Abstract
Polycystic kidney disease is an inherited degenerative disease in which the uriniferous tubules are replaced by expanding fluid-filled cysts that ultimately destroy organ function. Autosomal dominant polycystic kidney disease (ADPKD) is the most common form, afflicting approximately 1 in 1,000 people and is caused by mutations in the transmembrane proteins polycystin-1 (Pkd1) and polycystin-2 (Pkd2). The mechanisms by which polycystin mutations induce cyst formation are not well understood, however pro-proliferative signaling must be involved for tubule epithelial cell number to increase over time. We recently found that the stress-activated mitogen-activated protein kinase (MAPK) pathway c-Jun N-terminal kinase (JNK) pathway is activated in cystic disease and genetically removing JNK reduces cyst growth driven by a loss of Pkd2. This review covers the current state of knowledge of signaling in ADPKD with an emphasis on the JNK pathway.
Keywords
Polycystic kidney disease, Jun N Terminal kinase, Polycystin-1, Polycystin-2, Cilia, Mus musculus, Mitogen-activated protein kinase signaling
Abbreviations
ADPKD: Autosomal Dominant Polycystic Kidney Disease; AP-1: Activator Protein-1; cAMP: Cyclic Adenosine Monophosphate; CDCA: Cilia-Dependent Cyst-Activating; CFTR: Cystic Fibrosis Transmembrane conductance Regulator; ESRD: End Stage Renal Disease; GPCR: G-protein Coupled Receptor; HEK-293T: Human Embryonic Kidney 293T; JNK: Jun N-terminal Kinase; MAP: Mitogen-Activated Protein; MAPK: Mitogen-Activated Protein Kinase; MAP2K: Mitogen-Activated Protein Kinase Kinase; MAP3K: Mitogen-Activated Protein Kinase Kinase Kinase; MDCK: Madin-Darby Canine Kidney; PKD: Polycystic Kidney Disease
Overview and Prevalence of Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD) is a common inherited disorder characterized by slow-growing, fluid-filled cysts in both kidneys. Liver and pancreatic cysts as well as cardiac and vascular abnormalities variably occur [1,2]. ADPKD symptoms include hypertension, kidney pain, hematuria, cyst infection, and urinary tract infections [1]. Over time, deteriorating kidney function leads to end stage renal disease (ESRD). While most ADPKD patients progress to ESRD by age 70, many reach it at younger ages [3]. Despite recent advances in therapy, ADPKD remains incurable, and for most patients treatment is limited to symptom management [4]. For patients with ESRD, renal replacement therapy in the form of dialysis or transplant is inevitable. Unfortunately, renal transplantation does not cure ESRD and patients commonly experience acute rejection, cardiovascular diseases, infection, and malignancy [5].
ADPKD is the most common inherited kidney disease, afflicting approximately 1 in 1,000 people in the US. Prevalence was originally estimated by postmortem kidney cyst detection [6,7] but is supported by recent large-scale population sequencing where 0.93 pathogenic mutations were found per 1,000 people [8]. In contrast, healthcare databases report 0.43 ADPKD cases per 1,000 people in the US [9,10]. This suggests significant underdiagnosis likely due to late onset and variable clinical presentation. Nevertheless, as the fourth leading cause of ESRD in the US, ADPKD places a heavy burden on individuals and healthcare systems alike [11].
Polycystin-1 and Polycystin-2 Mutations are the Major Cause of PKD
Genetic mutations in either polycystin-1 (Pkd1) or polycystin-2 (Pkd2), comprise most ADPKD cases. Pkd1 mutations make up 78% of cases, Pkd2 mutations make up 15%. The remaining cases are due to unidentified or rare mutations [12], including DNAJB11 [13], GANAB [14], ALG9 [15], and IFT140 [16]. Pkd1 and Pkd2 phenotypes are qualitatively similar, but Pkd1 mutations typically cause more severe disease with Pkd1 patients progressing to ESRD twenty years earlier than Pkd2 patients [17-19].
Pkd1 is located on chromosome 16p13.3 within a region of highly homologous repeats [20,21] where it encodes a large integral membrane protein of 4,304 amino acids. The N-terminal extracellular domain contains multiple domains with putative roles in cell adhesion, followed by 11 transmembrane passes and a cytoplasmic C-terminus [20]. Pkd1 is widely expressed in different tissues, with higher levels during development than adulthood [22]. It localizes to the plasma membrane and is concentrated in primary cilia, antennae-like sensory organelles that project from the apical cell membrane [23-25]. Pkd1’s physiological contexts remains obscure although it has been hypothesized to function as an atypical adhesion G protein-coupled receptor (GPCR) [26] or a Wnt ligand receptor [27].
Pkd2 maps to chromosome 4q22.1 [28,29]. The protein, which is a member of the Trp channel family, consists of 968 amino acids with intracellular N- and C-terminal regions and 6 transmembrane passes. Pkd2 is ubiquitously expressed and localizes to the plasma membrane, ciliary membrane, and endoplasmic reticulum [25,30,31]. Pkd2 is a calciumpermeable non-selective cation channel [32-34] with activity in the cilium [35,36].
Pkd1 complexes with Pkd2 [37-41] as a heterotetrameric complex consisting of three Pkd2 molecules and one Pkd1 molecule. Oligomerization appears to depend on the extracellular polycystin domains shared by both molecules [42], although earlier studies found that Pkd1 and Pkd2 interact via intracellular C-terminal coiled-coiled domains [37]. The ciliary polycystin complex maintains tubule architecture through an undefined mechanism. Leading theories include fluid flow sensation [43] and ligand detection in the filtrate [27].
To a limited extent, genotype predicts the clinical course of ADPKD. At a particular age, Pkd1 patients have greater total kidney volume than Pkd2 patients, indicating greater cystic load. However, cyst growth rate over time is the same in Pkd1 and Pkd2 patient populations, suggesting that the difference in disease progression is due to cyst initiation [44]. More than 2,000 Pkd1 mutation variants and more than 250 variants of Pkd2 have been identified [http://pkdb.pkdcure.org]. Predicted truncations are associated with the most severe disease outcomes, while missense mutations produce milder disease [18,45,46]. This is consistent with research showing that functional Pkd1 protein levels correlate with disease severity [47].
Cyst Initiation Theories
A person with ADPKD carries the pathogenic mutation in every cell, but cysts develop focally along kidney tubules. As disease progresses, cysts increase in both size and number. What triggers individual cells to switch from normal to cystic phenotype? Analyses revealed that epithelial cells lining human cysts are clones harboring the same somatic mutation in trans with the inherited mutation [48]. These findings suggest that cyst initiation occurs when a tubule cell with one mutant polycystin allele loses its remaining functional copy. The loss of both copies triggers focal proliferation and cyst formation. Analogous to Knudson’s two-hit hypothesis of tumor progression [49], this theory of cyst initiation is known as the two-hit model of ADPKD progression (Figure 1) [50,51].
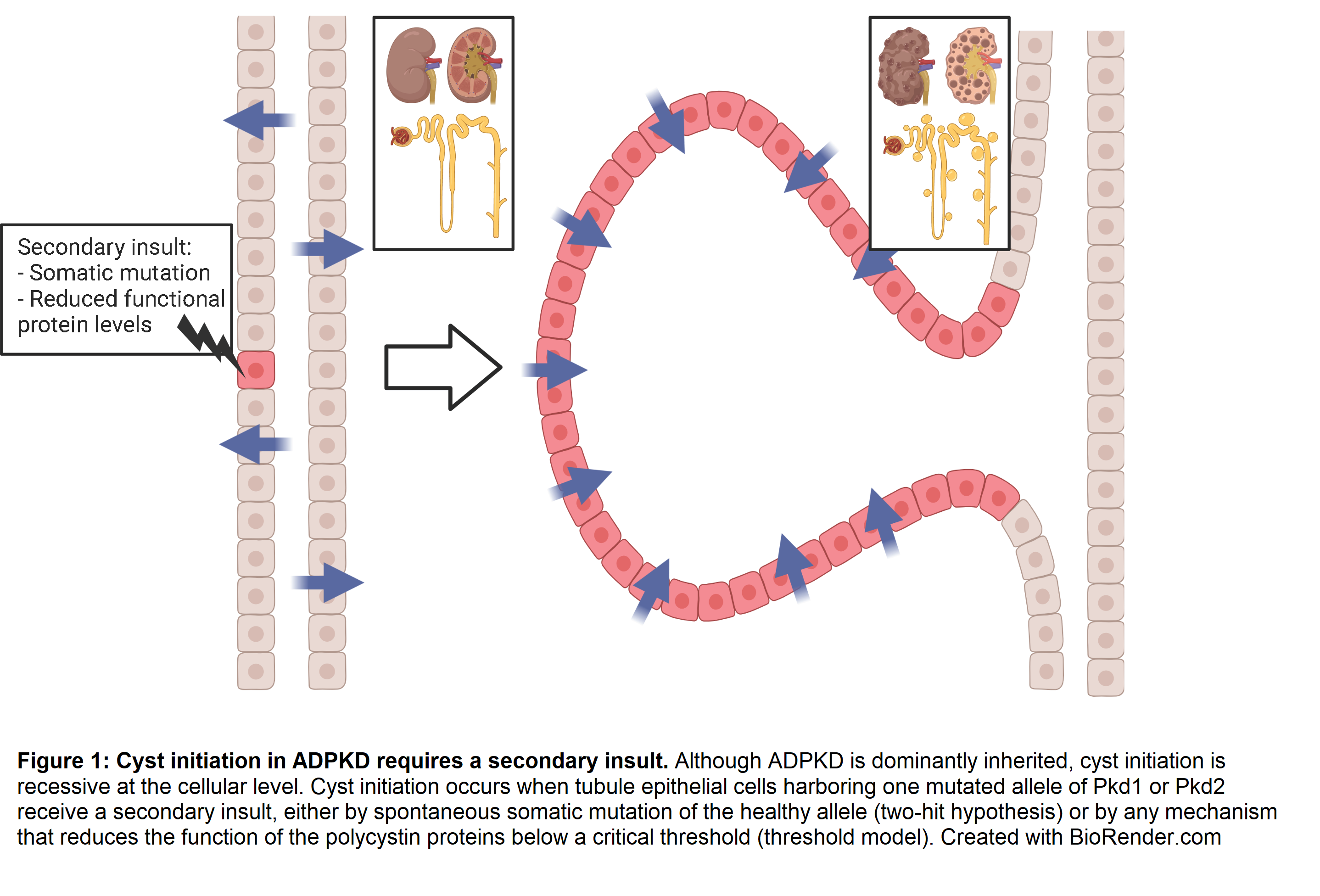
Additional evidence for the two-hit model comes from mouse genetic studies. Pkd2-/-mice die after embryonic day 14.5 with cardiac malformations, impaired right-left symmetry, and cysts in the kidney, liver, and pancreas [52]. Pkd1-/- mice also die in utero with massively cystic kidneys and pancreas, and their lungs are underdeveloped likely due to kidney malfunction causing oligohydramnios [53,54]. Early postnatal deletion of both alleles of Pkd1 or Pkd2 circumvents the embryonic lethality but still causes rapid cystic disease progression [55]. In sharp contrast to homozygous knockout mice, heterozygous mutations do not perturb normal development, lifespan, or produce significant cystic disease [47,56,57]. Wu et al. showed that increasing the somatic mutation rate in Pkd2+/- by introducing an unstable Pkd2 allele greatly increased cyst progression [56]. This evidence supports the idea that tubule cells function normally with one allele of Pkd1 or Pkd2, and only with a “second hit” do cysts form.
