Abstract
Fatty acid oxidation disorders unfortunately can result in the sudden unexplained death of infants. Mitochondrial trifunctional protein (MTP) deficiency is one such disease where long-chain fatty acids cannot be fully oxidized through beta-oxidation which, can lead to cardiac arrythmias in an infant. Furthermore, mothers who are carrying an MTP deficient fetus have a prevalence for pregnancy complications, especially AFLP, acute fatty liver of pregnancy and HELLP syndrome. To better understand the etiology of the potential pro-arrhythmic state the MTP deficient infants may enter, we developed an in vitro model of MTP deficiency in cardiomyocytes to elucidate the underpinning molecular mechanism of this disease. Using CRISPR/Cas9, we developed MTP deficient mutant and knockout pluripotent stem cell lines. Furthermore, we generated patient derived induced pluripotent stem cell lines harboring a so-called founder mutation, the most commonly identified alteration in MTP in the population. Upon differentiating these mutant stem cells into cardiomyocytes and then challenging with fatty acids, we observed pro-arrhythmic behavior, depressed mitochondrial energetics, and elevated hydroxylated long-chain fatty acids, all perhaps expected phenotypes due to MTP deficiency. However, unexpectedly, we also identified an inability of these disease cardiomyocytes to generate mature cardiolipin. Cardiolipin is a key pillar of the powerhouse of life, mitochondria. For the first time this disease-in-a-dish model revealed the key culprit for the dramatic MTP mutant mitochondrial defects and identified potentially a second role for the enzyme HADHA in MTP. HADHA is required for the biosynthesis of functional cardiolipin and therefore healthy mitochondria. However, in the disease, defective cardiolipin results in mitochondrial abnormalities and cardiac arrythmias in infants. These studies reveal an important target for sudden infant death syndrome therapy. With this foundational work on an in vitro model of MTP deficiency and potential avenues for therapy, the next important task is to extend this model to address fetal-maternal interactions towards better governing maternal health.
Commentary
Mitochondrial trifunctional protein (MTP) deficiency is a disease that manifests in the cardiac system after birth. This is thought to occur due to the change in newborn diet at birth where the infant begins consumption of a mother’s breast milk, which is full of many essential and important molecules, including antibodies. Unfortunately for MTP deficient infants, breast milk is also high in fats, especially in long-chain fatty acids that MTP patients can not break down. This process then precipitates in disease pathology.
MTP deficiency is caused by mutations in either hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase /enoyl-CoA hydratase subunit A (HADHA) or subunit B (HADHB). The result is a severe limitation in long-chain fatty acid oxidation (FAO) and is considered one of the more severe FAO disorders without pharmacological treatment [1]. Mitochondrial FAO disorders are recessively inherited, and defects in FAO are estimated to affect ~1 in 10,000 newborns [2]. This results in around 400 infants per year born in the USA with this condition that is potentially lethal and has no cure. In particular regions of Poland the frequency is over ten-fold higher [3]. Unfortunately, a mother carrying a MTP deficient fetus has a 30% chance of exhibiting pregnancy complications [4]. In particular, if the mother has a mutation in one copy of HADHA and the fetus has two copies of the HADHA mutation, the mother is more likely to have acute fatty liver of pregnancy (AFLP) and HELLP syndrome (hemolysis, elevated liver enzymes and low platelet count). Yet, little is known about how the mother develops AFLP and/or HELLP syndrome. MTP deficiency and human stem cell derived cell types provide an opportunity to better understand human cellto- cell communication in human physiology and disease pathology [5].
MTP deficiency results in sudden unexplained infant death, Reye-like syndrome, cardiomyopathy and/or skeletal myopathy [1,6,7]. A major phenotype of MTPdeficient newborns is sudden infant death syndrome (SIDS), which manifests after birth once the child begins nursing on lipid-rich breast milk. Defects in FAO have a role in promoting a pro-arrhythmic cardiac environment, however, the exact mechanism of action was not understood, and there are no current therapies [8,9]. We (Miklas et al.) recently used human induced pluripotent stem cells from a HADHA patient and HADHA CRISPR edited iPSCs to model human MTP deficient cardiomyocytes, heart cells, to better understand the cardiac pathology [5]. Two key findings were made. The first was that HADHA mutant cardiomyocytes, when challenged with fatty acids, had defective calcium dynamics and repolarization kinetics which resulted in a pro-arrhythmic state. Second, defective HADHA led to abnormal cardiolipin remodeling resulting in the inability to produce and possibly maintain the acylchain composition of mature cardiolipin [10].
