Abstract
Acute myeloid leukemia (AML) is a type of blood malignancies with high genetic heterogeneity. Epigenetic alterations including histone acetylation have been found to be the key genetic variations driving the occurrence of AML. However, targeting histone acetylation remains to be unsuccessful in leukemia therapy. Recently, acidic nuclear phosphoprotein 32 family member A (ANP32A) was shown to be a novel biomarker of unfavorable outcome for leukemia and promote AML by stimulating histone 3 (H3) acetylation and the abnormal expression of lipid metabolism genes. A small peptide that competitively binds to H3 to block the interaction of ANP32A and H3 exhibited therapeutic effect in vitro and in vivo. Here, we further explore the potential anti-leukemia strategy by targeting ANP32A that consequently lead to alteration of H3 acetylation. In view of the therapeutic significance of targeting ANP32A in AML, the intervention tactics as well as its clinical perspectives and challenges will be adequately discussed.
Keywords
ANP32A, AML, H3 Acetylation, H3BP, Targeted intervention
Background
Acute myeloid leukemia (AML) is a type of hematological malignancies that originate from hematopoietic stem/progenitor cells in myeloid lineage [1]. The genetic heterogeneity of AML causes varied responses to the existing treatment options, as well as drug resistance and refractory/recurrent, posing a challenge to personalized precision medicine[2]. Therefore, it is urgent to identify novel molecular targets, discover patient specific and disease specific risk factors, and explore effective combinations of modalities and drugs in the foreseeable future.
Disorders of histone modification patterns in acetylation, methylation, and phosphorylation are important features of epigenetic inheritance that closely related to aberrant gene expression profiles associated with different disease states in leukemic cells. Epigenetic modifications of histone play important roles in the initiation, development, drug resistance and even relapse or refractory of leukemia [3,4], of which histone acetylation was noted to be associated with transcriptionally active chromatin and the intervention of histone acetylation is a potential therapeutic strategy against AML [5]. However, acetylation modification of histone proteins is a highly regulated and reversible process and coupled with the overall metabolic state of a cell [6]. The role and mechanism of histone acetylation in the occurrence and treatment of leukemia have not been fully revealed that obstructs the utilization and development of optimal therapeutic strategies [5]. Targeting histone acetylation in anti-AML therapy still faces many challenges.
ANP32A is a H3 Acetylation Regulator and Promising Target for AML
Acidic nuclear phosphoprotein 32 family member A (ANP32A) is an important epigenetic regulatory “cofactor” in the acetylation of core histone H3 [7,8], which highly expressed in varieties of solid tumors and promotes tumor proliferation [9-14]. Despite whether and how the oncogenic activity of ANP32A depends on histone acetylation remains debating, ANP32A deficiency is associated with reduced levels of histone acetylation in prostatic cancer TSU-Pr1 cells and cervical carcinoma HeLa S3 cells [14,15]. Our previous studies revealed ANP32A as a novel target and biomarker of unfavorable outcome in AML [16,17]. We found ANP32A dysregulation contributes to abnormal megakaryopoiesis in acute megakaryoblastic leukemia (AMKL) cells by regulating ERK signaling [16]. What's more, abnormally high ANP32A is indispensable for the maintenance of malignant survival of AML cells and promotes leukemogenesis by increasing H3 acetylation and gene expression on key biological processes including the lipid metabolism [17]. A recent report evaluating the prognostic significance of ANP32A through multivariable analysis reaffirmed the significance of high level ANP32A as an unfavorable prognostic biomarker in AML risk stratification and a potential therapeutic target for AML patients [18]. Interestingly, Anp32a null mice exhibits apparently negligible impact on steady hematopoiesis [16,17]. Thus, the differential effect in normal and malignant blood cells indicates that targeting ANP32A will be a promising epigenetic strategy for the treatment of AML.