Scenarios beyond the two-hit model, such as hypomorphic mutations, compound heterozygosity, and polycystin overexpression, require alternative modes of cyst initiation. The dosage threshold model hypothesizes that epithelial cells function normally only when polycystin levels are within a limited range. Above or below that range, cells exhibit an abnormal cystic phenotype (Figure 2). Lantinga-van Leeuwen et al. provided evidence for the threshold model by inserting a neomycin cassette in a Pkd1 intron to create a hypomorphic allele. This insertion disrupted splicing and reduced the amount of correctly spliced Pkd1 to 15% of normal. Unlike mice homozygous for true null mutations, the hypomorphs survived development but developed significant cystic kidney disease by one month of age [47]. To further explore how Pkd1 dosage affects cyst formation, researchers used a hypomorphic mutation based on human disease variant p.R3277C (Pkd1RC). Like Pkd1+/+ mice, Pkd1RC/+ mice did not develop cysts within one year. Pkd1RC/RC mice were viable with slow disease progression and no change in survival compared to wild type. Pkd1RC/- mice were also viable but rapidly developed severe cystic disease associated with premature death. This contrasts with Pkd1-/- mice, which die in utero [58]. Pkd2nf3 hypomorphic mice exhibited similar results [59]. Furthermore, transgenic Pkd2 expression rescued the embryonic lethality phenotype in Pkd2-/- mice. Pkd2 transgene expression dose-dependently slowed cystic phenotype progression [60]. The findings show that functional polycystin protein dose inversely correlates with disease progression and severity.
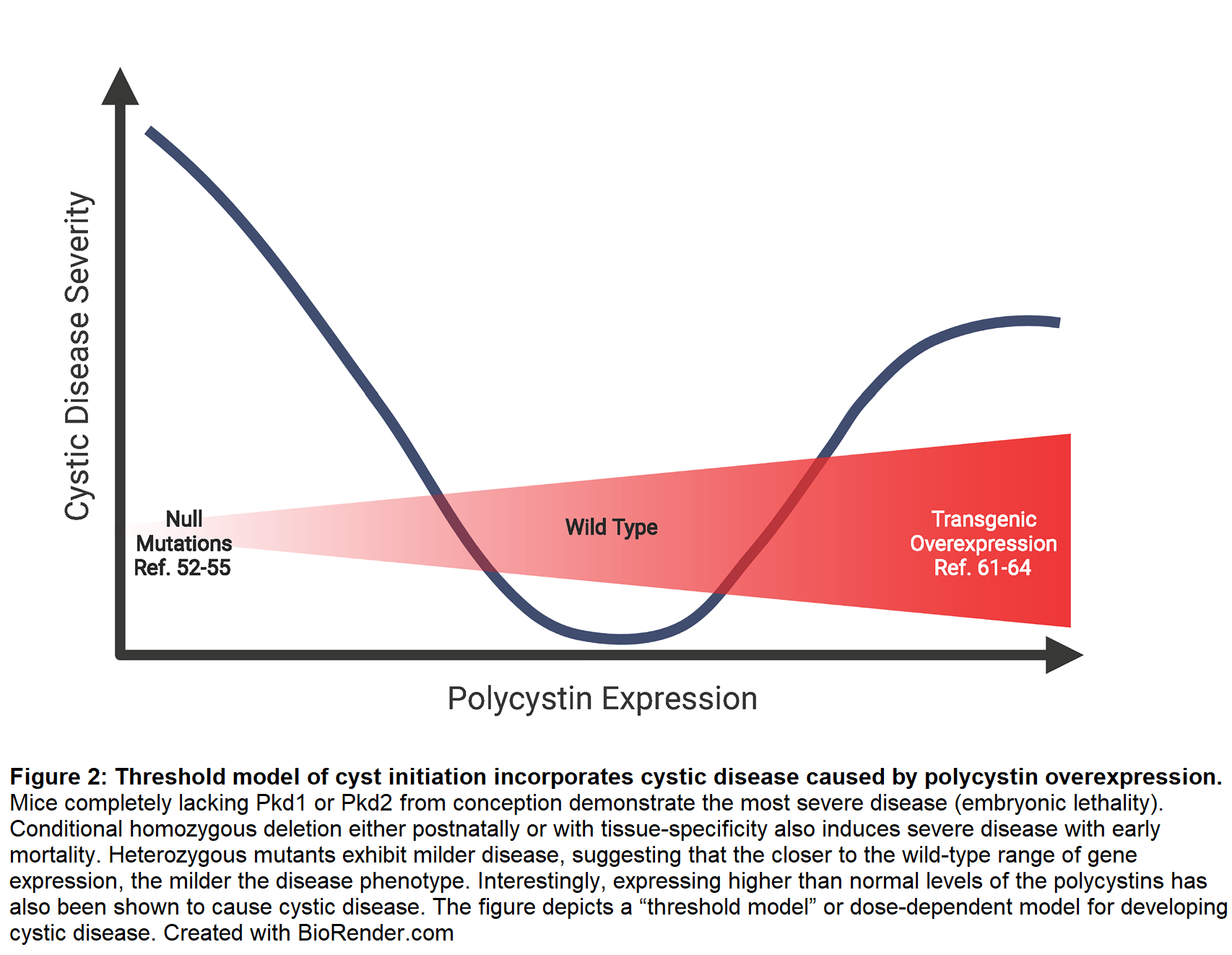
Paradoxically, overexpressing polycystins also induces cysts. In a rescue experiment, overexpressing human Pkd1 prevented embryonic lethality in Pkd1-/- mice but caused cystic kidney disease in Pkd1+/+ animals [61]. Subsequent models confirmed that Pkd1 overexpression [62,63] and, to a lesser extent, Pkd2 [64] cause cystic kidney disease. While most ADPKD cases involve loss-of-function mutations, high Pkd1 and Pkd2 levels have been detected in human cystic renal tissues [22,65]. While it is not known how overexpressed polycystins can cause cyst formation, it is possible that abnormally high levels of one of the two polycystins may disrupt the integrity of the polycystin complex, leading to loss of function.
Interestingly, mice homozygous for the Pkd1RC mutation developed focal cysts even though every cell in the tubule was genetically identical [58]. This suggests that additional stimuli are needed to convert Pkd1-deficient tubule epithelium into cystic epithelium. The additional stimuli, called the third hit, was proposed when it was found that deletions of Pkd1 or Ift88 in mice with fully developed kidneys were largely immune to cyst formation [66-68] unless they incurred kidney injury [69]. Acute kidney injury (AKI) represents the extreme end of potential renal challenges that ADPKD patients face and can occur due to hypovolemia, acute urine outflow obstruction, or nephrotoxic drug response. Studies in animal models show that cyst growth increases after AKI [70-72]. AKI also activates c-Jun N-terminal kinase (JNK) in animal studies and human tissue samples, and JNK inhibition reduces organ damage due to acute injury [73-77]. As discussed below, JNK appears to promote cyst growth in ADPKD by amplifying cellular response to injury.
Primary Cilia are Critical to Preventing Cystic Disease
The epithelial cells that form cysts in ADPKD each possess a single primary cilium projecting into the tubule lumen. Primary cilia are microtubule-supported, membrane-covered organelles that function as signal transduction centers for the cell [78,79]. Cilia mutations underlie syndromic forms of polycystic kidney disease [80], including Bardet-Biedl syndrome [81], Nephronophthisis [82,83], Joubert syndrome [84], and Meckel-Gruber syndrome [85, 86]. Mice with mutations in ciliary assembly genes (Kif3a, Ift88, Ift140, Ift20) develop renal cysts [66,80,87,88]. Pkd1 and Pkd2 co-localize in primary cilia membranes [25,31].
Accumulating evidence points to a requirement for ciliary localization of polycystins to prevent cyst formation [89-92]. Cai et al. identified missense mutations in Pkd1 and Pkd2 that impaired trafficking to cilia while preserving protein levels and other aspects of protein function such as the Pkd1-Pkd2 interaction. These trafficking mutations failed to rescue Pkd1-/- lethality or cystic disease caused by kidney-specific Pkd1 deletion [91]. In a similar study, Walker et al. studied the Pkd2lrm4/lrm4 mouse line, which carries a missense mutation in Pkd2 that preserves its channel properties but disrupts its ability to localize to cilia. Pkd2 levels in Pkd2lrm4/lrm4 embryonic kidneys were equivalent to Pkd2+/- mice, which do not develop severe disease. In contrast the Pkd2lrm4/lrm4 died in utero with kidney cysts similar to those observed in Pkd2-/-mice. They attributed the cystic phenotype to the failure of Pkd2 to localize to cilia in the mutant mice [90]. To strengthen the conclusions made by both studies, future experiments should retarget the mutant forms of Pkd1 and Pkd2 to the ciliary compartment to determine if relocalization rescues the cystic phenotype.