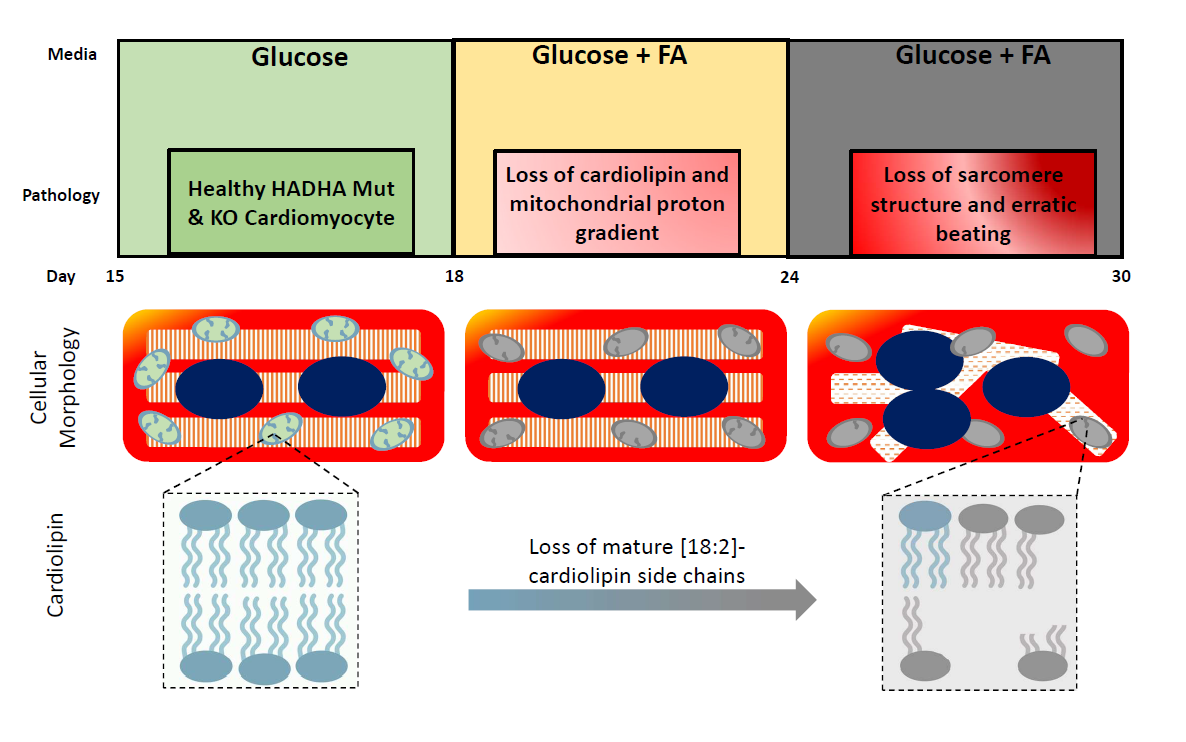
Cardiolipins are a critical component of the mitochondrial inner membrane. Cardiolipin is an atypical phospholipid composed of four (instead of two) acyl-chains that are connected with a glycerol moiety. This atypical structure of cardiolipin results in a conical shape that is thought to be critical for inner mitochondrial membrane structure and function [11]. In particular, cardiolipin has been shown to function in organizing the electron transport chain (ETC) higher order structure, important for ETC activity, and acts as a proton trap on the outer leaflet of the inner mitochondrial membrane [12]. Hence, the reduction of the mature form of cardiolipin results in mitochondrial abnormalities such as proton gradient loss, ETC depression resulting in depressed ATP production and abnormal mitochondrial architecture [13].
Pathological remodeling of cardiolipin has been implicated previously in the mitochondrial dysfunction observed in diabetes, heart failure, neurodegeneration, and aging [13,14]. However, the pattern and composition of abnormal cardiolipin species in the case of the HADHA mutant and knockout cardiomyocytes were more specific than seen previously in heart failure or diabetes, suggesting that HADHA may be directly involved in cardiolipin processing. Interestingly, previous studies using HeLa cells have suggested HADHA exhibits acyl-CoA transferase activity upon monolyso-cardiolipin for its remodeling into cardiolipin [15]. As such, these data suggest that defects in HADHA directly cause impaired cardiolipin remodeling resulting in the inability to produce and possibly maintain the acyl-chain composition of mature cardiolipin [10]. However, the exact contribution of this acyltransferase to physiological cardiolipin remodeling has been unclear [15]. Miklas et al. report that, FA challenged human HADHA mutant and knockout cardiomyocytes have reduced mature tetra[18:2]-cardiolipin and compromised mitochondrial activity (Figure 1). This is similar to previously seen findings in tafazzin mutants causing Barth’s syndrome [16], an X-linked cardiac and skeletal mitochondrial myopathy [16,17]. These data, for the first time, establish the exact contribution of HADHA acyltransferase to physiological cardiolipin remodeling in human cardiomyocytes and open the door for future treatments and therapies. In particular, cardiolipin has previously been considered as a druggable target and the existing and future molecules affecting and stabilizing cardiolipin have a strong potential to become therapeutic agents for SIDS.