Strategies on Targeting ANP32A to Intervene AML
ANP32A can bind to the disordered N-terminal tail of H3, and the sequence spanning 151-180 amino acids of ANP32A is necessary for its ability to bind H3 and regulate H3 acetylation in vitro [8,19]. We synthesized the peptide and termed it as H3-binding peptide (H3BP). We speculated that H3BP competed with ANP32A to interact with H3. Therefore, H3BP could be used to block ANP32A function and interpose leukemia (Figure 1). Indeed, our research verified that overexpression of H3BP-GFP fusion protein in AML cells competed with endogenous ANP32A to bind to H3, intervened ANP32A functionally, and suppressed leukemia cell proliferation in vitro. Then, we exploited the TAT, a cell penetrating peptide (Figure 1) derived from the α-helical domain of the TAT protein encoded by the human immunodeficiency virus type 1 (HIV1), to overcome the cell membrane barrier for macromolecular peptide delivery. This TAT-mediated drug delivery was studied in a large variety of pathological conditions including leukemia, solid tumor, ischemia and neurodegenerative diseases and more recently metabolic disorders and exhibited a good application prospect [19,20]. To explore the therapeutic potential, TAT-H3BP fusion peptide (Figure 1) was synthesized and its therapeutic effect was verified in AML cell lines, mouse model, as well as patient primary AML cells.
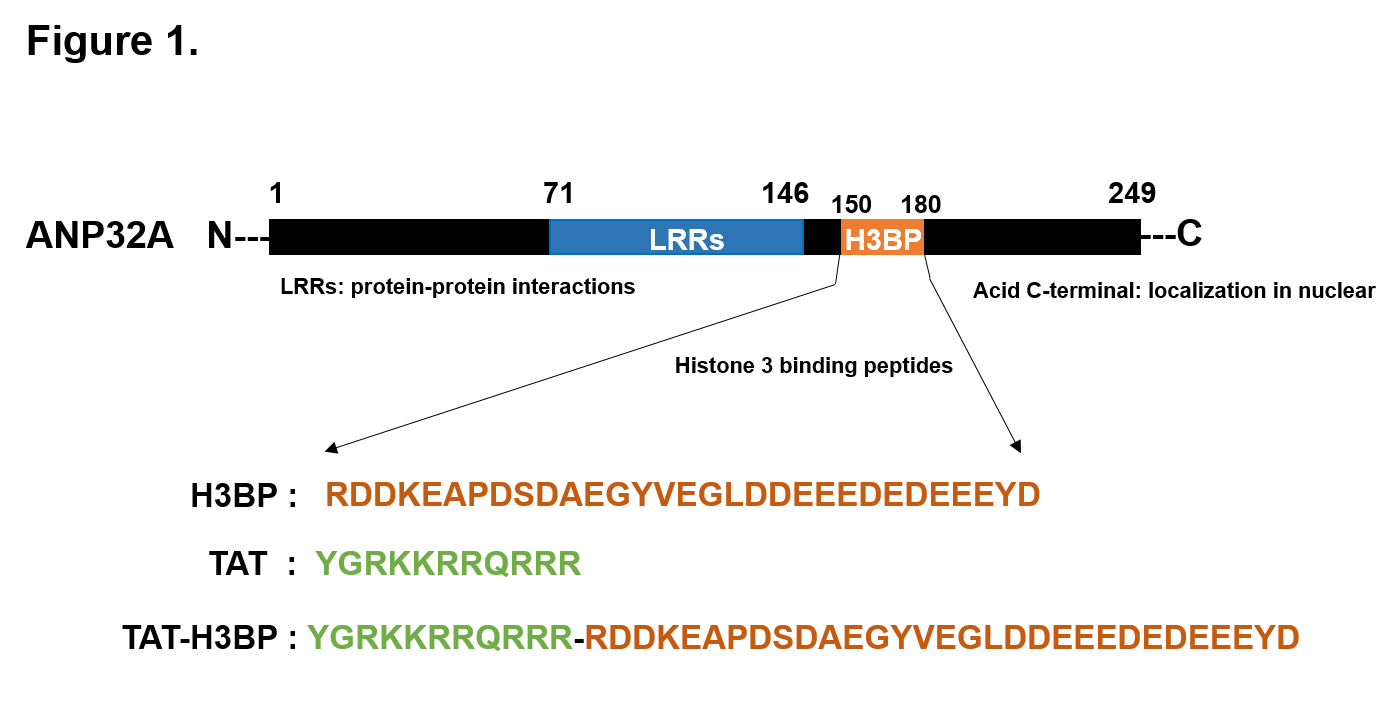
Figure 1. Key domain of ANP32A protein and the amino acid sequence of H3BP and TAT-H3BP fusion peptides. ANP32A protein consists of 249 amino acids (AA). The key structures include the highly conservative N-terminal LRR (Leucine-rich repeats) domain (71-146 AA) mainly mediating the interaction between ANP32A and other partners, the long acidic tail at the C-terminal carrying the nuclear localization signal, and the Histone 3 binding peptides (H3BP) which are indispensable for acetylation regulatory of H3 (150-180 AA) [7,43]. TAT-H3BP is synthesized by TAT delivery peptides fused on the N-terminal of H3BP.