Interpreting results from point mutations affecting polycystin trafficking is challenging because these mutations may affect other functions. An alternative is to disrupt the delivery of wild-type polycystins to cilia. Two approaches have been taken in this space. One focuses on the role Tulp3 plays in cystic disease [89,93]. Tulp3 is an intraflagellar transport associated protein necessary for trafficking transmembrane proteins including Pkd1 and Pkd2 to cilia, but its deletion does not alter cilia structure or whole cell Pkd1 or Pkd2 levels [94]. Tulp3 deletion caused polycystic kidney disease that was intermediate between Pkd1 deletion and cilia deletion, implying that trafficking to cilia is a critical factor in the ability of polycystins to prevent cyst formation. The second approach focuses on Bardet-Biedl syndrome (BBS) proteins that bind to the polycystins and deliver Pkd1 to cilia [95]. The observation that BBS patients develop cystic disease supports the idea that the polycystins must be trafficked to and enter primary cilia. However, a caveat to both the Tulp3 and BBS studies is that these proteins play roles in cilia biology beyond the polycystins, and these other functions could be important to prevent cystic disease.
Experiments involving animals lacking cilia and Pkd1 or Pkd2 revealed a complex relationship between cilia and polycystin signaling [92]. Mice with double mutations in Pkd1 and Kif3a or in Pkd2 and Ift20 exhibited an intermediate phenotype between the milder cilia-induced and more severe polycystininduced cystic kidneys. Furthermore, disease severity positively correlated with the time between polycystin removal and cilia disappearance. Cilia disruption reduced tubule epithelial cell proliferation in polycystin mutants. These findings lead to the hypothesis that cyst growth depends on propagating a pro-proliferative signal by intact cilia, which was termed the cilia-dependent cyst-activating (CDCA) signal. When Pkd1 and Pkd2 are present, they inhibit the CDCA, thereby inhibiting tubule cells from proliferating and forming cysts. The model predicts that CDCA activity would be low in control kidneys, greatly elevated in Pkd mutant kidneys, and brought back towards controls in Pkd and cilia double mutant kidneys. ERK, mTOR, and cAMP signaling were tested as candidates for propagating the CDCA signal. However, none of these pathways matched the activity pattern predicted, leaving the identity of the CDCA unknown [92]. To identify candidate pathways controlling the CDCA, transcription profiling was used to identify genes whose expression is elevated by the loss of Pkd2 and brought back towards controls in Pkd2, Ift88 double mutant kidneys. Genes that matched the CDCA profile were enriched in cell proliferation functions. Cdk1 stood out as a strong candidate as its expression was increased ~5 fold by the loss of Pkd2 and returned to normal in the Pkd2 cilia-minus kidneys. Importantly, the loss of Cdk1 reduced cyst growth in a Pkd1 mutant kidney, making the Cdk1 pathway a strong candidate for the CDCA [96].
Signal Transduction by the Polycystin Complex
The physiological purpose of ciliary-localized polycystins is not known. One popular model posits that polycystins function as mechanosensors for fluid flow through the tubule lumen. This idea grew from studies showing that intracellular calcium increased in response to cilia deflection [97]. Comparing calcium responses to shear stress in collecting duct cells from wild type and homozygous null Pkd1 (Pkd1del34/del34) mice found that cytosolic calcium was transiently increased in wild type cells, but not Pkd1 mutant cells. Blocking antibodies to Pkd2 also abolished the flow-induced calcium signal [43]. These findings supported a model in which polycystin mutations disrupt mechanosensation and dysregulate calcium signaling, leading to abnormal downstream signaling and cysts. However, the results left open the possibility that calcium changes start in the cytosol rather than the cilium of kidney tubule cells. To clarify the source of the initial calcium influx, Delling et al. examined collecting duct cells from a transgenic mouse expressing a fluorescent calcium reporter specific for changes in the ciliary calcium levels. These cells did not exhibit any flow-induced calcium fluctuations under physiologically relevant flow conditions that caused cilia bending. Surprisingly, cilia bending did not elicit cilia-derived changes in calcium in any tissue tested. Instead, they reported that elevated calcium in the cytoplasm could diffuse into the cilium in a relatively short time frame of less than 200 milliseconds, potentially explaining the discrepancy with earlier findings. They also noted that extreme flow rates could tear cilia tips, allowing extracellular calcium to flow into the cilium. In conclusion, they suggested that cilia may function as mechanosensors, but not through a transient increase in calcium concentration [98].
Recently, an intra vital technique to visualize renal cilia in a live mouse showed that under normal conditions, the tubule luminal cilia do not oscillate but rather remain bent flat against the tubule wall [99]. This suggests that, at least in mice, cilia are typically subjected to maximum deflection. An important caveat to this work is that the resting heart rate in mice is 500-700 beats per minute compared to 50-70 beats per minute in humans. In mice near death, the cilia did oscillate as heart rate slowed [100]. The studies also observed that tubule epithelium undergoes dynamic cytoplasmic calcium fluctuations under normal conditions, but they did not observe any correlation between calcium changes and cilia bending. It will be interesting to use this live imaging approach to see if mice carrying cilia or polycystin mutations display disrupted calcium signaling.
Alternatively, the ciliary polycystin complex could be detecting molecules in the filtrate. For example, Wnt ligands bind Pkd1 to effect changes in calcium signaling [27]. In addition, structural similarities [101,102] and a capacity to bind heterotrimeric G proteins [103-105] suggest that Pkd1 may be an atypical adhesion GPCR.
cAMP and Calcium Signaling Promote Cyst Growth
The fundamental factors underpinning cyst growth in ADPKD are tubule epithelial cell proliferation and fluid secretion. Healthy kidney tubules have a narrow lumen with a diameter of about 30 microns [106]. In contrast, cysts can reach over 70 millimeters in diameter [107]. Using scanning electron microscopy to directly count cells and measure their surface area, researchers determined that tubule cells proliferate rather than expand as the cystic lumen increases in surface area [108]. Microdissection and serial scanning electron microscopy showed that most cysts lacked connections to tubules suggesting that cyst fluid comes from transepithelial secretion rather than accumulated glomerular filtrate. Understanding what stimulates proliferative and secretory changes in cystic epithelium is critical to future therapy development.
The finding that cyst fluid and cystic tissues contained high cAMP levels led to the hypothesis that this ubiquitous second-messenger promotes cystic changes in tubule epithelium [109-111]. This idea is supported by in vitro studies showing that cAMP stimulated cyst growth in wildtype Madin-Darby Canine Kidney (MDCK) cells grown in a three-dimensional hydrated collagen matrix [112]. In control media, the cells formed small, slow-growing spheres with apical membranes toward the lumen. However, adding cAMP agonist prostaglandin-E1 to the media expanded the spheres into fluid-filled cysts [110]. Solute transport inhibitors blocked cyst expansion, suggesting that cyst growth depends on cAMP-stimulated chloride secretion. cAMP promotes secretion in tubule epithelial cells by activating cystic fibrosis transmembrane conductance regulator (CFTR) channels in the apical cell membrane (Figure 3) [113]. Further evidence that cyst growth depends on secretion comes from families with both cystic fibrosis and ADPKD. Patients with mutations in CFTR and PKD1 have less severe kidney and liver disease than patients with ADPKD alone [114,115].
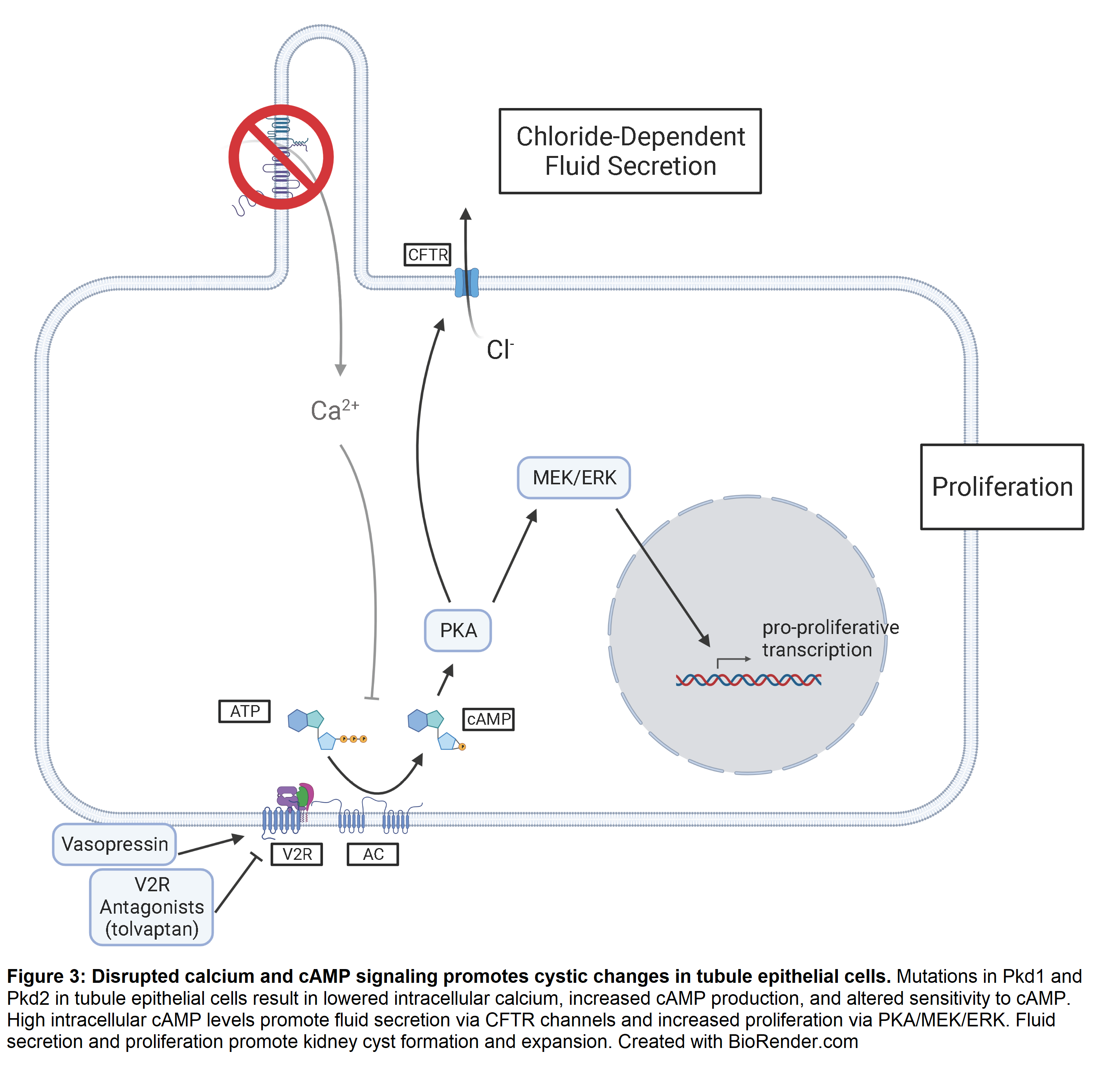
cAMP’s role in proliferation was less clear. In vitro studies showed that cAMP inhibited proliferation by blocking the ERK pathway [116,117]. In addition, vasopressin, a hormone that increases intracellular cAMP levels, reduced ERK activation and proliferation in kidney cells derived from rat [118] and dog [119]. Rats treated with folic acid to induce rapid kidney cell proliferation experienced decreased proliferation rates upon cAMP treatment as measured by radioactive thymidine incorporation [120]. However, several studies showed that cyst fluid from human and mouse kidneys stimulated increased intracellular cAMP levels in cultured kidney cells and promoted secretion and proliferation. Specific cAMP agonists recapitulated secretion but had no significant effect on proliferation, suggesting that mitogens in the cyst fluid stimulated growth by additional mechanisms [121,122].