Model maternal pregnancy complications using MTP deficient stem cell derived placental cells
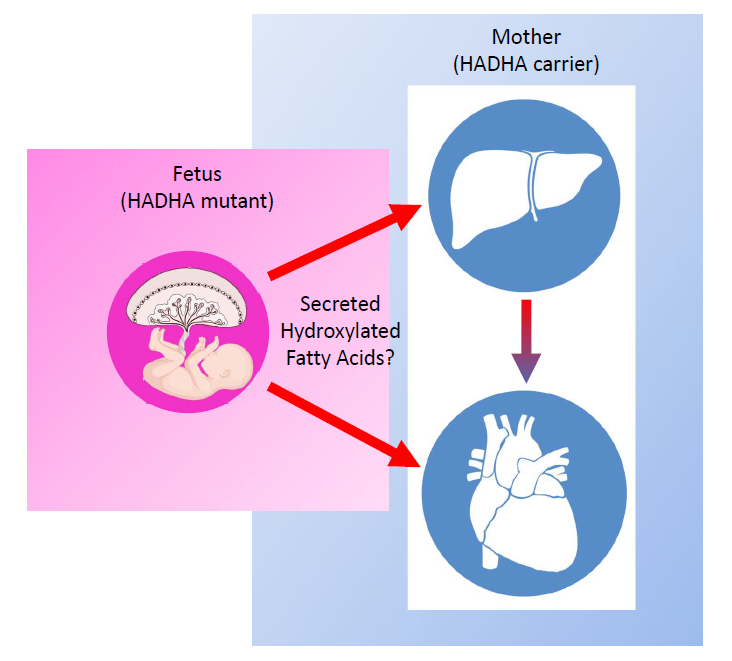
Now that the foundational cell culture framework has been generated to study MTP deficiency in vitro in cardiomyocytes [5], it is important in the future to extend this in vitro model to better understand AFLP and HELLP syndrome that occur during pregnancy [18]. Using recent advances in stem cell biology, it is possible to stably culture trophoblast stem cells, the cells that form the placenta, either directly from an embryo [19] or by reprogramming embryonic stem cells [20]. It has been shown that the placenta has a large amount of fatty acid oxidation enzymes present, including mitochondrial tri-functional protein [21]. It is hypothesized that placental tissue may take up free fatty acids from the mother’s blood and incorrectly process those fatty acids in a similar manner as shown in our work and patients. As a result, an intermediate fatty acid, such as hydroxylated long-chain fatty acids, may accumulate and be exported back into the mother’s blood from the placental tissue. However, the specific lipid substrates the placental tissue uses for fatty acid oxidation, what fatty acid substrates might be transported to the fetus, and whether or not processed fatty acids can be exported back into the mother’s blood remain unknown. By using trophoblast stem cells to generate the working cells found in the placenta, such as the villous cytotrophoblast and/ or extravillous trophoblast [22], one could use global lipidomics techniques to assess fatty acid content of these cells and their supernatant in the wild type and HADHA knockout state. By better understanding the fundamental fatty acid metabolism underlying placental biology, during health and disease, has the potential to better understand the fundamental way in which development occurs and the role the placenta has in being a gatekeeper of certain metabolic substrates for the fetus.
Finally, leveraging human-on-a-chip co-culture in vitro platforms [23], organ-organ lipid communication can be modeled to understand how, placental tissue lacking the HADHA gene may impact the health of a heterozygous mother’s liver to result in AFLP and HELLP syndrome (Figure 2). In doing so, ideally a causal lipid or set of lipids may be identified that result in exacerbating the mother’s liver’s limited ability to process long-chain fattyacids resulting in these complications. Furthermore, using such a platform, novel drug, protein or gene interventions can be tested to identify means of rescuing aberrant lipid molecule signaling. These findings could have generalizable uses in the wider array of AFLP and HEELP syndromes complications that unfortunately arise during so many pregnancies worldwide.
Funding
This work was partly supported by P01 GM081619 for HRB.