The potential utility of H3BP in treating leukemia is supported by the fact that disruption of physical interaction of ANP32A and H3 by small molecule peptide TAT-H3BP mimics the biological activity of ANP32A deficiency in leukemia cells [21], significantly suppresses cell survival by decreasing acetyl-H3 enrichment and mRNA expression in genes of lipid metabolism including APOC1, PCSK9, and LPPR3, etc. Spectacularly, micromole concentration of TAT-H3BP peptide exhibits a sensitively inhibitory effect on samples from newly diagnosed AML patients and relapsed or refractory elderly patients above the age of 60 years that with high ANP32A expression (Table 1), but a little effect on samples isolated from normal cord blood with lower ANP32A level at the same micromole concentration. The addiction of leukemia cell to superfluous ANP32A may provide a therapeutic window and basis for the clinical application of TAT-H3BP peptide. Since the median overall survival after 5 years in 18–60 year AML patients is roughly 40%, however, with only around 10% surviving older patients above the age of 60 years under current standard chemotherapy that is far from being satisfactory [22,23], targeting ANP32A through H3BP may be an alternative way for therapy in those elderly AML patients. Thus, our works affirmed the effectiveness and feasibility of intervening H3 acetylation by targeting ANP32A in the treatment of leukemia.
| Patient ID | FBA classification | Gender | Source | Age (years) |
Status | Efficacy | ANP32A level (AML/Normal) |
|---|---|---|---|---|---|---|---|
| 33 | M2 | M | BM | 74 | Refractory | - | 0.691 |
| 75 | M1 | M | PB | 70 | Diagnosis | + | 1.680 |
| 85 | M1 | F | PB | 65 | Diagnosis | + | 2.477 |
| 37 | M2 | F | PB | 62 | Diagnosis | - | 1.030 |
| 60 | M2 | M | PB | 60 | Diagnosis | + | 5.096 |
| 84 | M2 | M | PB | 55 | Relapse | + | 1.266 |
| 57 | M2 | M | PB | 51 | Diagnosis | + | 2.024 |
| 83 | M2 | M | PB | 49 | Diagnosis | - | 1.980 |
| 19 | M2 | M | BM | 48 | Refractory | + | 1.200 |
| 22 | M2 | M | PB | 47 | Diagnosis | - | 0.924 |
| 20 | M1 | F | BM | 45 | Diagnosis | - | 0.901 |
| M: Male; F: Female; PB: Peripheral Blood; BM: Bone Marrow; + indicates inhibition and - indicates no inhibition. | |||||||
Table 1: TAT-H3BP treatment in AML patient samples.