The experiments documenting cAMP inhibiting proliferation used wild-type cells, whereas the cells that form cysts in ADPKD are mutated in at least one allele of Pkd1 or Pkd2. To determine how cAMP effects mutated cells, researchers obtained cystlining epithelial cell cultures from human ADPKD kidneys. Comparing these ADPKD cells to cells derived from healthy kidney cortex, they made the surprising discovery that ADPKD cells proliferated in response to cAMP while wild-type cells did not [123,124]. cAMP agonists enhanced ERK activity in ADPKD cells but not wild-type kidney cells [124].
Polycystin mutations are associated with reduced resting intracellular calcium levels [125]. Reducing calcium levels in wild-type cells using channel blockers switched their response to cAMP from quiescent to proliferative [126]. Calcium restriction also led to cAMP-dependent ERK activation (Figure 3). Transfecting wild-type cells with the C-terminus of Pkd1 also produced the proliferative response to cAMP, which was reversed by replenishing calcium levels by calcium ionophore treatment [126]. Importantly, elevating calcium levels in ADPKD cells induced normal response to cAMP and reduced cyst formation in an organoid model [125]. The pro-secretory and pro-proliferative effects on ADPKD cells suggested that reducing cAMP levels would be a viable option for slowing cyst growth.
Two important treatments to reduce cAMP in kidney tubule epithelial cells have been tested in pre-clinical and clinical trials. Somatostatin analogues reduce adenylyl cyclase activity by binding to somatostatin receptors on the cell surface, which reduces intracellular cAMP production [127,128]. Somatostatin analogues effectively reduced cyst growth in initial clinical trials [129]. However, subsequent larger randomized clinical trials showed that somatostatin analogues did not preserve kidney function enough to justify the risks [130]. The larger trials enrolled later stage ADPKD patients with significantly reduced kidney function at baseline and it is possible that a therapeutic effect may be seen if used in patients with milder disease. Notably, somatostatin analogues show promise in reducing liver cysts [131,132].
Healthy mammalian kidneys experience high tonic exposure to vasopressin to prevent excessive water loss. The hormone acts on vasopressin 2 receptors in the collecting ducts and distal nephron [133]. Vasopressin 2 receptor activates adenylyl cyclase 6 through Gs proteins, thereby increasing cAMP production [134]. Vasopressin 2 receptor antagonists such as tolvaptan prevent vasopressin from binding to vasopressin 2 receptor, which effectively decreases intracellular cAMP (Figure 3). Vasopressin 2 receptor antagonism reduced cyst growth in animal models [135,136] and human ADPKD cells in a collagen matrix [137]. Tolvaptan successfully reduced the rate of decline in kidney function in clinical trials and was approved for treating ADPKD in 2018.
JNK is a MAP Kinase Pathway Activated by PKD
The mitogen-activated protein kinase (MAPK) superfamily includes the ERK1/2, p38, JNK, and ERK5 signaling pathways, all of which are activated through multi-tiered phosphorylation cascades in response to various stimuli [138]. MAPK signaling broadly impacts human health and disease due to its roles in cell proliferation, survival, and differentiation [139]. Our recent work showed that Pkd2 loss activates JNK signaling and that genetic reduction of JNK activity reduced cyst growth triggered by loss of Pkd2 [140].
JNKs become activated by dual phosphorylation of threonine and tyrosine in the T-P-Y motif within the activation loop [141]. JNK phosphorylation is mediated by two MAP kinase kinases (MAP2K), MKK4 and MKK7. In turn, MAP2Ks are activated through phosphorylation by MAP kinase kinase kinases (MAP3K). The mouse and human genomes encode 24 MAP3Ks that feed into the ERK1/2, JNK, p38, and ERK5 pathways, with at least 14 known to phosphorylate MKK4 or MKK7 upstream of JNKs [142,143]. MAP3Ks are activated by various stimuli including cellular stress, inflammatory cytokines, growth factors, and GPCR agonists. These signals are typically detected by receptor tyrosine kinases, GPCRs, and other membrane receptors, and the signal is propagated to the MAP3Ks through the actions of Rac1 and Cdc42, although a variety of other mechanisms exist [144]. For example, reactive oxygen species activate the MAP3K, ASK1 through a mechanism involving thioredoxin binding [145]. It is thought that the specificity of JNK activation in different contexts is mediated by MAP3K activation (Figure 4).
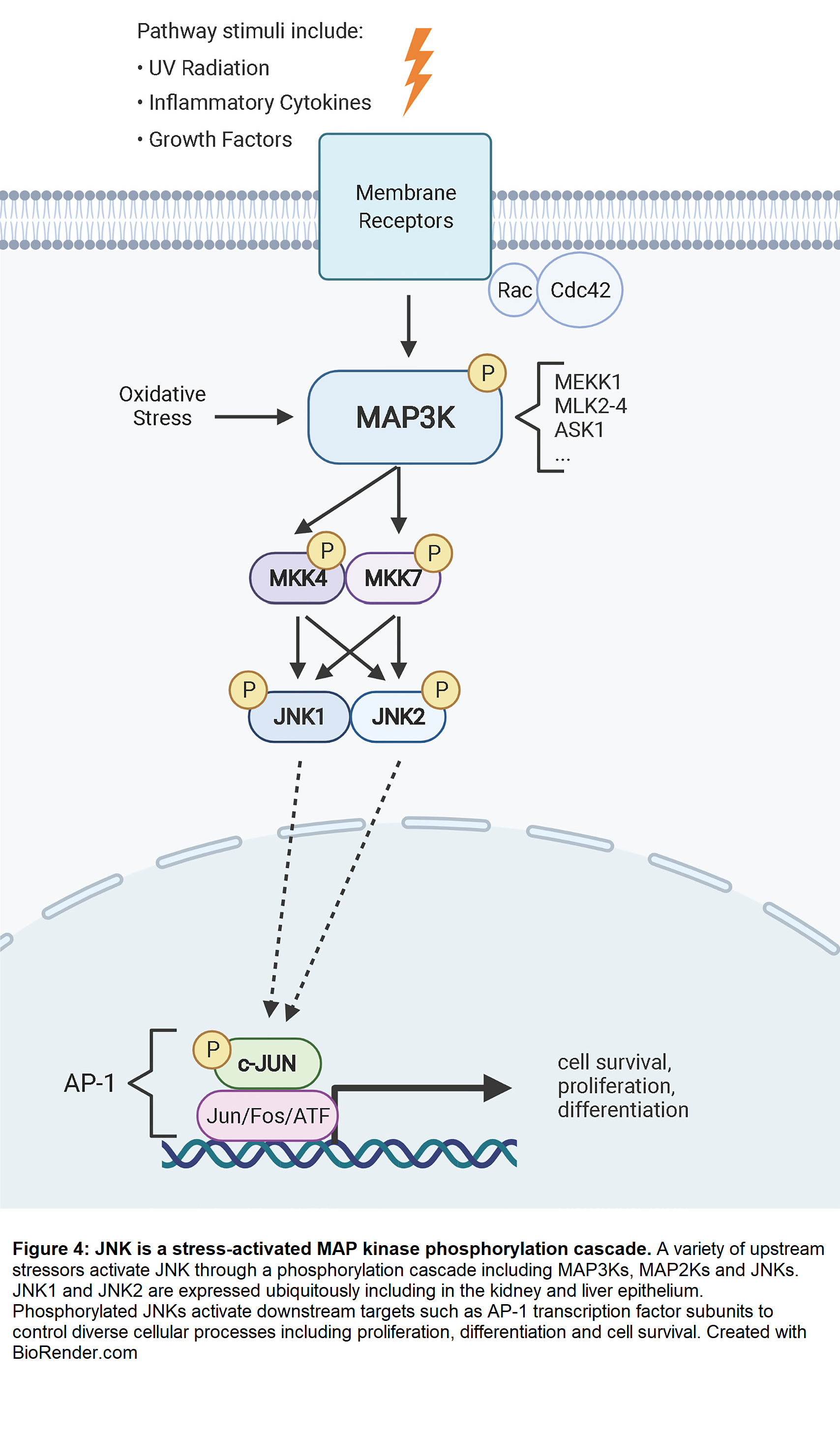
Three highly homologous genes encode Jun N-terminal kinases: JNK1/MAPK8, JNK2/MAPK9, and JNK3/MAPK10 [146]. JNK1 and JNK2 are widely expressed in most tissues, including the kidneys, while JNK3 expression is primarily restricted to the central nervous system [142]. The sequence identity between JNK1 and JNK2 is 73% [146,147]. However, the ATP binding site is 98% conserved between the proteins, presenting a challenge to finding selective JNK inhibitors for research and therapeutics [148,149].