References
2. Zytkovicz TH, Fitzgerald EF, Marsden D, Larson CA, Shih VE, Johnson DM, Strauss AW, Comeau AM, Eaton RB, Grady GF. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clinical Chemistry. 2001 Nov 1;47(11):1945-55.
3. Nedoszytko B, Siemińska A, Strapagiel D, Dąbrowski S, Słomka M, Sobalska-Kwapis M, et al. High prevalence of carriers of variant c. 1528G> C of HADHA gene causing long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) in the population of adult Kashubians from North Poland. PloS one. 2017 Nov 2;12(11):e0187365.
4. Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, et al. A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. New England Journal of Medicine. 1999 Jun 3;340(22):1723-31.
5. Miklas JW, Clark E, Levy S, Detraux D, Leonard A, Beussman K, et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nature Communications. 2019;10:4671.
6. Tonin AM, Amaral AU, Busanello EN, Grings M, Castilho RF, Wajner M. Long-chain 3-hydroxy fatty acids accumulating in long-chain 3-hydroxyacyl-CoA dehydrogenase and mitochondrial trifunctional protein deficiencies uncouple oxidative phosphorylation in heart mitochondria. Journal of Bioenergetics and Biomembranes. 2013 Feb 1;45(1-2):47-57.
7. Tyni T, Pihko H. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatrica. 1999 Mar;88(3):237-45.
8. Vishwanath VA. Fatty acid beta-oxidation disorders: a brief review. Annals of Neurosciences. 2016;23(1):51-5.
9. Wajner M, Amaral AU. Mitochondrial dysfunction in fatty acid oxidation disorders: insights from human and animal studies. Bioscience Reports. 2016 Feb 1;36(1).
10. Li J, Romestaing C, Han X, Li Y, Hao X, Wu Y, et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metabolism. 2010 Aug 4;12(2):154-65.
11. Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. Journal of Cell Biology. 2011 Jan 10;192(1):7-16.
12. Arnarez C, Marrink SJ, Periole X. Identification of cardiolipin binding sites on cytochrome c oxidase at the entrance of proton channels. Scientific Reports. 2013 Feb 12;3:1263.
13. Claypool SM, Koehler CM. The complexity of cardiolipin in health and disease. Trends in Biochemical Sciences. 2012 Jan 1;37(1):32-41.
14. Lane RK, Hilsabeck T, Rea SL. The role of mitochondrial dysfunction in age-related diseases. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2015 Nov 1;1847(11):1387-400.
15. Taylor WA, Mejia EM, Mitchell RW, Choy PC, Sparagna GC, Hatch GM. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PloS one. 2012 Nov 9;7(11):e48628.
16. Spencer CT, Byrne BJ, Gewitz MH, Wechsler SB, Kao AC, Gerstenfeld EP, Merliss AD, Carboni MP, Bryant RM. Ventricular arrhythmia in the X-linked cardiomyopathy Barth syndrome. Pediatric cardiology. 2005 Oct 1;26(5):632-7.
17. Schlame M, Ren M. Barth syndrome, a human disorder of cardiolipin metabolism. FEBS Letters. 2006 Oct 9;580(23):5450-5.
18. Goel A, Jamwal KD, Ramachandran A, Balasubramanian KA, Eapen CE. Pregnancy-related liver disorders. Journal of Clinical and Experimental Hepatology. 2014 Jun 1;4(2):151-62.
19. Okae H, Toh H, Sato T, Hiura H, Takahashi S, Shirane K, et al. Derivation of human trophoblast stem cells. Cell Stem Cell. 2018 Jan 4;22(1):50-63.
20. Dong C, Beltcheva M, Gontarz P, Zhang B, Popli P, Fischer LA, Khan SA, Park KM, Yoon EJ, Xing X, Kommagani R. Derivation of trophoblast stem cells from naïve human pluripotent stem cells. Elife. 2020 Feb 12;9:e52504.
21. Shekhawat P, Bennett MJ, Sadovsky Y, Nelson DM, Rakheja D, Strauss AW. Human placenta metabolizes fatty acids: implications for fetal fatty acid oxidation disorders and maternal liver diseases. American Journal of Physiology-Endocrinology and Metabolism. 2003 Jun 1;284(6):E1098-105.
22. Vento-Tormo R, Efremova M, Botting RA, Turco MY, Vento-Tormo M, Meyer KB, et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature. 2018 Nov;563(7731):347-53.
23. Herland A, Maoz BM, Das D, Somayaji MR, Prantil- Baun R, Novak R, et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nature Biomedical Engineering. 2020 Apr;4(4):421-36.