Blocking ANP32A expression may be an alternative way to target ANP32A. The mechanism of commanding ANP32A overexpression in AML has not been reported. Exploring the key upstream signaling and corresponding drug that controls the ascent of ANP32A in AML may be another effective method to target ANP32A. It is worth mentioning that the transcription factor ChIP-sequence database in the UCSC Genome Browser exhibits a classic SP1 binding motif located on CpG island (hg19_chr15:69109709-69114001) of ANP32A gene promoter in K562 and HepG2 cell (Figure 2). Coincidentally, our preliminary experiment shown that Mithramycin A, a clinical anti-leukemia antibiotic as the canonical inhibitor of SP1, which decreases SP1 protein by inducing proteasome-dependent degradation and displaces SP1 from GC-rich regions of genes regulatory motif, efficiently suppressed ANP32A expression leading to apoptosis in AML cells. The combination of Mithramycin A and low-dose TAT-H3BP pre-treatment optimized the inhibitory effects of Mithramycin A on leukemia cells (data not shown). Therefore, we speculated that SP1 might be one upstream regulatory signal that triggers ANP32A overexpression in AML. The transcription factor SP1 (Specificity protein 1) belongs to the Sp/Kruppel (KLF) family, binds to GC-rich regions of regulatory motif of various genes and thus controls the transcription of genes responsible for proliferation, cell cycle regulation, and differentiation [24-26]. It has been reported as a metastasis risk and poor prognostic factor in multiple solid tumors, and an undertaking tumor therapeutic target [27-30]. Several studies have also uncovered the oncogenicity of SP1 in leukemia. Paranormal SP1 contributes to the drug resistance of leukemia stem cells by inducing the expression of Survivin through ERK-MSK MAPK signaling pathway and drives the expression of oncogene DHX15 in acute lymphoblastic leukemia [31,32]. Mithramycin A consequently exhibited anti-cancer effect through a SP1-sensitive action both as monotherapy and in combination with antitumor drugs [33,34] (Figure 3). It is likely that combination of TAT-H3BP and clinical agent Mithramycin A may provide a solution to alleviate dose-toxicity, off-target effects and drug resistance caused by single drug treatment, and will be an alternative tactic in targeted therapy in leukemia (Figure 3) [35].
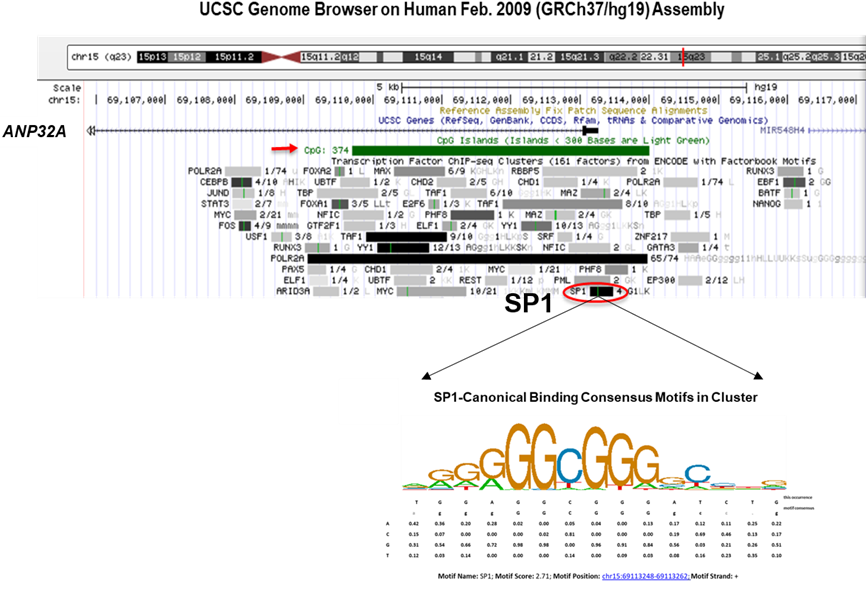
Figure 2. ANP32A promoter contains canonical SP1-binding motif. The potential SP1 binding site located on CpG island of ANP32A promoter and the matrix of SP1 consensus motifs in UCSC Database.