Alternative splicing adds complexity to the JNK pathway. Two independent splicing events produce at least four distinct isoforms of both JNK1 and JNK2 with differences at the C-terminal end and within the kinase domain [146]. The functional consequences of C-terminal splicing are not known. However, in the kinase domain, JNK containing exon 7b is more active in phosphorylating c-Jun than isoforms containing 7a [150]. JNK splicing regulation in the kidney has not been described, but it is possible that differential splicing could yield proteins with differing substrate specificities.
Activator protein-1 (AP-1) transcription factors are the most studied JNK substrates. JNK activates c-Jun by binding the transactivating domain and phosphorylating N-terminal serines 63 and 73 [141,151]. JNK similarly activates Activating Transcription Factor 2 (ATF2) [152]. This activation is critical for proliferation as fibroblasts expressing a mutant c-Jun that is incapable of JNK-mediated phosphorylation exhibited significantly reduced proliferation [153]. Interestingly, mice carrying this mutation developed normally, though they are smaller than controls. In contrast, c-Jun knockout mice die by embryonic day 13 with heart and liver malformations [154]. Overexpressing c-Jun in mice causes multi-organ fibrosis, including in the kidney and liver [155]. Although it is not discussed in the publication, c-Jun overexpression in kidney also caused cystic disease (see Figure 2E in [155]). Elevated levels of c-Jun have been detected in cancers including sarcomas [156] and Hodgkin lymphoma [157]. However, JNK and c-Jun/AP-1 can also have anti-tumor effects by participating in programmed cell death in response to DNA damage [158]. Cell type and stimulus are contextual factors that influence the outcomes of JNK activation.
JNK Involvement in Kidney Disease
JNK activation has been detected in many forms of kidney disease [159]. In animal models, JNK inhibition prior to ischemiareperfusion or tubule obstruction reduces inflammation and fibrosis, and preserves kidney function [73-77]. Interestingly, acute kidney injury exacerbates polycystic kidney disease [69-72]. In chronic kidney insult, progressive interstitial fibrosis contributes to organ failure. JNK inhibition reduces profibrotic factors in the kidney [76,77]. Furthermore, researchers produced severe kidney fibrosis in mice by overexpressing the JNK target c-Jun [155]. Whether JNK contributes to fibrosis primarily through activation in tubule cells, interstitial cells, or a combination is yet to be determined.
JNK Signaling in ADPKD
As with tumor growth, ADPKD is characterized by abnormally sustained proliferation. Elevated AP-1 components in cystic epithelium from ADPKD patients and mice suggest a proproliferative role for JNK/AP-1 signaling in ADPKD pathology [160]. To better understand the signaling pathways regulated by Pkd1, Arnould et al. targeted a construct containing Pkd1’s C-terminal intracellular tail to the cell membrane in human embryonic kidney 293T (HEK-293T) cells expressing an AP-1 luciferase reporter [161]. Expressing the Pkd1 construct induced 5-fold greater AP-1 reporter activity compared to a control construct. They further showed that JNK1 kinase activity was elevated in cells expressing the Pkd1 C-terminus, but surprisingly neither p38 nor p44/ERK1 were affected. Finally, they showed that Pkd1-induced AP-1 and JNK activity were blocked by expressing dominant-negative forms of the Rho GTPases, Cdc42 and Rac1 as well as a dominant-negative form of calcium-dependent PKC-alpha. The researchers hypothesized that high Pkd1 expression during renal development drives proliferation via Rac1/Cdc42 and PKCalpha. Under normal conditions, the reduction in Pkd1 levels that occur once development is complete would reduce the pathway activity and stop proliferation in mature tissues [161].
In a subsequent publication, the same authors showed that expressing full-length Pkd2 in HEK-293T cells robustly activated AP-1 and c-Jun reporters [162]. Pkd2-dependent activation was reduced by dominant-negative Cdc42, Rac1, and RhoA. Unlike Pkd1, which did not activate p38 or ERK, Pkd2 expression activated p38 in addition to JNK. Unique to Pkd2, AP-1 activation depended on calcium-independent PKC-epsilon and not PKC-alpha. Interested in the combined function of Pkd1 and Pkd2, the researchers co-expressed the C-terminal 112 base pairs of Pkd1 together with full length Pkd2. This resulted in higher AP-1 activity than with Pkd2 alone. The augmentation could be reversed by dominantnegative PKC-alpha or treatment with a calcium chelator. The authors concluded that Pkd1 and Pkd2 stimulate AP-1 activity in an additive manner via PKC-alpha and PKCepsilon, respectively [162]. In their discussion, the authors speculated that Pkd1/Pkd2 induction of AP-1 activity might be important in directing proliferation and differentiation during nephrogenesis. However, they did not address how their findings apply to ADPKD, in which low or absent polycystin proteins promote proliferation in mature tubule cells. We now know that overexpressing Pkd1 or Pkd2 in mice induces a cystic phenotype [61,62]. While the mechanism is not known, it is possible that overexpression disrupts the polycystin complex, mimicking a loss-of-function mutation.
An independent study similarly found that AP-1 and JNK activity were increased in HEK-293T cells upon transfection with membrane-targeted Pkd1 constructs [163]. They additionally showed that dominant negative heterotrimeric G proteins abrogated the activation, while wild-type G protein expression augmented it. They hypothesized that Pkd1 regulated JNK activity by modulating the availability of heterotrimeric G proteins for downstream signaling. In a subsequent study, Pkd1-overexpressing MDCK cells were resistant to thrombin-induced JNK activation and c-Jun phosphorylation while cells lacking Pkd1 had increased JNK activation in response to thrombin [104].
While an immortalized epithelial cell line derived from an ADPKD patient exhibited reduced c-Jun phosphorylation compared to control cells [164], cyst-lining epithelium with Pkd1 mutations in human and mouse kidneys expressed high levels of phosphorylated c-Jun and ATF2 [160]. These findings suggest that in vivo context is important for JNK/AP-1 activation in ADPKD. Although the mechanism by which Pkd1 and Pkd2 regulate JNK/AP-1 signaling in vivo is not known, cell culture studies showed that activation depends on Rho GTPases [161,162] or heterotrimeric G proteins [104,163,165].
The evidence linking Pkd1 and Pkd2 to JNK/AP-1 activity led us to ask what role JNK signaling plays in cyst formation and disease progression downstream of polycystin mutations [140]. To do this, we crossed mice carrying floxed alleles of Pkd2 and Jnk1, Jnk2 null alleles, and a tamoxifen-inducible Cre. The offspring, which segregated these four alleles, were treated with tamoxifen in the early post-natal period through maternal delivery. Pkd2 loss led to rapid cyst formation along with increased JNK and c-Jun phosphorylation. Phospho c-Jun was concentrated in the nuclei of cells lining cysts and pre-cystic tubules, suggesting that it might drive cyst formation rather than occur as a response to tissue disruption. Mice segregating both Pkd2 and Jnk1/Jnk2 alleles showed significantly reduced cystic disease compared to mice only missing Pkd2. Whereas Pkd2 mutant kidneys showed extensive cysts throughout the organ, Pkd2/Jnk1/Jnk2 triple mutants retained cysts only at the cortical-medullary boundary and showed less proliferation and less fibrosis. While Jnk1 and Jnk2 are thought to be largely interchangeable, organ-specific functions have been observed. Our studies found that Jnk1 drove the protective effect, as the loss of Jnk1 was almost as protective as Jnk1 and Jnk2 double loss. As the early postnatal delivery of tamoxifen produces rapid cystic disease progression, we sought to determine if JNK loss was protective in slow-progressing disease by treating mice with tamoxifen in early adulthood. The experiment had to be terminated before the kidneys became cystic because the loss of Pkd2 caused serious liver cysts. However, the loss of JNK activity dramatically reduced the severity of the liver cysts suggesting that the protective effects of JNK loss extend beyond the early post-natal period and protect the liver.
Prospective
Our finding that removal of JNK activity is protective against cystic disease suggests that drugs targeting this branch of MAPK signaling should be explored as therapeutic targets. Drugs active against JNK are in development as anti-cancer treatments and several candidates have been identified. For example, SP600125 [166] is a potent inhibitor of JNK with activity against numerous cancers [167]. With relevance to PKD, SP600125 is protective against ischemic/reperfusion injury of murine kidneys [168]. Other drugs include the small molecules JNK-in-8 [169] and CC-90001 (NCT02510937) as well as a TAT-JIP1 peptide inhibitor Brimapitide [170] that has been used to prevent the development of diabetes in mice [171]. The extensive functions of JNK in regulating cellular physiology may limit its effectiveness as a long-term drug. Therefore, upstream kinases such as MAP3Ks may be better targets with less adverse effects. Further work will be required to identify the MAP3K that functions between the polycystins and JNK. However, assuming that the specific MAP3K can be identified, there is precedence for developing specific MAP3K inhibitors that are effective and safe. These include the MLK2/MLK3 inhibitor URMC-099 [172] that has been shown to suppress murine non-alcoholic steatohepatitis [173] and shows neuroprotection in a murine model of Alzheimer’s disease [174,175]. The ASK1 inhibitor GS-444217 is effective for the treatment of diabetic kidney disease in rodents [176]. Selonsertib, which also inhibits ASK1, has been tested in clinical trials for diabetic kidney disease [177] and hepatic fibrosis in patients with non-alcoholic steatohepatitis [178,179].
