There is need for additional investigations to identify candidate genes, pathways, and ultimately, epigenetic markers associated with adaptive plasticity of rice under varying environmental conditions [59]. The prospect of utilizing these epimarkers for epigenome editing to improve adaptability of rice to changing climatic conditions highlights the potential/practical implications of the research [60,61], which might be useful in improving DSR cultivars with minimized biosafety issues [62] making them more adaptable, water-productive, and ecologically efficient, which is the need of the day for climate-resilient agriculture.
Acknowledgements
KS gratefully acknowledges the financial support (Research Associate) from Extramural Research grant [18(3)/2018-O&P], Indian Council of Agricultural Research, Govt. of India, New Delhi, India.
Funding
The research work was carried out with the funding from the National Agricultural Science Fund (NASF/ABP-70161/2018–19) as well as the financial support from Extramural Research Grant [NASF/EMR(OG)/06/2021], Indian Council of Agricultural Research, Govt. of India, New Delhi, India.
Ethics Statement
Not applicable.
Data Availability Statement
Raw sequence data for whole genome bisulfite sequencing (SUB11780954) and RNA-sequencing (SUB9557360) utilized in the present study are available at NCBI database (https://www.ncbi.nlm.nih.gov/sra).
Author Contribution
SK and TM conceived and supervised the experiment. KS and AG carried out the experiments, performed analyses of data, and prepared the initial draft manuscript. SK and TM revised the manuscript. All the authors read and approved the final manuscript for its publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Tuong TP, BAM B, Mortimer M. More rice, less water—integrated approaches for increasing water productivity in irrigated Rice-Based systems in Asia—. Plant Production Science. 2005;8(3):231-41.
3. Kumar S. Saving water for ecological integrity: Agricultural perspective of Per drop more crop. Int. J. Environ. Sci. Nat. Res. 2021;28:1-7.
4. Tyagi A, Kumar S, Mohapatra T. Biochemical, physiological and molecular responses of rice to terminal drought stress: Transcriptome profiling of leaf and root reveals the key stress-responsive genes. J. Plant Biochem. Biotechnol. 2023;33.
5. Tabbal DF, Bouman BA, Bhuiyan SI, Sibayan EB, Sattar MA. On-farm strategies for reducing water input in irrigated rice; case studies in the Philippines. Agricultural Water Management. 2002 Jul 30;56(2):93-112.
6. Liu H, Hussain S, Zheng M, Sun L, Fahad S, Huang J, et al. Progress and constraints of dry direct-seeded rice in China. J. Food Agric. Environ. 2014 Apr;12(2):465-72.
7. Azmi M, Chin DV, Vongsaroj P, Johnson DE. Emerging issues in weed management of direct-seeded rice in Malaysia, Vietnam and Thailand. In: Rice is life: Scientific perspectives for the 21stcentury. Proc World Rice Res Conf Tsukuba, Japan. 2004, 196-7.
8. Rao AN, Johnson DE, Sivaprasad B, Ladha JK, Mortimer AM. Weed management in direct-seeded rice. Advances in agronomy. 2007 Jan 1;93:153-255.
9. Sagare DB, Abbai R, Jain A, Jayadevappa PK, Dixit S, Singh AK, et al. More and more of less and less: Is genomics-based breeding of dry direct-seeded rice (DDSR) varieties the need of hour? Plant Biotechnol J. 2020 Nov;18(11):2173-86.
10. Liu H, Hussain S, Zheng M, Peng S, Huang J, Cui K, Nie L. Dry direct-seeded rice as an alternative to transplanted-flooded rice in Central China. Agron. Sustain. Dev. 2015 Jan;35:285-94.
11. Kumar S. The role of biopesticides in sustainably feeding the nine billion global populations. J Biofertil Biopestici. 2013;4:e114.
12. Kumar S, Agrawal A, Seem K, Kumar S, Vinod KK, Mohapatra T. Transcriptome analysis of a near-isogenic line and its recurrent parent reveals the role of Pup1 QTL in phosphorus deficiency tolerance of rice at tillering stage. Plant Mol Biol. 2022 May;109(1-2):29-50.
13. Reddy ChS, Babu AP, Swamy BP, Kaladhar K, Sarla N. ISSR markers based on GA and AG repeats reveal genetic relationship among rice varieties tolerant to drought, flood, or salinity. J Zhejiang Univ Sci B. 2009 Feb;10(2):133-41.
14. Vikram P, Swamy BP, Dixit S, Ahmed HU, Teresa Sta Cruz M, Singh AK,et al. qDTY₁.₁, a major QTL for rice grain yield under reproductive-stage drought stress with a consistent effect in multiple elite genetic backgrounds. BMC Genet. 2011 Oct 18;12:89.
15. Casartelli A, Riewe D, Hubberten HM, Altmann T, Hoefgen R, Heuer S. Exploring traditional aus-type rice for metabolites conferring drought tolerance. Rice (N Y). 2018 Jan 25;11(1):9.