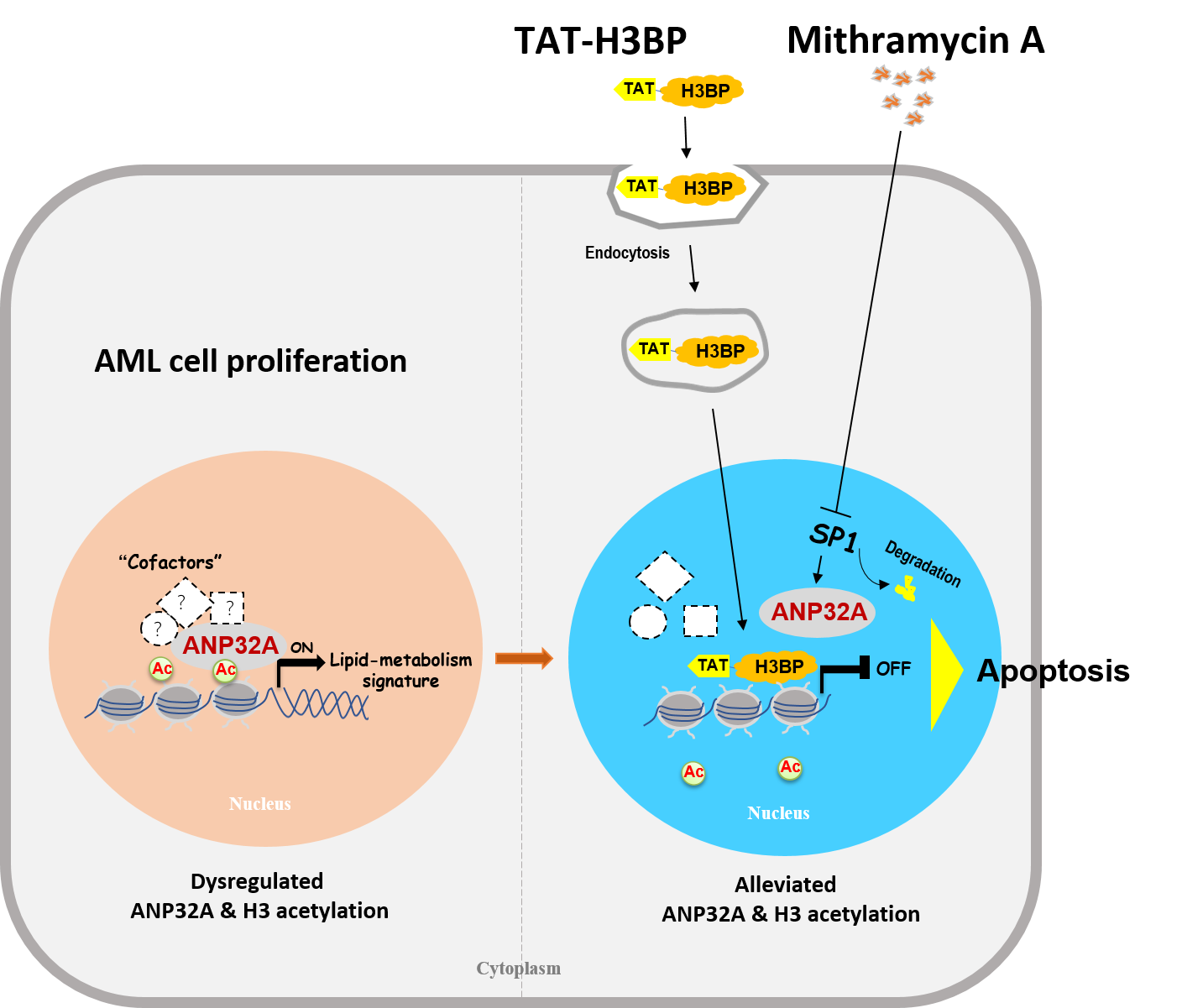
Figure 3. Combination of TAT-H3BP and Mithramycin A synergistically targets ANP32A to induce apoptosis of AML cell. Disruption of physical interaction of ANP32A and H3 by small peptide TAT-H3BP to alleviate disordered H3 acetylation and gene expression of lipid metabolism genes or/and inhibiting ANP32A expression by targeting the oncogene SP1 with the clinical drug Mithramycin A can be new strategy for AML therapy targeting ANP32A.