Acknowledgements
This work was supported by National Institute of Health grant DK103632 to GJP and DK107220 to RJD.
Disclosures
The authors have no conflicts to disclose.
References
2. Kuo IY, Chapman AB. Polycystins, ADPKD, and Cardiovascular Disease. Kidney Int Rep. 2020;5(4):396-406.
3. McEwan P, Bennett Wilton H, Ong ACM, Orskov B, Sandford R, Scolari F, et al. A model to predict disease progression in patients with autosomal dominant polycystic kidney disease (ADPKD): the ADPKD Outcomes Model. BMC Nephrol. 2018;19(1):37.
4. Testa F, Magistroni R. ADPKD current management and ongoing trials. J Nephrol. 2020;33(2):223-37.
5. Parajuli S, Mandelbrot DA, Aziz F, Garg N, Muth B, Mohamed M, et al. Characteristics and Outcomes of Kidney Transplant Recipients with a Functioning Graft for More than 25 Years. Kidney Dis (Basel). 2018;4(4):255-61.
6. Dalgaard OZ. Bilateral polycystic disease of the kidneys; a follow-up of 284 patients and their families. Dan Med Bull. 1957;4(4):128-33.
7. Iglesias CG, Torres VE, Offord KP, Holley KE, Beard CM, Kurland LT. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935-1980. Am J Kidney Dis. 1983;2(6):630-9.
8. Lanktree MB, Haghighi A, Guiard E, Iliuta IA, Song X, Harris PC, et al. Prevalence Estimates of Polycystic Kidney and Liver Disease by Population Sequencing. J Am Soc Nephrol. 2018;29(10):2593- 600.
9. Willey C, Kamat S, Stellhorn R, Blais J. Analysis of Nationwide Data to Determine the Incidence and Diagnosed Prevalence of Autosomal Dominant Polycystic Kidney Disease in the USA: 2013- 2015. Kidney Dis (Basel). 2019;5(2):107-17.
10. Suwabe T, Shukoor S, Chamberlain AM, Killian JM, King BF, Edwards M, et al. Epidemiology of Autosomal Dominant Polycystic Kidney Disease in Olmsted County. Clin J Am Soc Nephrol. 2020;15(1):69-79.
11. Cloutier M, Manceur AM, Guerin A, Aigbogun MS, Oberdhan D, Gauthier-Loiselle M. The societal economic burden of autosomal dominant polycystic kidney disease in the United States. BMC Health Serv Res. 2020;20(1):126.
12. Heyer CM, Sundsbak JL, Abebe KZ, Chapman AB, Torres VE, Grantham JJ, et al. Predicted Mutation Strength of Nontruncating PKD1 Mutations Aids Genotype-Phenotype Correlations in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27(9):2872-84.
13. Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. American Journal of Human Genetics. 2018;102(5):832-44.
14. Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, et al. Mutations in GANAB, Encoding the Glucosidase IIalpha Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. American Journal of Human Genetics. 2016;98(6):1193-207.
15. Besse W, Chang AR, Luo JZ, Triffo WJ, Moore BS, Gulati A, et al. ALG9 Mutation Carriers Develop Kidney and Liver Cysts. J Am Soc Nephrol. 2019;30(11):2091-102.
16. Senum SR, Li YSM, Benson KA, Joli G, Olinger E, Lavu S, et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. American Journal of Human Genetics. 2021.
17. Hateboer N, v Dijk MA, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353(9147):103-7.
18. Hwang YH, Conklin J, Chan W, Roslin NM, Liu J, He N, et al. Refining Genotype-Phenotype Correlation in Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol. 2016;27(6):1861-8.
19. Cornec-Le Gall E, Audrezet MP, Renaudineau E, Hourmant M, Charasse C, Michez E, et al. PKD2-Related Autosomal Dominant Polycystic Kidney Disease: Prevalence, Clinical Presentation, Mutation Spectrum, and Prognosis. Am J Kidney Dis. 2017;70(4):476-85.
20. Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet. 1995;10(2):151-60.
21. EPKDC. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell. 1994;78(4):725.
22. Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, et al. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc Natl Acad Sci U S A. 1996;93(4):1524-8.
23. Ibraghimov-Beskrovnaya O, Dackowski WR, Foggensteiner L, Coleman N, Thiru S, Petry LR, et al. Polycystin: in vitro synthesis, in vivo tissue expression, and subcellular localization identifies a large membrane-associated protein. Proc Natl Acad Sci U S A. 1997;94(12):6397-402.
24. Scheffers MS, van der Bent P, Prins F, Spruit L, Breuning MH, Litvinov SV, et al. Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum Mol Genet. 2000;9(18):2743-50.
25. Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13(10):2508-16.
26. Maser RL, Calvet JP. Adhesion GPCRs as a paradigm for understanding polycystin-1 G protein regulation. Cell Signal. 2020;72:109637.
27. Kim S, Nie H, Nesin V, Tran U, Outeda P, Bai CX, et al. The polycystin complex mediates Wnt/Ca(2+) signalling. Nat Cell Biol. 2016;18(7):752-64.
28. Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339-42.
29. Schneider MC, Rodriguez AM, Nomura H, Zhou J, Morton CC, Reeders ST, et al. A gene similar to PKD1 maps to chromosome 4q22: a candidate gene for PKD2. Genomics. 1996;38(1):1-4.
30. Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, et al. Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem. 1999;274(40):28557-65.
31. Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol. 2002;12(11):R378-80.
32. Giamarchi A, Padilla F, Coste B, Raoux M, Crest M, Honore E, et al. The versatile nature of the calcium-permeable cation channel TRPP2. EMBO Rep. 2006;7(8):787-93.
33. Gonzalez-Perrett S, Kim K, Ibarra C, Damiano AE, Zotta E, Batelli M, et al. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+- permeable nonselective cation channel. Proc Natl Acad Sci U S A. 2001;98(3):1182-7.
34. Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, et al. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca(2+) homeostasis in polycystic kidney disease. Biochem Biophys Res Commun. 2001;282(1):341-50.
35. Liu X, Vien T, Duan J, Sheu SH, DeCaen PG, Clapham DE. Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. Elife. 2018;7.
36. Kleene SJ, Kleene NK. The native TRPP2-dependent channel of murine renal primary cilia. Am J Physiol Renal Physiol. 2017;312(1):F96-F108.
37. Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 1997;16(2):179-83.
38. Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A. 1997;94(13):6965-70.
39. Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, et al. Co-assembly of polycystin-1 and -2 produces unique cationpermeable currents. Nature. 2000;408(6815):990-4.
40. Newby LJ, Streets AJ, Zhao Y, Harris PC, Ward CJ, Ong AC. Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J Biol Chem. 2002;277(23):20763-73.
41. Grimm DH, Cai Y, Chauvet V, Rajendran V, Zeltner R, Geng L, et al. Polycystin-1 distribution is modulated by polycystin-2 expression in mammalian cells. J Biol Chem. 2003;278(38):36786- 93.
42. Su Q, Hu F, Ge X, Lei J, Yu S, Wang T, et al. Structure of the human PKD1-PKD2 complex. Science. 2018;361(6406).
43. Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129-37.
44. Bae KT, Zhou W, Shen C, Landsittel DP, Wu Z, Tao C, et al. Growth Pattern of Kidney Cyst Number and Volume in Autosomal Dominant Polycystic Kidney Disease. Clin J Am Soc Nephrol. 2019;14(6):823-33.
45. Cornec-Le Gall E, Audrezet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24(6):1006-13.
46. Carrera P, Calzavara S, Magistroni R, den Dunnen JT, Rigo F, Stenirri S, et al. Deciphering Variability of PKD1 and PKD2 in an Italian Cohort of 643 Patients with Autosomal Dominant Polycystic Kidney Disease (ADPKD). Sci Rep. 2016;6:30850.
47. Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13(24):3069-77.
48. Qian F, Watnick TJ, Onuchic LF, Germino GG. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87(6):979-87.
49. Knudson AG, Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820-3.
50. Pei Y. A "two-hit" model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med. 2001;7(4):151-6.
51. Reeders ST. Multilocus polycystic disease. Nat Genet.1992;1(4):235-7.
52. Wu G, Markowitz GS, Li L, D'Agati VD, Factor SM, Geng L, et al. Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet. 2000;24(1):75-8.
53. Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, et al. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet. 1997;17(2):179-81.
54. Muto S, Aiba A, Saito Y, Nakao K, Nakamura K, Tomita K, et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. 2002;11(15):1731-42.
55. Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJ. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet. 2007;16(24):3188-96.
56. Wu G, D'Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93(2):177-88.
57. Lu W, Fan X, Basora N, Babakhanlou H, Law T, Rifai N, et al. Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat Genet. 1999;21(2):160-1.
58. Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122(11):4257-73.
59. Kim I, Li C, Liang D, Chen XZ, Coffy RJ, Ma J, et al. Polycystin-2 expression is regulated by a PC2-binding domain in the intracellular portion of fibrocystin. J Biol Chem. 2008;283(46):31559-66.
60. Li A, Tian X, Zhang X, Huang S, Ma Y, Wu D, et al. Human polycystin-2 transgene dose-dependently rescues ADPKD phenotypes in Pkd2 mutant mice. Am J Pathol. 2015;185(10):2843- 60.
61. Pritchard L, Sloane-Stanley JA, Sharpe JA, Aspinwall R, Lu W, Buckle V, et al. A human PKD1 transgene generates functional polycystin-1 in mice and is associated with a cystic phenotype. Hum Mol Genet. 2000;9(18):2617-27.
62. Thivierge C, Kurbegovic A, Couillard M, Guillaume R, Cote O, Trudel M. Overexpression of PKD1 causes polycystic kidney disease. Mol Cell Biol. 2006;26(4):1538-48.