16. Kumar S, Kumar S, Krishnan GS, Mohapatra T. Molecular basis of genetic plasticity to varying environmental conditions on growing rice by dry/direct-sowing and exposure to drought stress: Insights for DSR varietal development. Front Plant Sci. 2022 Oct 24;13:1013207.
17. Shrestha R, Al-Shugeairy Z, Al-Ogaidi F, Munasinghe M, Radermacher M, Vandenhirtz J, et al. Comparing simple root phenotyping methods on a core set of rice genotypes. Plant Biol (Stuttg). 2014 May;16(3):632-42.
18. Lenka SK, Katiyar A, Chinnusamy V, Bansal KC. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol J. 2011 Apr;9(3):315-27.
19. Sandhu N, Subedi SR, Singh VK, Sinha P, Kumar S, Singh SP, et al. Deciphering the genetic basis of root morphology, nutrient uptake, yield, and yield-related traits in rice under dry direct-seeded cultivation systems. Sci Rep. 2019 Jun 27;9(1):9334.
20. Wang X, Li Q, Yuan W, Cao Z, Qi B, Kumar S, et al. The cytosolic Fe-S cluster assembly component MET18 is required for the full enzymatic activity of ROS1 in active DNA demethylation. Sci Rep. 2016 May 19;6:26443.
21. Li Y, Kumar S, Qian W. Active DNA demethylation: mechanism and role in plant development. Plant Cell Rep. 2018 Jan;37(1):77-85.
22. Zhang M, Xu C, von Wettstein D, Liu B. Tissue-specific differences in cytosine methylation and their association with differential gene expression in sorghum. Plant Physiol. 2011 Aug;156(4):1955-66.
23. Kumar S, Singh A. Epigenetic regulation of abiotic stress tolerance in plants. Advances in Plants and Agriculture Research. 2016;5:517-521.
24. Kumar S, Chinnusamy V, Mohapatra T. Epigenetics of Modified DNA Bases: 5-Methylcytosine and Beyond. Front Genet. 2018 Dec 18;9:640.
25. Kumar S, Seem K, Kumar S, Mohapatra T. RNA-seq analysis reveals the genes/pathways responsible for genetic plasticity of rice to varying environmental conditions on direct-sowing and transplanting. Sci Rep. 2022 Feb 10;12(1):2241.
26. Wada Y, Miyamoto K, Kusano T, Sano H. Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Mol Genet Genomics. 2004 Jul;271(6):658-66.
27. Secco D, Wang C, Shou H, Schultz MD, Chiarenza S, Nussaume L, et al. Stress induced gene expression drives transient DNA methylation changes at adjacent repetitive elements. Elife. 2015 Jul 21;4:e09343.
28. Zheng X, Chen L, Xia H, Wei H, Lou Q, Li M, et al. Transgenerational epimutations induced by multi-generation drought imposition mediate rice plant's adaptation to drought condition. Sci Rep. 2017 Jan 4;7:39843.
29. Chu S, Zhang X, Yu K, Lv L, Sun C, Liu X, et al. Genome-Wide Analysis Reveals Dynamic Epigenomic Differences in Soybean Response to Low-Phosphorus Stress. Int J Mol Sci. 2020 Sep 17;21(18):6817.
30. Kumar S, Seem K, Kumar S, Vinod KK, Chinnusamy V, Mohapatra T. Pup1 QTL Regulates Gene Expression Through Epigenetic Modification of DNA Under Phosphate Starvation Stress in Rice. Front Plant Sci. 2022 May 31;13:871890.
31. Kumar S, Seem K, Kumar S, Singh A, Krishnan SG, Mohapatra T. DNA methylome analysis provides insights into gene regulatory mechanism for better performance of rice under fluctuating environmental conditions: epigenomics of adaptive plasticity. Planta. 2023 Nov 22;259(1):4.
32. Schulze WX, Altenbuchinger M, He M, Kränzlein M, Zörb C. Proteome profiling of repeated drought stress reveals genotype-specific responses and memory effects in maize. Plant Physiol Biochem. 2021 Feb;159:67-79.
33. Kumar S, Seem K, Mohapatra T. Biochemical and Epigenetic Modulations under Drought: Remembering the Stress Tolerance Mechanism in Rice. Life (Basel). 2023 May 10;13(5):1156.
34. Smith S, De Smet I. Root system architecture: insights from Arabidopsis and cereal crops. Philos Trans R Soc Lond B Biol Sci. 2012 Jun 5;367(1595):1441-52.
35. Koevoets IT, Venema JH, Elzenga JT, Testerink C. Roots Withstanding their Environment: Exploiting Root System Architecture Responses to Abiotic Stress to Improve Crop Tolerance. Front Plant Sci. 2016 Aug 31;7:1335.
36. Karlova R, Boer D, Hayes S, Testerink C. Root plasticity under abiotic stress. Plant Physiol. 2021 Nov 3;187(3):1057-70.
37. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011 Jun 1;27(11):1571-2.