The Challenges and Perspectives of H3BP in Future
Our TAT-H3BP peptide basing on the molecular, cellular, and structural data with more explicit information shows great superiority on targeting ANP32A to disturb leukemia. However, the TAT delivered system still has some drawbacks including poor stability in blood circulation, potential immunogenicity, and non-organ/tissue/cell specificity. In addition, the reversible activity of TAT to move out cells may cause lower intra-cellular concentration of cargo [19,36]. That may partly limit the action of TAT-H3BP and the clinical applications of TAT-H3BP in the future. Novel drug delivery systems integrating modification of TAT with nanoparticles [37], specific targeting antibody or ligand [38,39], and liposome [40] with targeting or/and stimuli-responsive properties (triggered manually by pH, light, magnetic field, enzymes etc.) may facilitate the application of H3BP in leukemia therapy. Nevertheless, it is necessary to create a less toxic and leukemia-cell-targeted carriers for small H3BP delivery in future studies.
In comparison to the immaturity of peptide drug and the difficulty for clinical use in the future, small molecule inhibitors seem to be a better choice. Over the last decades, amplified luminescent proximity homogeneous assay (AlphaLISA) has emerged as one of the most reliable screening technologies for competitive blocking agent because of its versatility, sensitivity, and homogeneous format without the need for plate evacuation or washing [41,42]. The AlphaLISA screening system can be established based on the H3BP-dependent binding activity of ANP32A with H3. Hopefully, small molecule compounds disrupting the interaction may be identified for drug development.
Conclusions
In summary, our studies revealed that ANP32A is a key epigenetic regulators and therapeutic target for leukemia, provided the competitive intervention strategies of small peptide H3BP, and confirmed the effectiveness and feasibility of targeting ANP32A against leukemia. It is a novel treatment option different from the previously reported inhibitors specific to the "writers" or "readers" of H3 acetylation. These researches provide new scheme and idea for targeted therapy and precision medicine for AML.
Author Contributions Statement
Manman Wang and Zan Huang drafted and reviewed the manuscript.
Conflict of Interest
The authors declare that they have no conflicts of interest in this article.
References
2. Newell LF, Cook RJ. Advances in acute myeloid leukemia. The BMJ. 2021 Oct 6;375.
3. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New England Journal of Medicine. 2013 May 30;368(22):2059-74.
4. Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harbor Perspectives in Biology. 2016 Apr 1;8(4):a019521.
5. Benton CB, Fiskus W, Bhalla KN. Targeting histone acetylation: readers and writers in leukemia and cancer. The Cancer Journal. 2017 Sep 1;23(5):286-91.
6. Dhall A, Zee BM, Yan F, Blanco MA. Intersection of epigenetic and metabolic regulation of histone modifications in acute myeloid leukemia. Frontiers in Oncology. 2019 May 22;9:432.
7. Seo SB, Macfarlan T, McNamara P, Hong R, Mukai Y, Heo S, et al. Regulation of histone acetylation and transcription by nuclear protein pp32, a subunit of the INHAT complex. Journal of Biological Chemistry. 2002 Apr 19;277(16):14005-10.
8. Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell. 2001 Jan 12;104(1):119-30.
9. Velmurugan BK, Yeh KT, Lee CH, Lin SH, Chin MC, Chiang SL, et al. Acidic leucine-rich nuclear phosphoprotein-32A (ANP32A) association with lymph node metastasis predicts poor survival in oral squamous cell carcinoma patients. Oncotarget. 2016 Mar 8;7(10):10879-90.