63. Kurbegovic A, Cote O, Couillard M, Ward CJ, Harris PC, Trudel M. Pkd1 transgenic mice: adult model of polycystic kidney disease with extrarenal and renal phenotypes. Hum Mol Genet. 2010;19(7):1174-89.
64. Burtey S, Riera M, Ribe E, Pennekamp P, Passage E, Rance R, et al. Overexpression of PKD2 in the mouse is associated with renal tubulopathy. Nephrol Dial Transplant. 2008;23(4):1157-65.
65. Ong AC, Ward CJ, Butler RJ, Biddolph S, Bowker C, Torra R, et al. Coordinate expression of the autosomal dominant polycystic kidney disease proteins, polycystin-2 and polycystin-1, in normal and cystic tissue. Am J Pathol. 1999;154(6):1721-9.
66. Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17(18):1586-94.
67. Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13(12):1490-5.
68. Takakura A, Contrino L, Beck AW, Zhou J. Pkd1 inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19(12):2351-63.
69. Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18(14):2523-31.
70. Happe H, Leonhard WN, van der Wal A, van de Water B, Lantinga-van Leeuwen IS, Breuning MH, et al. Toxic tubular injury in kidneys from Pkd1-deletion mice accelerates cystogenesis accompanied by dysregulated planar cell polarity and canonical Wnt signaling pathways. Hum Mol Genet. 2009;18(14):2532-42.
71. Kurbegovic A, Trudel M. Acute kidney injury induces hallmarks of polycystic kidney disease. Am J Physiol Renal Physiol. 2016;311(4):F740-F51.
72. Prasad S, McDaid JP, Tam FW, Haylor JL, Ong AC. Pkd2 dosage influences cellular repair responses following ischemiareperfusion injury. Am J Pathol. 2009;175(4):1493-503.
73. Grynberg K, Ozols E, Mulley WR, Davis RJ, Flavell RA, Nikolic- Paterson DJ, et al. JNK1 signaling in the proximal tubule causes cell death and acute renal failure in rat and mouse models of renal ischaemia/reperfusion injury. Am J Pathol. 2021.
74. Kanellis J, Ma FY, Kandane-Rathnayake R, Dowling JP, Polkinghorne KR, Bennett BL, et al. JNK signalling in human and experimental renal ischaemia/reperfusion injury. Nephrol Dial Transplant. 2010;25(9):2898-908.
75. Ma FY, Flanc RS, Tesch GH, Bennett BL, Friedman GC, Nikolic- Paterson DJ. Blockade of the c-Jun amino terminal kinase prevents crescent formation and halts established anti-GBM glomerulonephritis in the rat. Lab Invest. 2009;89(4):470-84.
76. Ma FY, Flanc RS, Tesch GH, Han Y, Atkins RC, Bennett BL, et al. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J Am Soc Nephrol. 2007;18(2):472-84.
77. de Borst MH, Prakash J, Sandovici M, Klok PA, Hamming I, Kok RJ, et al. c-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J Pharmacol Exp Ther. 2009;331(3):896-905.
78. Pazour GJ, Quarmby L, Smith AO, Desai PB, Schmidts M. Cilia in cystic kidney and other diseases. Cell Signal. 2020;69:109519.
79. Pazour GJ, Witman GB. The vertebrate primary cilium is a sensory organelle. Curr Opin Cell Biol. 2003;15(1):105-10.
80. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151(3):709-18.
81. Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet- Biedl syndrome. Nature. 2003;425(6958):628-33.
82. Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34(4):413-20.
83. Fliegauf M, Horvath J, von Schnakenburg C, Olbrich H, Muller D, Thumfart J, et al. Nephrocystin specifically localizes to the transition zone of renal and respiratory cilia and photoreceptor connecting cilia. J Am Soc Nephrol. 2006;17(9):2424-33.
84. Damerla RR, Cui C, Gabriel GC, Liu X, Craige B, Gibbs BC, et al. Novel Jbts17 mutant mouse model of Joubert syndrome with cilia transition zone defects and cerebellar and other ciliopathy related anomalies. Hum Mol Genet. 2015;24(14):3994-4005.
85. Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, et al. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38(2):155-7.
86. Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38(2):191-6.
87. Jonassen JA, SanAgustin J, Baker SP, Pazour GJ. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J Am Soc Nephrol. 2012;23(4):641-51.
88. Jonassen JA, San Agustin J, Follit JA, Pazour GJ. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol. 2008;183(3):377-84.
89. Hwang SH, Somatilaka BN, Badgandi H, Palicharla VR, Walker R, Shelton JM, et al. Tulp3 Regulates Renal Cystogenesis by Trafficking of Cystoproteins to Cilia. Curr Biol. 2019;29(5):790-802 e5.
90. Walker RV, Keynton JL, Grimes DT, Sreekumar V, Williams DJ, Esapa C, et al. Ciliary exclusion of Polycystin-2 promotes kidney cystogenesis in an autosomal dominant polycystic kidney disease model. Nat Commun. 2019;10(1):4072.
91. Cai Y, Fedeles SV, Dong K, Anyatonwu G, Onoe T, Mitobe M, et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J Clin Invest. 2014;124(12):5129-44.
92. Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45(9):1004-12.
93. Legue E, Liem KF, Jr. Tulp3 Is a Ciliary Trafficking Gene that Regulates Polycystic Kidney Disease. Curr Biol. 2019;29(5):803-12e5.
94. Badgandi HB, Hwang SH, Shimada IS, Loriot E, Mukhopadhyay S. Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J Cell Biol. 2017;216(3):743-60.
95. Su X, Driscoll K, Yao G, Raed A, Wu M, Beales PL, et al. Bardet-Biedl syndrome proteins 1 and 3 regulate the ciliary trafficking of polycystic kidney disease 1 protein. Hum Mol Genet. 2014;23(20):5441-51.
96. Zhang C, Balbo B, Ma M, Zhao J, Tian X, Kluger Y, et al. Cyclin- Dependent Kinase 1 Activity Is a Driver of Cyst Growth in Polycystic Kidney Disease. J Am Soc Nephrol. 2021;32(1):41-51.
97. Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184(1):71-9.
98. Delling M, Indzhykulian AA, Liu X, Li Y, Xie T, Corey DP, et al. Primary cilia are not calcium-responsive mechanosensors. Nature. 2016;531(7596):656-60.
99. Revell DZ, Yoder BK. Intravital visualization of the primary cilium, tubule flow, and innate immune cells in the kidney utilizing an abdominal window imaging approach. Methods Cell Biol. 2019;154:67-83.
100. O'Connor AK, Malarkey EB, Berbari NF, Croyle MJ, Haycraft CJ, Bell PD, et al. An inducible CiliaGFP mouse model for in vivo visualization and analysis of cilia in live tissue. Cilia. 2013;2(1):8.
101. Kurbegovic A, Kim H, Xu H, Yu S, Cruanes J, Maser RL, et al. Novel functional complexity of polycystin-1 by GPS cleavage in vivo: role in polycystic kidney disease. Mol Cell Biol. 2014;34(17):3341-53.
102. Trudel M, Yao Q, Qian F. The Role of G-Protein-Coupled Receptor Proteolysis Site Cleavage of Polycystin-1 in Renal Physiology and Polycystic Kidney Disease. Cells. 2016;5(1).
103. Parnell SC, Magenheimer BS, Maser RL, Rankin CA, Smine A, Okamoto T, et al. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun. 1998;251(2):625-31.
104. Yu W, Kong T, Beaudry S, Tran M, Negoro H, Yanamadala V, et al. Polycystin-1 protein level determines activity of the Galpha12/ JNK apoptosis pathway. J Biol Chem. 2010;285(14):10243-51.
105. Delmas P, Nomura H, Li X, Lakkis M, Luo Y, Segal Y, et al. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J Biol Chem. 2002;277(13):11276-83.
106. Li Q, Onozato ML, Andrews PM, Chen CW, Paek A, Naphas R, et al. Automated quantification of microstructural dimensions of the human kidney using optical coherence tomography (OCT). Opt Express. 2009;17(18):16000-16.
107. Grantham JJ, Mulamalla S, Grantham CJ, Wallace DP, Cook LT, Wetzel LH, et al. Detected renal cysts are tips of the iceberg in adults with ADPKD. Clin J Am Soc Nephrol. 2012;7(7):1087-93.
108. Grantham JJ, Geiser JL, Evan AP. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987;31(5):1145-52.
109. Grantham JJ, Mangoo-Karim R, Uchic ME, Grant M, Shumate WA, Park CH, et al. Net fluid secretion by mammalian renal epithelial cells: stimulation by cAMP in polarized cultures derived from established renal cells and from normal and polycystic kidneys. Trans Assoc Am Physicians. 1989;102:158-62.
110. Grantham JJ, Uchic M, Cragoe EJ, Jr., Kornhaus J, Grantham JA, Donoso V, et al. Chemical modification of cell proliferation and fluid secretion in renal cysts. Kidney Int. 1989;35(6):1379-89.
111. Mangoo-Karim R, Uchic M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci U S A. 1989;86(15):6007-11.
112. McAteer JA, Evan AP, Gardner KD. Morphogenetic clonal growth of kidney epithelial cell line MDCK. Anat Rec. 1987;217(3):229-39.
113. Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, et al. Early embryonic renal tubules of wildtype and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(-) Co-transporter-dependent cystic dilation. J Am Soc Nephrol. 2006;17(12):3424-37.
114. O'Sullivan DA, Torres VE, Gabow PA, Thibodeau SN, King BF, Bergstralh EJ. Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1998;32(6):976-83.
115. Persu A, Devuyst O, Lannoy N, Materne R, Brosnahan G, Gabow PA, et al. CF gene and cystic fibrosis transmembrane conductance regulator expression in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2000;11(12):2285-96.
116. Cook SJ, McCormick F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993;262(5136):1069-72.
117. Hordijk PL, Verlaan I, Jalink K, van Corven EJ, Moolenaar WH. cAMP abrogates the p21ras-mitogen-activated protein kinase pathway in fibroblasts. J Biol Chem. 1994;269(5):3534-8.