38. Wang L, Cao S, Wang P, Lu K, Song Q, Zhao FJ, Chen ZJ. DNA hypomethylation in tetraploid rice potentiates stress-responsive gene expression for salt tolerance. Proc Natl Acad Sci U S A. 2021 Mar 30;118(13):e2023981118.
39. Wang X, Hao D, Kadarmideen HN. GeneDMRs: An R Package for Gene-Based Differentially Methylated Regions Analysis. J Comput Biol. 2021 Mar;28(3):304-16.
40. Kumar S, Mohapatra T. Dynamics of DNA Methylation and Its Functions in Plant Growth and Development. Front Plant Sci. 2021 May 21;12:596236.
41. Borgel J, Guibert S, Li Y, Chiba H, Schübeler D, Sasaki H, et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet. 2010 Dec;42(12):1093-100.
42. Hashimoto K, Otero M, Imagawa K, de Andrés MC, Coico JM, Roach HI, Oreffo ROC, Marcu KB, Goldring MB. Regulated transcription of human matrix metalloproteinase 13 (MMP13) and interleukin-1β (IL1B) genes in chondrocytes depends on methylation of specific proximal promoter CpG sites. J Biol Chem. 2013 Apr 5;288(14):10061-72.
43. Bhatia H, Khemka N, Jain M, Garg R. Genome-wide bisulphite-sequencing reveals organ-specific methylation patterns in chickpea. Sci Rep. 2018 Jun 26;8(1):9704.
44. Kang JG, Park JS, Ko JH, Kim YS. Regulation of gene expression by altered promoter methylation using a CRISPR/Cas9-mediated epigenetic editing system. Sci Rep. 2019 Aug 19;9(1):11960.
45. Grzybkowska D, Nowak K, Gaj MD. Hypermethylation of Auxin-Responsive Motifs in the Promoters of the Transcription Factor Genes Accompanies the Somatic Embryogenesis Induction in Arabidopsis. Int J Mol Sci. 2020 Sep 18;21(18):6849.
46. Gutierrez-Arcelus M, Ongen H, Lappalainen T, Montgomery SB, Buil A, Yurovsky A, et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015 Jan 29;11(1):e1004958.
47. Wang P, Xia H, Zhang Y, Zhao S, Zhao C, Hou L, et al. Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across embryo and endosperm in Maize (Zea may). BMC Genomics. 2015 Jan 23;16(1):21.
48. Yong-Villalobos L, González-Morales SI, Wrobel K, Gutiérrez-Alanis D, Cervantes-Peréz SA, Hayano-Kanashiro C, et al. Methylome analysis reveals an important role for epigenetic changes in the regulation of the Arabidopsis response to phosphate starvation. Proc Natl Acad Sci U S A. 2015 Dec 29;112(52):E7293-302.
49. Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, et al. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics. 2011 May 11;12(1):231.
50. Dubin MJ, Zhang P, Meng D, Remigereau MS, Osborne EJ, Paolo Casale F, et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife. 2015 May 5;4:e05255.
51. Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007 Jan;39(1):61-9.
52. Li X, Zhu J, Hu F, Ge S, Ye M, Xiang H, et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics. 2012 Jul 2;13:300.
53. Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, et al. CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013 Apr;23(4):628-37.
54. Vining KJ, Romanel E, Jones RC, Klocko A, Alves-Ferreira M, Hefer CA, et al. The floral transcriptome of Eucalyptus grandis. New Phytol. 2015 Jun;206(4):1406-22.
55. Anastasiadi D, Esteve-Codina A, Piferrer F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics & Chromatin. 2018 Jun 29;11(1):37.
56. Hu G, Huang B, Wang K, Frasse P, Maza E, Djari A, et al. Histone posttranslational modifications rather than DNA methylation underlie gene reprogramming in pollination-dependent and pollination-independent fruit set in tomato. New Phytol. 2021 Jan;229(2):902-19.
57. Jabre I, Chaudhary S, Guo W, Kalyna M, Reddy ASN, Chen W, et al. Differential nucleosome occupancy modulates alternative splicing in Arabidopsis thaliana. New Phytol. 2021 Feb;229(4):1937-45.
58. Kumar S, Kaur S, Seem K, Kumar S, Mohapatra T. Understanding 3D Genome Organization and Its Effect on Transcriptional Gene Regulation Under Environmental Stress in Plant: A Chromatin Perspective. Front Cell Dev Biol. 2021 Dec 8;9:774719.
59. Kumar S, Mohapatra T. Deciphering Epitranscriptome: Modification of mRNA Bases Provides a New Perspective for Post-transcriptional Regulation of Gene Expression. Front Cell Dev Biol. 2021 Mar 16;9:628415.
60. Kumar S. Genome editing to epigenome editing: Towards unravelling the enigmas in developmental biology. Trends Dev. Biol. 2019:31-8.
61. Seem K, Kaur S, Kumar S, Mohapatra T. Epigenome editing for targeted DNA (de)methylation: a new perspective in modulating gene expression. Crit Rev Biochem Mol Biol. 2024 Mar 5:1-30.
62. Kumar S. Biosafety issues of genetically modified organisms. Biosafety. 2014;3(2): e150.