10. Yan W, Bai Z, Wang J, Li X, Chi B, Chen X. ANP32A modulates cell growth by regulating p38 and Akt activity in colorectal cancer. Oncology Reports. 2017 Sep 1;38(3):1605-12.
11. Tian Z, Liu Z, Fang X, Cao K, Zhang B, Wu R, et al. ANP32A promotes the proliferation, migration and invasion of hepatocellular carcinoma by modulating the HMGA1/STAT3 pathway. Carcinogenesis. 2021 Mar;42(3):493-506.
12. Reilly PT, Yu Y, Hamiche A, Wang L. Cracking the ANP32 whips: important functions, unequal requirement, and hints at disease implications. Bioessays. 2014 Nov;36(11):1062-71.
13. Zhu BD, Li XL, Liu Y, Chang J, Liu Y, Zhang DD, et al. Involvement of hepatopoietin Cn in the development of human hepatocellular carcinoma. Clinical & Experimental Metastasis. 2010 Dec;27(8):571-80.
14. Brody JR, Kadkol SS, Hauer MC, Rajaii F, Lee J, Pasternack GR. pp32 reduction induces differentiation of TSU-Pr1 cells. The American Journal of Pathology. 2004 Jan 1;164(1):273-83.
15. Kadota S, Nagata K. pp32, an INHAT component, is a transcription machinery recruiter for maximal induction of IFN-stimulated genes. Journal of Cell Science. 2011 Mar 15;124(6):892-9.
16. Sun X, Lu B, Han C, Qiu W, Jin Q, Li D, et al. ANP32A dysregulation contributes to abnormal megakaryopoiesis in acute megakaryoblastic leukemia. Blood Cancer Journal. 2017 Dec 22;7(12):661.
17. Yang X, Lu B, Sun X, Han C, Fu C, Xu K, et al. ANP32A regulates histone H3 acetylation and promotes leukemogenesis. Leukemia. 2018 Jul;32(7):1587-97.
18. Huang S, Huang Z, Ma C, Luo L, Li YF, Wu YL, et al. Acidic leucine-rich nuclear phosphoprotein-32A expression contributes to adverse outcome in acute myeloid leukemia. Annals of Translational Medicine. 2020 Mar;8(6):345.
19. Berillo D, Yeskendir A, Zharkinbekov Z, Raziyeva K, Saparov A. Peptide-Based Drug Delivery Systems. Medicina. 2021 Nov;57(11):1209.
20. Rapoport M, Lorberboum-Galski H. TAT-based drug delivery system–new directions in protein delivery for new hopes?. Expert Opinion on Drug Delivery. 2009 May 1;6(5):453-63.
21. Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. Journal of Biological Chemistry. 1997 Jun 20;272(25):16010-7.
22. Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997-2002. Cancer Causes & Control. 2008 May;19(4):379-90.
23. Schlenk RF, Döhner H. Genomic applications in the clinic: use in treatment paradigm of acute myeloid leukemia. Hematology 2013, the American Society of Hematology Education Program Book. 2013 Dec 6;2013(1):324-30.
24. Wierstra I. Sp1: emerging roles-beyond constitutive activation of TATA-less housekeeping genes. Biochem. Biophys. Res. Commun.. 2008;372:1-13.
25. Wierstra I, Alves J. FOXM1c and Sp1 transactivate the P1 and P2 promoters of human c-myc synergistically. Biochemical and Biophysical Research Communications. 2007 Jan 5;352(1):61-8.
26. De Borja PF, Collins NK, Du P, Azizkhan-Clifford J, Mudryj M. Cyclin A–CDK phosphorylates Sp1 and enhances Sp1-mediated transcription. The EMBO Journal. 2001 Oct 15;20(20):5737-47.
27. Wang X, Yan Z, Fulciniti M, Li Y, Gkotzamanidou M, Amin SB, et al. Transcription factor-pathway coexpression analysis reveals cooperation between SP1 and ESR1 on dysregulating cell cycle arrest in non-hyperdiploid multiple myeloma. Leukemia. 2014 Apr;28(4):894-903.