118. Heasley LE, Senkfor SI, Winitz S, Strasheim A, Teitelbaum I, Berl T. Hormonal regulation of MAP kinase in cultured rat inner medullary collecting tubule cells. Am J Physiol. 1994;267(3 Pt 2):F366-73.
119. Yamada T, Terada Y, Homma MK, Nonoguchi H, Sasaki S, Yuasa Y, et al. AVP inhibits EGF-stimulated MAP kinase cascade in Madin-Darby canine kidney cells. Kidney Int. 1995;48(3):745-52.
120. Matousovic K, Tsuboi Y, Walker H, Grande JP, Dousa TP. Inhibitors of cyclic nucleotide phosphodiesterase isozymes block renal tubular cell proliferation induced by folic acid. J Lab Clin Med. 1997;130(5):487-95.
121. Yamaguchi T, Nagao S, Takahashi H, Ye M, Grantham JJ. Cyst fluid from a murine model of polycystic kidney disease stimulates fluid secretion, cyclic adenosine monophosphate accumulation, and cell proliferation by Madin-Darby canine kidney cells in vitro. Am J Kidney Dis. 1995;25(3):471-7.
122. Ye M, Grant M, Sharma M, Elzinga L, Swan S, Torres VE, et al. Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J Am Soc Nephrol. 1992;3(4):984-94.
123. Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11(7):1179-87.
124. Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signalregulated kinase pathway. Kidney Int. 2000;57(4):1460-71.
125. Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17(1):178-87.
126. Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279(39):40419- 30.
127. Winkler SN, Torikai S, Levine BS, Kurokawa K. Effect of somatostatin on vasopressin-induced antidiuresis and renal cyclic AMP of rats. Miner Electrolyte Metab. 1982;7(1):8-14.
128. Friedlander G, Amiel C. Somatostatin and alpha 2-adrenergic agonists selectively inhibit vasopressin-induced cyclic AMP accumulation in MDCK cells. FEBS Lett. 1986;198(1):38-42.
129. Ruggenenti P, Remuzzi A, Ondei P, Fasolini G, Antiga L, Ene- Iordache B, et al. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68(1):206-16.
130. Meijer E, Visser FW, van Aerts RMM, Blijdorp CJ, Casteleijn NF, D'Agnolo HMA, et al. Effect of Lanreotide on Kidney Function in Patients With Autosomal Dominant Polycystic Kidney Disease: The DIPAK 1 Randomized Clinical Trial. JAMA. 2018;320(19):2010-9.
131. Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3',5'-cyclic monophosphate. Gastroenterology. 2007;132(3):1104- 16.
132. Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Gradilone SA, Huang B, et al. Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology. 2013;58(1):409-21.
133. Mutig K, Paliege A, Kahl T, Jons T, Muller-Esterl W, Bachmann S. Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol. 2007;293(4):F1166-77.
134. Hoffert JD, Chou CL, Fenton RA, Knepper MA. Calmodulin is required for vasopressin-stimulated increase in cyclic AMP production in inner medullary collecting duct. J Biol Chem. 2005;280(14):13624-30.
135. Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH, 2nd. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med. 2004;10(4):363-4.
136. Wang X, Gattone V, 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005;16(4):846-51.
137. Reif GA, Yamaguchi T, Nivens E, Fujiki H, Pinto CS, Wallace DP. Tolvaptan inhibits ERK-dependent cell proliferation, Cl(-) secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol. 2011;301(5):F1005-13.
138. Schaeffer HJ, Weber MJ. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol. 1999;19(4):2435-44.
139. Lee S, Rauch J, Kolch W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int J Mol Sci. 2020;21(3).
140. Smith AO, Jonassen JA, Preval KM, Davis RJ, Pazour GJ. c-Jun N-terminal kinase (JNK) signaling contributes to cystic burden in polycystic kidney disease. PLoS Genet. 2021;17(12):e1009711.
141. Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, et al. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76(6):1025-37.
142. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239-52.
143. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19(2):142-9.
144. Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J Biol Chem. 1996;271(44):27225-8.
145. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596-606.
146. Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15(11):2760-70.
147. Park H, Iqbal S, Hernandez P, Mora R, Zheng K, Feng Y, et al. Structural basis and biological consequences for JNK2/3 isoform selective aminopyrazoles. Sci Rep. 2015;5:8047.
148. Koch P, Gehringer M, Laufer SA. Inhibitors of c-Jun N-terminal kinases: an update. J Med Chem. 2015;58(1):72-95.
149. Duong MTH, Lee JH, Ahn HC. C-Jun N-terminal kinase inhibitors: Structural insight into kinase-inhibitor complexes. Comput Struct Biotechnol J. 2020;18:1440-57.
150. Vernia S, Edwards YJ, Han MS, Cavanagh-Kyros J, Barrett T, Kim JK, et al. An alternative splicing program promotes adipose tissue thermogenesis. Elife. 2016;5.
151. Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7(11):2135-48.
152. Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science. 1995;267(5196):389-93.
153. Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21(3):326-9.
154. Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath I, Guertl B, et al. Functions of c-Jun in liver and heart development. J Cell Biol. 1999;145(5):1049-61.
155. Wernig G, Chen SY, Cui L, Van Neste C, Tsai JM, Kambham N, et al. Unifying mechanism for different fibrotic diseases. Proc Natl Acad Sci U S A. 2017;114(18):4757-62.
156. Mariani O, Brennetot C, Coindre JM, Gruel N, Ganem C, Delattre O, et al. JUN oncogene amplification and overexpression block adipocytic differentiation in highly aggressive sarcomas. Cancer Cell. 2007;11(4):361-74.
157. Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J. 2002;21(15):4104-13.
158. Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 2000;103(6):897-907.
159. De Borst MH, Prakash J, Melenhorst WB, van den Heuvel MC, Kok RJ, Navis G, et al. Glomerular and tubular induction of the transcription factor c-Jun in human renal disease. J Pathol. 2007;213(2):219-28.
160. Le NH, van der Wal A, van der Bent P, Lantinga-van Leeuwen IS, Breuning MH, van Dam H, et al. Increased activity of activator protein-1 transcription factor components ATF2, c-Jun, and c-Fos in human and mouse autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2005;16(9):2724-31.
161. Arnould T, Kim E, Tsiokas L, Jochimsen F, Gruning W, Chang JD, et al. The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinasedependent activation of the transcription factor AP-1. J Biol Chem. 1998;273(11):6013-8.
162. Arnould T, Sellin L, Benzing T, Tsiokas L, Cohen HT, Kim E, et al. Cellular activation triggered by the autosomal dominant polycystic kidney disease gene product PKD2. Mol Cell Biol. 1999;19(5):3423-34.
163. Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf AM, Calvet JP. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J Biol Chem. 2002;277(22):19566-72.
164. Le NH, van der Bent P, Huls G, van de Wetering M, Loghman- Adham M, Ong AC, et al. Aberrant polycystin-1 expression results in modification of activator protein-1 activity, whereas Wnt signaling remains unaffected. J Biol Chem. 2004;279(26):27472-81.
165. Parnell SC, Magenheimer BS, Maser RL, Pavlov TS, Havens MA, Hastings ML, et al. A mutation affecting polycystin-1 mediated heterotrimeric G-protein signaling causes PKD. Hum Mol Genet. 2018;27(19):3313-24.
166. Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98(24):13681-6.
167. Wu Q, Wu W, Jacevic V, Franca TCC, Wang X, Kuca K. Selective inhibitors for JNK signalling: a potential targeted therapy in cancer. J Enzyme Inhib Med Chem. 2020;35(1):574-83.
168. Wang Y, Ji HX, Xing SH, Pei DS, Guan QH. SP600125, a selective JNK inhibitor, protects ischemic renal injury via suppressing the extrinsic pathways of apoptosis. Life Sci. 2007;80(22):2067-75.
169. Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, et al. Discovery of potent and selective covalent inhibitors of JNK. Chem Biol. 2012;19(1):140-54.
170. Chiquet C, Aptel F, Creuzot-Garcher C, Berrod JP, Kodjikian L, Massin P, et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am J Ophthalmol. 2017;174:76-84.
171. Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka TA, Matsuhisa M, et al. Possible novel therapy for diabetes with cellpermeable JNK-inhibitory peptide. Nat Med. 2004;10(10):1128-32.
172. Goodfellow VS, Loweth CJ, Ravula SB, Wiemann T, Nguyen T, Xu Y, et al. Discovery, synthesis, and characterization of an orally bioavailable, brain penetrant inhibitor of mixed lineage kinase 3. J Med Chem. 2013;56(20):8032-48.
173. Tomita K, Kohli R, MacLaurin BL, Hirsova P, Guo Q, Sanchez LHG, et al. Mixed-lineage kinase 3 pharmacological inhibition attenuates murine nonalcoholic steatohepatitis. JCI Insight. 2017;2(15).
174. Dong W, Embury CM, Lu Y, Whitmire SM, Dyavarshetty B, Gelbard HA, et al. The mixed-lineage kinase 3 inhibitor URMC-099 facilitates microglial amyloid-beta degradation. J Neuroinflammation. 2016;13(1):184.
175. Kiyota T, Machhi J, Lu Y, Dyavarshetty B, Nemati M, Zhang G, et al. URMC-099 facilitates amyloid-beta clearance in a murine model of Alzheimer's disease. J Neuroinflammation. 2018;15(1):137.
176. Liles JT, Corkey BK, Notte GT, Budas GR, Lansdon EB, Hinojosa- Kirschenbaum F, et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J Clin Invest. 2018;128(10):4485-500.
177. Chertow GM, Pergola PE, Chen F, Kirby BJ, Sundy JS, Patel UD, et al. Effects of Selonsertib in Patients with Diabetic Kidney Disease. J Am Soc Nephrol. 2019;30(10):1980-90.
178. Younossi ZM, Stepanova M, Lawitz E, Charlton M, Loomba R, Myers RP, et al. Improvement of hepatic fibrosis and patientreported outcomes in non-alcoholic steatohepatitis treated with selonsertib. Liver Int. 2018;38(10):1849-59.
179. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology. 2018;67(2):549-59.