28. Wang L, Wei D, Huang S, Peng Z, Le X, Wu TT, et al. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clinical Cancer Research. 2003 Dec 15;9(17):6371-80.
29. Jiang NY, Woda BA, Banner BF, Whalen GF, Dresser KA, Lu D. Sp1, a new biomarker that identifies a subset of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiology and Prevention Biomarkers. 2008 Jul 1;17(7):1648-52.
30. Lin RK, Wu CY, Chang JW, Juan LJ, Hsu HS, Chen CY, et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Research. 2010 Jul 15;70(14):5807-17.
31. Zhang Y, Chen HX, Zhou SY, Wang SX, Zheng K, Xu DD, et al. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Molecular Cancer. 2015 Dec;14(1):56.
32. Chen XL, Cai YH, Liu Q, Pan LL, Shi SL, Liu XL, et al. ETS 1 and SP 1 drive DHX 15 expression in acute lymphoblastic leukaemia. Journal of Cellular and Molecular Medicine. 2018 May;22(5):2612-21.
33. Vagapova ER, Lebedev TD, Tikhonova AD, Goikhman BV, Ivanenko KA, Spirin PV, et al. High Expression Level of SP1, CSF1R, and PAK1 Correlates with Sensitivity of Leukemia Cells to the Antibiotic Mithramycin. Molecular Biology. 2020 May;54(3):522-8.
34. Choi ES, Nam JS, Jung JY, Cho NP, Cho SD. Modulation of specificity protein 1 by mithramycin A as a novel therapeutic strategy for cervical cancer. Scientific Reports. 2014 Nov 24;4(1):1-8.
35. Kayser S, Levis MJ. Advances in targeted therapy for acute myeloid leukaemia. British Journal of Haematology. 2018 Feb;180(4):484-500.
36. Jones SW, Christison R, Bundell K, Voyce CJ, Brockbank SM, Newham P, et al. Characterisation of cell-penetrating peptide-mediated peptide delivery. British Journal of Pharmacology. 2005 Aug 29;145(8):1093-102.
37. Ding Y, Sun Z, Tong Z, Zhang S, Min J, Xu Q, et al. Tumor microenvironment-responsive multifunctional peptide coated ultrasmall gold nanoparticles and their application in cancer radiotherapy. Theranostics. 2020;10(12):5195-208.
38. Kanazawa T, Taki H, Okada H. Nose-to-brain drug delivery system with ligand/cell-penetrating peptide-modified polymeric nano-micelles for intracerebral gliomas. European Journal of Pharmaceutics and Biopharmaceutics. 2020 Jul 1;152:85-94.
39. Glinsky GV, Godugu K, Sudha T, Rajabi M, Chittur SV, Hercbergs AA, et al. Effects of Anticancer Agent P-bi-TAT on Gene Expression Link the Integrin Thyroid Hormone Receptor to Expression of Stemness and Energy Metabolism Genes in Cancer Cells. Metabolites. 2022 Apr 4;12(4):325.
40. Sun Y, Li X, Zhang L, Liu X, Jiang B, Long Z, et al. Cell permeable NBD peptide-modified liposomes by hyaluronic acid coating for the synergistic targeted therapy of metastatic inflammatory breast cancer. Molecular Pharmaceutics. 2019 Jan 22;16(3):1140-55.
41. Bielefeld-Sevigny M. AlphaLISA immunoassay platform—the “no-wash” high-throughput alternative to ELISA. Assay and Drug Development Technologies. 2009 Feb 1;7(1):90-2.
42. Xiong Y, Wu Y, Luo S, Gao Y, Xiong Y, Chen D, et al. Development of a novel immunoassay to detect interactions with the transactivation domain of p53: application to screening of new drugs. Scientific Reports. 2017 Aug 23;7(1):9185.
43. Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Current Opinion in Structural Biology. 2001 Dec 1;11(6):725-32.